

Abstract
Mutant alleles at the dilute unconventional myosin heavy chain locus cause diluted coat color, opisthotonic seizures, and death. The dilute coat color phenotype is caused by irregular clumping of pigment in the hair, but amounts of melanin are unchanged from wild-type controls. The melanocyte phenotype has been described as adendritic, since hair bulb and Harderian gland melanocytes appear to be rounded in tissue sections. These observations do not exclude the possibility that the processes lack pigment, since the melanocyte shape was judged by the distribution of melanin. We have tested this hypothesis by culturing primary melanocytes from dilute mutant and wild-type mice. The mutant melanocytes do not lack processes; instead, they exhibit a concentrated perinuclear distribution of melanosomes, while wild-type melanocytes have a very uniform cytoplasmic distribution of melanosomes. Electron micrographs show no detectable differences in melanosome morphology or maturation between dilute and wild-type melanocytes. Immunofluorescence experiments indicate that the dilute protein is concentrated in regions of the cytoplasm that contain melanosomes. These experiments show that the dilute myosin is necessary for the localization of melanosomes, either by active transport or tethering.
Keywords: myosin, actin, transport, lysosome
Many mouse coat color mutations have pleiotropic effects (1). All mutant alleles of the dilute locus cause a dilution in the coat color of homozygotes. Null dilute mutations cause a neurological phenotype consisting of opisthotonus and death by three weeks of age (2), in addition to the coat color dilution phenotype. Mice homozygous for dilute-lethal mutations have no early gross morphological abnormalities in the nervous system that would help to direct anatomical or physiological studies (J.A.M., N. Copeland, and N. Jenkins, unpublished results). Therefore, we are concentrating on the melanocyte system to study dilute myosin function on a cellular level.
Physiologically, dilute mice have normal levels of melanin but show a lightening of their coat color due to the clumping and irregular distribution of melanin in the hair shaft, decreasing light absorption (3). Melanocytes produce melanin within melanosomes, specialized membrane-bound organelles (MBOs). Melanocytes are derived from the neural crest, migrate to the dermis, and form dendritic processes that extend to the hair bulb. The melanosomes are transported through the processes into the growing hair shaft. Previous studies have described dilute melanocytes as adendritic and that this loss of dendrites results in an inefficient transport of melanosomes (4). This interpretation relied on examination of melanin distribution in sections of Harderian gland and hair bulbs. Since the melanocytes were characterized as adendritic by the distribution of their melanosomes, these data do not exclude the possibility that dilute melanocytes do form processes but that melanosome localization is altered.
Many unconventional myosins have been shown to have roles in vesicle transport, actin organization, and cell motility (5). Several hypotheses can be advanced to explain the original observations of an adendritic melanocyte morphology: (i) dilute function is required for the formation or maintenance of dendritic processes (the historical explanation); (ii) dilute function is only required at an early stage of melanosome maturation, and further maturation is required for distribution throughout the cell; (iii) dilute function is required for transport of melanosomes throughout the cell; or (iv) melanosome transport is accomplished by other means, and dilute merely acts as a tether after transport has occurred.
In this report, we present data that argue strongly against hypotheses i and ii (process formation and melanosome maturation) listed above and establish that dilute function is necessary for localization of melanosomes within the cytoplasm, consistent with hypotheses iii and iv (transport or tethering) above.
MATERIALS AND METHODS
Mice.
The dilute mutant mice (C57BL/6J dvse/dvse and C57BL/6J dvse/dl20J) were a gift from Nancy Jenkins and Neal Copeland (National Cancer Institute–Frederick Cancer Research and Development Center, Frederick, MD). Mutant dilute suppressor (a/a dv/dv dsu/dsu) and leaden (C57BL/6J fz ln/ln) mice were purchased from the Jackson Laboratory.
Tissue Culture.
