

Abstract
Expression of BAX, without another death stimulus, proved sufficient to induce a common pathway of apoptosis. This included the activation of interleukin 1β-converting enzyme (ICE)-like proteases with cleavage of the endogenous substrates poly(ADP ribose) polymerase and D4-GDI (GDP dissociation inhibitor for the rho family), as well as the fluorogenic peptide acetyl-Asp-Glu-Val-Asp-aminotrifluoromethylcoumarin (DEVD-AFC). The inhibitor benzyloxycarbonyl-Val-Ala-Asp-fluoromethyl ketone (zVAD-fmk) successfully blocked this protease activity and prevented FAS-induced death but not BAX-induced death. Blocking ICE-like protease activity prevented the cleavage of nuclear and cytosolic substrates and the DNA degradation that followed BAX induction. However, the fall in mitochondrial membrane potential, production of reactive oxygen species, cytoplasmic vacuolation, and plasma membrane permeability that are downstream of BAX still occurred. Thus, BAX-induced alterations in mitochondrial function and subsequent cell death do not apparently require the known ICE-like proteases.
Keywords: apoptosis, cysteine protease, FAS, mitochondria, reactive oxygen species
Bax, a Bcl-2 family member, functions as a death agonist within a common apoptotic pathway (1). BAX forms homodimers and also heterodimerizes with death antagonists, BCL-2 and BCL-XL (1, 2). The ratio of BCL-2 family death agonists to antagonists dictates the susceptibility of cells to an apoptotic stimulus (3). When BAX is in excess, multiple death stimuli including withdrawal of survival factors, γ-irradiation, and dexamethasone result in apoptosis. Bax-deficient mice confirmed this role displaying an excess of lymphocytes, granulosa cells, spermatogonia, and select neurons that also demonstrate marked resistance to neurotrophic factor deprivation (4, 5). Important questions remained as to whether the BAX/BCL-2 ratio represents a passive checkpoint requiring an additional apoptotic signal or whether BAX itself could initiate death, and if so, how.
A family of interleukin 1β-converting enzyme (ICE)-like cysteine proteases homologous to the ICE are clearly activated in apoptosis and appear required for certain aspects of cell death (6). The ICE homolog in Caenorhabditis elegans ced-3, is required for cell death in that extra cells accumulate in ced-3 mutants. In mammals the number of ICE-like proteases has expanded and includes CPP32/APOPAIN/YAMA, MCH2, MCH3/LAP-3, ICErel-III, ICH-1/NEDD-2, and ICH-2/TX/ICErel-II (6, 7). The activated ICE-like proteases have a unique recognition site cleaving at a novel P1 aspartic acid (8, 9). The subset of proteases most similar to the original ICE prefers Tyr-Val-Ala-Asp (YVAD), while the CPP32 subset prefers Asp-Glu-Val-Asp (DEVD). Substrates of ICE-like proteases include poly(ADP ribose) polymerase (PARP), D4-GDI (GDP dissociation inhibitor for the rho family), sterol regulatory element-binding proteins SREBP-1 and SREBP-2, GAS2, 70-kDa component of U1 small nuclear ribonucleoprotein, catalytic subunit of DNA protein kinase, and protein kinase Cδ (10). Some of the targeted proteins may prove to be death substrates that upon cleavage ensure the inevitability of death. In mammals, FAS-induced death appears to require ICE-like proteases, since protease inhibitors can prevent this cell death (11). Whether other pathways of apoptosis in mammalian cells absolutely require ICE-like proteases to induce death is less certain.
MATERIALS AND METHODS
Inducible Expression System.
Jurkat cells (clone E6-1, American Type Culture Collection) were cultured in medium RPMI 1640 supplemented with 10% fetal calf serum (FCS) and penicillin/streptomycin (100 units/ml). To establish the Jurkat-rtTA-Bax cell line, Jurkat cells were first transfected with rtTA transactivator plasmid (PUHD172-1). Stable transfectants (referred to Jt-1) were screened and selected as described (12). Murine Bax cDNA was cloned into the EcoRI site of pUHD10-3 and cotransfected into Jt-1 with PGK-Hygromycin B. For induction of BAX protein, doxycyline (Sigma) at 1 μg/ml was added to the culture for various periods of time as indicated.
Antibodies and Immunoblot Analysis.
Anti-human Fas antibody (IgM, clone CH-11) was purchased from Upstate Biotechnology. Anti-mouse BAX polyclonal antibody (651) was made against murine BAX amino acids 43–61. Anti-CPP32 and ICH-1L antibodies were from Transduction Lab (Lexington, KY). Anti-LAP3 antibody was a gift from V. M. Dixit (University of Michigan). Anti-β-actin antibody was from Sigma. Anti-PARP antibody was a gift from N. A. Berger (Case Western Univ., Cleveland, OH). Anti-D4-GDI antibody was provided by G. M. Bokoch (Scripps Research Institute, La Jolla, CA). For Western blot analysis, cells were lysed in buffer containing 20 mM Hepes (pH 7.4), 0.25% Nonidet P-40, leupeptin at 10 μg/ml, aprotinin at 10 μg/ml, and trypsin inhibitor at 10 μg/ml. Thirty to 50 μg of protein was separated by SDS/PAGE and transferred to poly(vinylidene difluoride) membranes. The filters were first blocked with 2% milk, followed by 1-hr incubations with primary and secondary antibodies and finally developed with ECL (Amersham).