Primary melanocytes were cultured from 1- to 3-day-old pups by the basic method of Boissy et al. (6). Pups were euthanized and rinsed in 70% ethanol and sterile phosphate-buffered saline (PBS). The dorsal skin was removed aseptically, rinsed in sterile PBS, and placed in 3 ml of 0.05% trypsin-EDTA (GIBCO/BRL). The skin was shaken in an incubator at 37°C for 4 hr. The epidermis was peeled away from the dermis and the dermis was shredded with a scalpel followed by vortex mixing for 3–5 min. The cell suspension was plated in MGM-2 medium (Clonetics, San Diego). Cell cultures were maintained at 37°C in a humidified 5% CO2/95% air atmosphere.
Thin-Section Electron Microscopy.
Primary melanocytes were fixed by a 15-min incubation in 3% glutaraldehyde. Fixed melanocytes were dehydrated through an alcohol series, embedded in Epon, and sectioned (500 nm). The sections were stained in 2% uranyl acetate and lead citrate. Electron micrographs were produced with a Jeol 120 CX electron microscope.
Immunofluorescence.
Primary melanocytes from dilute (C57BL/6J dvse/dvse), dilute suppressor (a/a dv/dv dsu/dsu), leaden (C57BL/6J fz ln/ln), and wild-type (C57BL/6J) mice were grown on glass coverslips, rinsed in PBS, fixed in methanol for 5 min at −20°C, and rinsed three times in PBS. The fixed cells were incubated with a dilution of primary antibody or antiserum in PBS for 1 hr, rinsed three times in PBS, incubated with a 1:200 dilution of tetramethylrhodamine isothiocyanate-labeled goat anti-rabbit IgG (Jackson ImmunoResearch) in PBS for 1 hr, rinsed three times in PBS, and mounted for examination using Fluoromount-G (Southern Biotechnology Associates). The primary dilute antiserum (1:500 or 1:1000 dilution) was raised in rabbits against a peptide from the dilute tail domain (DLMEQLEKQDKTVRK; amino acids 1411–1425, GenBank accession no. X57377X57377) synthesized as a multiple antigenic peptide (Research Genetics, Huntsville, AL). On Western blots, this antibody recognizes a single higher molecular weight (approximately 200 kDa) band in extracts from B16 melanoma cells, plus several lower molecular weight bands. Only the lower bands are seen with control extracts from S91 (dilute mutant) melanoma cells (data not shown). Antibody against the carboxyl-terminal region of tyrosinase (αPEP7) (7) was a gift from Vincent J. Hearing (National Institutes of Health) and was used at a 1:200 dilution in PBS. Except where underexposures in printing are specifically noted, photographic exposures were done under equivalent conditions.
RESULTS
Primary Melanocyte Morphology in dilute and Wild-Type Mice.
The overall shapes of wild-type and dilute melanocytes are similar, although not identical (Fig. 1). Both spread across the substrate and form multiple dendritic processes. Primary melanocytes from dilute and wild-type mice can easily be distinguished from each other by their distributions of melanosomes. Wild-type melanocytes have a very uniform distribution of melanosomes with virtually no region of cytoplasm without melanosomes (Fig. 1A). This uniform distribution includes the dendritic processes that are formed in culture. Melanocytes from dilute mutant mice also have dendritic processes (Fig. 1B, same magnification as A). However, they have an accumulation of melanosomes in the perinuclear region with occasional pockets of melanosomes in other regions, including processes. The periphery of the cytoplasm of dilute melanocytes is nearly devoid of melanosomes; the dilute cells shown in a colony in Fig. 1B are contacting each other. There is a difference in the growth rate of dilute and wild-type melanocytes; primary cultures of dilute melanocytes double at approximately half of the rate of wild-type cells (1 week; data not shown).
Figure 1.
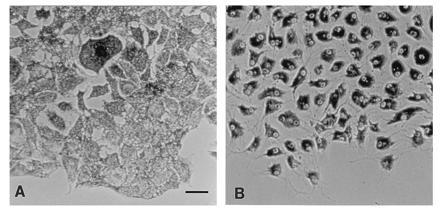
Phase-contrast micrograph of wild-type (A) and dv/dv (B) primary melanocytes, shown at the same magnification. The regions between the accumulated melanosomes in the dv/dv cells (B) contain cytoplasm lacking melanosomes. (Bar = 50 μm.)