CPP32 and ICE Activity Assay.
Cells were lysed in buffer A containing 25 mM Hepes (pH 7.4), 5 mM EDTA, 2 mM DTT, and 10 μM digitonin. The lysates were clarified by centrifugation and the supernatants were used for enzyme assays. Enzymatic reactions were carried out in buffer A containing 20 μg of protein and 50 μM acetyl-Asp-Glu-Val-Asp-aminotrifluoromethylcoumarin (DEVD-AFC) or 12 μM acetyl-Tyr-Val-Ala-Asp-aminotrifluoromethylcoumarin (YVAD-AFC). The reaction mixtures were incubated at 37°C for 15 min, and the fluorescent AFC formation was measured at excitation 400 nm and emission 505 nm using a FL500 microplate fluorescence reader (Bio-Tek, Burlington, VT). Sensitivity of the enzyme assay with AFC (Enzyme Systems Products, Livermore, CA) is equivalent to aminomethylcoumarin (8) in purified systems.
Fluorescence and Electron Microscopy.
Apoptotic nuclei were visualized by H33258 staining and the DNA fragmentation was detected by TdT-mediated dUTP-cyanine-3 nick end-labeling (TdT-TUNEL) (13). For electron microscopy, 5 × 106 cells were fixed with 3% glutaraldehyde in cocadylate buffer and analyzed by transmission electron micrographs.
Mitochondrial Potential and Reactive Oxygen Species (ROS) Measurement.
For mitochondrial potential and intracellular ROS measurement, 5 × 105 cells were incubated for 15 min at 37°C with 3,3′-dihexyloxacarbocynine iodide [DiOC6(3), 40 nM], hydroethidine (2 μM), or 2′,7′-dichlorofluorescin diacetate (DCFH-DA, 5 μM; Molecular Probes), followed by FACScan (Becton Dickinson) analysis. The extracellular ROS was measured by electron paramagnetic resonance (EPR) using 5,5-dimethyl-1-pyrolline-N-oxide as the spin-trapping agent (14, 15).
RESULTS
While Bax required an additional death stimulus to promote apoptosis in stably transfected cells, the toxicity of BAX appeared to limit its level of expression (1). To determine whether BAX was sufficient to induce apoptosis, we established a reverse tetracycline (rtTA)-inducible system in Jurkat T cells (Jt-Bax) (12). BAX was induced within 2 hr of exposure to doxycycline (Fig. 1A), which resulted in cell death with the onset of membrane permeability by 8–10 hr, when BAX was half-maximal (Fig. 1B). While slower in time course than FAS-mediated death, BAX-induced death also displayed the hallmark features of apoptosis: nuclear condensation and DNA fragmentation as measured by H33258 nuclear staining and TdT-mediated dUTP-cyanine-3 nick end-labeling [TUNEL (13)] (Fig. 1C), membrane blebbing, and cell shrinkage.
Figure 1.
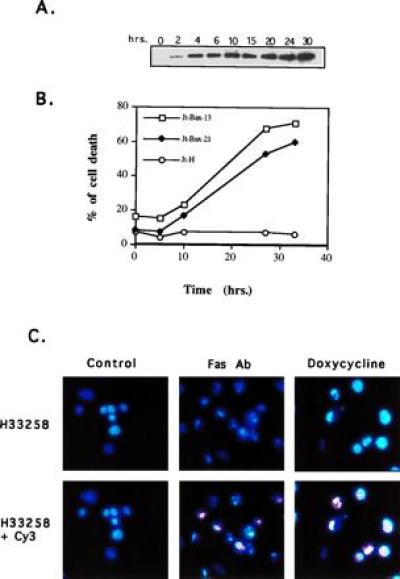
BAX-induced apoptosis in Jt-Bax cells. (A) Jt-Bax-21 cells were incubated with doxycycline (Sigma) at 1 μg/ml for various times. BAX expression was examined by immunoblot analysis with anti-BAX antibody 651. BAX levels were not substantially greater than that obtained in stable clones. (B) Cell viability was assayed by propidium iodide exclusion. Jt-Bax-13 and -21 were two independent Jt-Bax clones, while Jt-H was an empty vector containing control clone. (C) Jt-Bax-21 cells were treated with anti-FAS antibody (100 ng/ml, Upstate Biotechnology) for 4 hr or doxycycline (1 μg/ml) for 20 hr or left untreated (control). Apoptotic nuclei were visualized by H33258 or H33258 plus dUTP-cyanine-3-TdT-TUNEL (Cy3). The cells with blue or blue plus pink fragmented and condensed nuclei represent apoptotic cells.