Thin-Section Electron Microscopy of dilute and Wild-Type Melanocytes.
Four stages of melanosome maturation have been described from electron micrographic studies (8, 9). Stage 1 melanosomes are spherical vacuoles, lacking matrix filaments. Stage 2 melanosomes are elliptical in shape with well-defined matrix filaments. Stage 3 melanosomes are formed from the fusion of stage 2 melanosomes with coated Golgi-derived vesicles containing tyrosinase. Stage 3 melanosomes have deposits of dense melanin on the filaments. Stage 4 melanosomes are the mature melanosome and are completely electron-dense. Numerous markers are common to both melanosomes and lysosomes including LAMP-1, LAMP-2, acid phosphatase, and other lysosomal hydrolases (10–12) (LAMP is lysosome-associated membrane protein). It has been hypothesized that proteins phagocytosed into the lysosomal pathway may be the source of the melanin precursor tyrosine (13).
Thin-section electron micrographs demonstrate that melanosomes of all stages are contained within dilute melanocytes; a representative electron micrograph of a dv/dv melanocyte is shown in Fig. 2. The electron micrographs also were analyzed by Raymond Boissy (University of Cincinnati), who agreed with our conclusions (personal communication). These results indicate that the biogenesis of dilute melanosomes is proceeding normally despite their abnormal distribution. Comparison of the perinuclear regions of dilute and wild-type cells suggests that melanosomes accumulate in dilute cells, causing an increase in the volume of the cell where melanosomes are present (data not shown).
Figure 2.
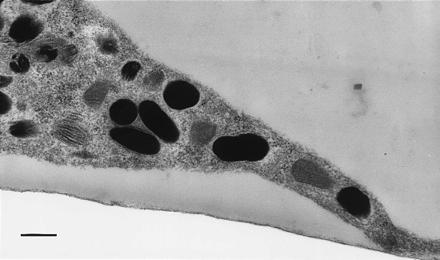
Thin-section electron micrograph of dv/dv melanocyte showing the presence of Stage 2, 3, and 4 melanosomes. (Bar = 500 nm.)
Immunofluorescence Localization of the dilute Myosin.
Immunofluorescence studies using a polyclonal antiserum against a peptide from the dilute tail domain show a punctate labeling pattern throughout the cytoplasm of wild-type melanocytes (Fig. 3 B and C). Preimmune serum and secondary antibody controls were negative (data not shown). There is no indication that the anti-dilute antiserum is reacting with the entire surface of melanosomes; immunofluorescence localization of melanosomes using an antibody to the carboxyl terminus of tyrosinase (αPEP7) (7) is different from the dilute localization (Fig. 3C), since it shows melanosomes as ring-like structures (Fig. 3D, arrowheads). The ring-like appearance is enhanced by the presence of melanin in the more mature melanosomes. Ring-like fluorescence also was observed when antibodies that recognize two lysosomal markers, LAMP-1 and LAMP-2, were used (data not shown).
Figure 3.
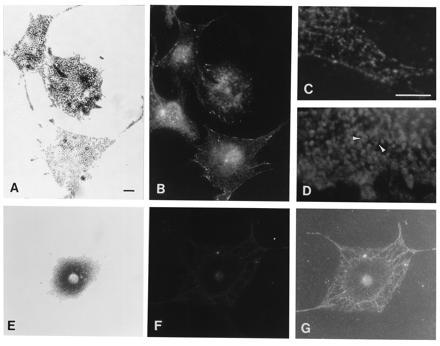
Bright-field (showing melanosomal distribution) (A and E) and immunofluorescence (B–D, F, and G) photomicrographs of wild-type (A–D) and dv/dv (E–G) primary melanocytes. Antiserum to a dilute tail peptide was used as the primary antibody in all panels except D, for which an antibody to the carboxyl terminus of tyrosinase (αPEP7) was used. G is an underexposure of the same negative shown in F; it shows the background labeling and the extent of the cell’s cytoplasm. The cell at the middle of the left margin of B is a fibroblast that cannot be seen in A. (Bars: A, 10 μm; C, 5 μM. C and D have the same magnification; the remaining panels have the same magnification as A.) The difference between the punctate staining of the anti-dilute antibody and the ring-like staining of the melanosome periphery by αPEP7 (arrowheads) can be seen in at higher magnification in C and D.
When the negative genetic control (melanocytes from dilute mutant mice) is used, very little reactivity is observed (Fig. 3 E–G). The background cytoskeletal labeling and the extent of the melanosome-free cytoplasm can be observed in an underexposure of the same negative (Fig. 3G). It is unlikely that the antibody is cross-reacting with the second member of the dilute/myosin-V family in vertebrates, myr 6 (14), since only 6 of the 14 residues of the antigenic peptide are shared between dilute and myr 6.
Since it is possible that fluorescence may be present in the dilute mutant melanocytes but is quenched by the perinuclear accumulation of melanosomes, we performed a second negative control. Melanocytes from dilute suppressor (a/a dv/dv dsu/dsu) mice (15) lack the punctate staining observed in wild-type melanocytes (Fig. 4 B and C), despite their more normal distribution of melanosomes (Fig. 4A) that nearly restores wild-type coat color in animals mutant at the dilute, leaden, or ashen loci (16). As a positive genetic control, we have performed immunofluorescence on melanocytes from leaden (ln/ln) mutant mice, whose coat color and melanocyte phenotypes are similar to those of dilute mutant mice (17). The ln/ln melanocytes exhibit perinuclear fluorescence with the dilute antiserum, coincident with their melanosome distribution (Fig. 4 D–F). The “fried egg” morphology of leaden and dilute melanocytes can be seen in Fig. 4F, in which the melanosome-rich perinuclear region is in sharp focus, while the flat dendritic region is out of the focal plane.
Figure 4.
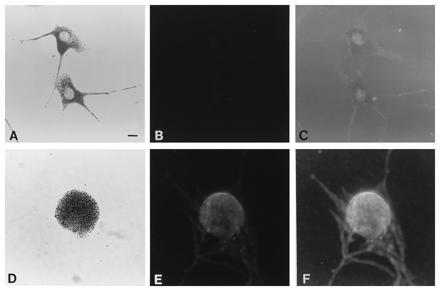
Bright-field (showing melanosomal distribution) (A and D) and immunofluorescence (B–F) photomicrographs of dilute suppressor (a/a dv/dv dsu/dsu) (A–C) and leaden (C57BL/6J fz ln/ln) (D–F) primary melanocytes, using the anti-dilute tail peptide antiserum as primary antibody. C and F are underexposures of the same negatives shown in B and E, respectively, to show the extent of the cytoplasm of the cells. (Bar = 10 μm.)
Antibodies against bacterially expressed tail fragments of both the likely chicken ortholog of dilute, chicken brain myosin-V (p190; provided by Roy Larson, University of Sao Paulo), and dilute labeled melanosomes weakly (data not shown). However, these antibodies reacted with melanosomes of dilute mutant melanocytes to a greater extent (although significantly less than to wild-type melanosomes) than did the anti-peptide antibody shown in Figs. 3 and 4. The difference may be due to the presence of other members of the dilute/myosin-V family in melanocytes, since the myosin-V and dilute tail antibodies were made to the entire protein or half of the protein, respectively, while our anti-peptide antibody was made to a sequence that is not well conserved between dilute and a new member of the dilute/myosin-V family, myr 6 (14). In support of this possibility, we have observed myr 6 transcripts in B16 melanoma cells by Northern blot analysis (unpublished results). Finally, it is possible that the truncated dilute transcripts found in dv/dv skin and melanoma cells (18) may produce a truncated protein that is recognized by the anti-dilute tail and myosin-V antisera but not by the antipeptide antiserum (since the peptide is from a region 3′ of the truncation).
DISCUSSION
We have observed a striking difference in the distribution of melanosomes in dilute and wild-type melanocytes. The melanosomes of dilute melanocytes are concentrated in the perinuclear region, while the melanosomes of wild-type melanocytes are evenly distributed throughout the cytoplasm and into dendrites. Dendritic processes are formed by dilute melanocytes in culture but in the rare cases where melanosomes are present, they are in small clumps. Electron micrographs show that melanosome biogenesis is not affected in dilute melanocytes, supporting previous studies (19) and arguing against a role for dilute in melanosome maturation (hypothesis ii, Introduction).
A localization of the dilute myosin to regions containing melanosomes by immunofluorescence, while not definitive, argues strongly for the transport and tethering (hypotheses iii or iv) and against the morphology (hypothesis i) hypotheses stated in the Introduction. An intriguing aspect of the immunolocalization data is the difference observed between the dilute and melanosomal or lysosomal markers: The dilute immunofluorescence is extremely punctate (Fig. 3C), while melanosomal and lysosomal markers have a ring-like appearance (Fig. 3D).
We advance three hypotheses for the difference in localization between dilute and melanosomal/lysosomal markers: the first, and most likely, is that our anti-peptide antibody only recognizes a subset of the dilute molecules present in a particular conformation, such as those that are engaged with local actin filaments; our unpublished results with other anti-dilute and myosin-V antibodies showing labeling of entire melanosomes are consistent with this hypothesis. The second hypothesis is that the dilute myosin is localized to a particular point on the surface of the melanosome, such as its point of interaction with actin filaments; and the third is that dilute interacts with a different MBO that is closely associated with melanosomes. The results from our collection of genetic controls (which give us confidence that we are actually seeing the localization of dilute) support the first hypothesis, since dilute immunoreactivity is always associated with melanosomes, even when they are mislocalized by another mutation (ln).
The peptide used to raise our antibody to dilute is from a region predicted to be in an α-helical coiled-coil region in chicken brain myosin-V (p190) by Espreafico et al. (20). However, the dilute tail-region splicing pattern in melanocytes differs from that in the brain (18), and insertion of the melanocyte-specific exons lowers the predicted probability that the region containing the peptide is in the coiled-coil conformation significantly (data not shown). In summary, our antibody has not been shown to recognize the entire distribution of the dilute myosin within the cell; however, our genetic controls show that the immunofluorescence represents the dilute myosin and no other. More stringent tests of these hypotheses will be provided by comparing fixatives and by immunoelectron microscopy experiments.
It is difficult to compare our data with the dilute/myosin-V immunolocalization data produced by other laboratories, since the experiments have been performed with different cell types and antibodies. The Mooseker laboratory has observed dendritic neuronal staining and colocalization with wheat germ agglutinin and actin (20, 21). Immunofluorescence with a monoclonal antibody raised against the human dilute ortholog (22) showed an unexpected colocalization with intermediate filaments. These studies did not use the genetic controls provided by the dilute system and, therefore, may be recognizing myr 6 (14) or other undiscovered members of the dilute/myosin-V family.
To transport melanosomes, dilute must interact with melanosomes and be a functional motor. An interaction between dilute and melanosomes is supported by our observation of colocalization of dilute with melanosomes by immunofluorescence. Biochemical studies with the likely chicken ortholog of dilute, myosin-V (p190), demonstrate that these myosins have an actin-activated ATPase activity characteristic of other myosins (21, 23, 24). Other evidence for the reliance of mammalian melanosome distribution on myosins comes from studies in avian melanocytes, in which disruption of actin filaments by cytochalasin B treatment causes a redistribution of melanosomes to the perinuclear region (25). The presence of dilute, an unconventional myosin, could explain the reliance of melanosome dispersion on coherent actin filaments. To discriminate between the transport and tethering hypotheses (hypotheses iii and iv) listed above, we are studying the disruption and recovery of melanosome distribution in the presence and absence of inhibitors of actin, myosin, and microtubule function.
We predict that dilute interacts with the melanosome through its tail domain. An emerging paradigm in the myosin field is that the tail domain, which exhibits extreme sequence divergence among myosin families, dictates cargo specificity and therefore function (5). The tail of dilute contains at least one α-helical coiled-coil domain and a globular domain (26). The coiled-coil domain allows dimerization of the heavy chains as seen in rotary-shadowed electron micrographs of chicken brain myosin-V (24). Interaction between dilute and melanosomes would most likely occur through the two globular domains, juxtaposed at the carboxyl terminus. The dilute sequence lacks the motifs that have been shown to directly bind phospholipids in the myosin-I class (27), suggesting that there is an intermediary protein or proteins through which dilute interacts with melanosomes.
We hypothesize that the function of the dilute myosin in neurons is the transport or localization of lysosomes or their derivatives, given the close relationship between the two types of organelles (11) based on phagocytic studies (13), the presence of lysosomal markers on melanosomes (10–12), and the lysosomal abnormalities found in some mouse pigmentation mutants (28). A fourth coat color mouse mutation that causes a lightening of coat color, beige (bg), a mouse model of Chediak–Higashi syndrome (29), also affects lysosomes. Melanocytes from bg/bg mice have a diluted coat color caused by multiple fusion events between premelanosomes. Within other cell types, large granular vesicles are formed by fusion of lysosomes (30). Aged bg/bg mice exhibit a neurological phenotype with a loss of cerebellar Purkinje cells (31), demonstrating a further correlation between the alterations of melanosome function and neurological phenotypes.
The neurological phenotype of dilute is subtle, considering that dilute is expressed in neurons in virtually all regions of the brain and that its high prenatal expression begins more than 3 weeks before the onset of the neurological phenotype (26). With the rapid increase in the number of unconventional myosins identified, it has been hypothesized that there are overlapping functions between members of the same class (32). Such a situation suggests that the loss of an individual myosin may take a long time to manifest itself phenotypically. If that is the case, then the presence of myr 6, the second member of the dilute/myosin V family (14), may be responsible for the delayed onset of the dilute neurological phenotype. We are targeting the myr 6 gene to produce mice mutant at both the myr6 and dilute loci to directly test this hypothesis.
Acknowledgments
We thank Chris McGregor, Alexander Zhadanov, Bill Crain, George Carlson, and Doug Coffin for reviewing the manuscript. This work was supported by National Institutes of Health Grant R01 NS30848. J.A.M. is an Established Investigator of the American Heart Association. M.W. was supported by the Summer Undergraduate Research Program at the University of Texas Southwestern Medical Center.
Footnotes
Abbreviation: LAMP, lysosome-associated membrane protein.
References
- 1.Silvers W K. The Coat Colors of Mice. New York: Springer; 1979. [Google Scholar]
- 2.Searle A G. Heredity. 1952;6:395–401. [Google Scholar]
- 3.Russell E S. Genetics. 1949;34:146–166. doi: 10.1093/genetics/34.2.146. [DOI] [PubMed] [Google Scholar]
- 4.Markert C L, Silvers W K. Genetics. 1956;41:429–450. doi: 10.1093/genetics/41.3.429. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Mooseker M S, Cheney R E. Annu Rev Cell Dev Biol. 1995;11:633–675. doi: 10.1146/annurev.cb.11.110195.003221. [DOI] [PubMed] [Google Scholar]
- 6.Boissy R E, Beato K E, Nordlund J J. Am J Pathol. 1991;138:1511–1525. [PMC free article] [PubMed] [Google Scholar]
- 7.Aroca P, Urabe K, Kobayashi T, Tsukamoto K, Hearing V J. J Biol Chem. 1993;268:25650–25655. [PubMed] [Google Scholar]
- 8.Seiji M, Shimao K, Birbeck M S C, Fitzpatrick T B. Ann NY Acad Sci. 1963;100:497–533. [PubMed] [Google Scholar]
- 9.Novikoff A B, Albala A, Biempica L. J Histochem Cytochem. 1968;16:299–319. doi: 10.1177/16.5.299. [DOI] [PubMed] [Google Scholar]
- 10.Hollyfield J G, Ward A. Invest Ophthalmol. 1974;13:1016–1023. [PubMed] [Google Scholar]
- 11.Novikoff A B, Neuenberger P M, Novikoff P M, Quintana N. Lab Invest. 1979;40:155–165. [PubMed] [Google Scholar]
- 12.Orlow S J, Boissy R E, Moran D J, Pifko-Hirst S. J Invest Dermatol. 1993;100:55–64. doi: 10.1111/1523-1747.ep12354138. [DOI] [PubMed] [Google Scholar]
- 13.Schraermeyer U. Pigm Cell Res. 1995;8:209–214. doi: 10.1111/j.1600-0749.1995.tb00665.x. [DOI] [PubMed] [Google Scholar]
- 14.Zhao L-P, Koslovsky J S, Reinhard J, Bähler M, Witt A E, Provance D W, Mercer J A. Proc Natl Acad Sci USA. 1996;93:10826–10831. doi: 10.1073/pnas.93.20.10826. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Sweet H O. J Hered. 1983;74:305–306. doi: 10.1093/oxfordjournals.jhered.a109794. [DOI] [PubMed] [Google Scholar]
- 16.Moore K J, Swing D A, Rinchik E M, Mucenski M L, Buchberg A M, Copeland N G, Jenkins N A. Genetics. 1988;119:933–941. doi: 10.1093/genetics/119.4.933. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Murray J M. Am Nat. 1933;67:278–283. [Google Scholar]
- 18.Seperack P K, Mercer J A, Strobel M C, Copeland N G, Jenkins N A. EMBO J. 1995;14:2326–2332. doi: 10.1002/j.1460-2075.1995.tb07227.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Hearing V J, Phillips P, Lutzner M A. J Ultrastruct Res. 1973;43:88–106. doi: 10.1016/s0022-5320(73)90072-5. [DOI] [PubMed] [Google Scholar]
- 20.Espreafico E M, Cheney R E, Matteoli M, Nascimento A A, De Camilli P V, Larson R E, Mooseker M S. J Cell Biol. 1992;119:1541–1557. doi: 10.1083/jcb.119.6.1541. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Espindola F S, Espreafico E M, Coelho M V, Martins A R, Costa F R, Mooseker M S, Larson R E. J Cell Biol. 1992;118:359–368. doi: 10.1083/jcb.118.2.359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Engle L J, Kennett R H. Genomics. 1994;19:407–416. doi: 10.1006/geno.1994.1088. [DOI] [PubMed] [Google Scholar]
- 23.Larson R E, Espindola F S, Espreafico E M. J Neurochem. 1990;54:1288–1294. doi: 10.1111/j.1471-4159.1990.tb01961.x. [DOI] [PubMed] [Google Scholar]
- 24.Cheney R E, O’Shea M K, Heuser J E, Coelho M V, Wolenski J S, Espreafico E M, Forscher P, Larson R E, Mooseker M S. Cell. 1993;75:13–23. doi: 10.1016/S0092-8674(05)80080-7. [DOI] [PubMed] [Google Scholar]
- 25.Mayerson P L, Brumbaugh J A. J Cell Sci. 1981;51:25–51. doi: 10.1242/jcs.51.1.25. [DOI] [PubMed] [Google Scholar]
- 26.Mercer J A, Seperack P K, Strobel M C, Copeland N G, Jenkins N A. Nature (London) 1991;349:709–713. doi: 10.1038/349709a0. [DOI] [PubMed] [Google Scholar]
- 27.Doberstein S K, Pollard T D. J Cell Biol. 1992;117:1241–1249. doi: 10.1083/jcb.117.6.1241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Novak E K, Swank R T. Genetics. 1979;92:189–204. doi: 10.1093/genetics/92.1.189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Lutzner M A, Lowrie C T, Jordan H W. J Hered. 1967;58:299–300. doi: 10.1093/oxfordjournals.jhered.a107620. [DOI] [PubMed] [Google Scholar]
- 30.Willingham M C, Spicer S S, Vincent R A. Exp Cell Res. 1981;136:157–168. doi: 10.1016/0014-4827(81)90047-1. [DOI] [PubMed] [Google Scholar]
- 31.Murphy E D, Roths J B. Jackson Lab Ann Rep. 1978;49:108–109. [Google Scholar]
- 32.Bement W M, Hasson T, Wirth J A, Cheney R E, Mooseker M S. Proc Natl Acad Sci USA. 1994;91:6549–6553. doi: 10.1073/pnas.91.14.6549. [DOI] [PMC free article] [PubMed] [Google Scholar]