A variety of apoptotic signals induce cell death by activating ICE-like cysteine proteases. ICE-like proteases are activated in a sequential cascade of cleavages from their inactive proforms (16, 17). To determine whether the ICE-like proteases were involved in BAX-induced apoptosis, the cleavage of specific fluorogenic peptide substrates YVAD-AFC for proteases most resembling ICE and DEVD-AFC for the CPP32-like subset was measured (Fig. 2A). Induction of BAX resulted in cleavage of DEVD, not YVAD, but in a delayed time course as compared with FAS-induced death. This enzymatic activity was blocked by pretreatment with the ICE-like protease inhibitor benzyloxycarbonyl-Val-Ala-Asp-fluoromethyl ketone (zVAD-fmk) (Fig. 2A). Western blot analysis identified CPP32 and LAP3 as participating proteases after BAX induction (Fig. 2B), while there was no change in the proenzyme levels of ICE or ICH-1L (data not shown). Activation of CPP32 and LAP3 was delayed and less complete in BAX than FAS-induced death.
Figure 2.
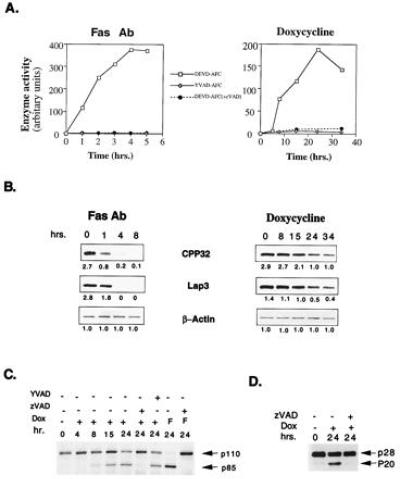
Activation of ICE-like proteases in Jt-Bax cells. (A) Jt-Bax-21 or Jt-H cells were treated with doxycycline or anti-FAS antibody in the presence (solid circle) or absence (open square and diamond) of zVAD-fmk (50 μM, Enzyme Systems Products). ICE and CPP32-like activities were measured using fluorogenic substrates YVAD-AFC and DEVD-AFC, respectively. (B) The cleavage of ICE-like proteases was examined by immunoblot analysis with the antibodies indicated and quantitated by densitometer scanning (Ultroscan XL) using β-actin as a control. Apoptotic degradation of PARP (C) or D4-GDI (D) was analyzed by immunoblot. p85, the apoptotic fragment of PARP; p20, the apoptotic fragment of D4-GDI; Dox, doxycycline; F, anti-FAS antibody.
The ICE-like proteases activated by BAX also cleaved endogenous substrates including PARP, a nuclear apoptotic landmark cleaved by several ICE-like proteases (9). Concomitant with the onset of cell death, PARP was cleaved to the expected 85-kDa apoptotic fragment (Fig. 2C). Moreover, a cytosolic substrate for the CPP32-like proteases, D4-GDI (18) was cleaved to the expected 20-kDa apoptotic fragment during BAX-induced apoptosis. Cleavage of PARP and D4-GDI was eliminated by zVAD-fmk, which blocks the enzymatic activity of the CPP32-like proteases, but not by YVAD-CMK, an ICE-specific peptide inhibitor (Fig. 2 C and D).
If the activated proteases were required for BAX to cause cell death, inhibition of the protease activity by zVAD-fmk should also block cell death, as noted for FAS-mediated apoptosis (11, 19). Surprisingly, the inhibition of ICE-like proteases only prevented DNA fragmentation, while membrane permeability cell death proceeded to approximately the same extent at the same time points (Fig. 3A). Ultrastructural analysis by electron microscopy revealed that zVAD-fmk prevented the apoptotic features of chromatin condensation, cell membrane blebbing, and cytoplasmic vacuolation in FAS-induced cell death (Fig. 3B). However, partial chromatin condensation, membrane blebbing, and dramatic cytoplasmic vacuolation still occurred in zVAD-fmk-treated cells undergoing BAX-induced death (Fig. 3B). Since zVAD-fmk, an effective inhibitor of ICE and all tested ICE-like proteases (19, 20), eliminates proteolytic activity (Fig. 2A) and substrate cleavage (Fig. 2 C and D), it is unlikely that residual ICE-like protease activity is responsible. Moreover, another effective ICE-like protease inhibitor t-butoxycarbonyl-Asp-fluoromethyl ketone (BD-fmk) also failed to prevent BAX-induced death. Other protease inhibitors, including serine protease inhibitors (3,4-DCI and l-1-tosylamido-2-phenylethyl chloromethyl ketone), cysteine protease inhibitors (calpain inhibitor I and II, E64), granzyme B inhibitor (zAAD-fmk), and proteosome inhibitor (lactacystin) all failed to prevent BAX-induced cell death (data not shown). These data suggest that BAX, unlike FAS-mediated apoptosis, may require a nonproteolytic pathway.
Figure 3.
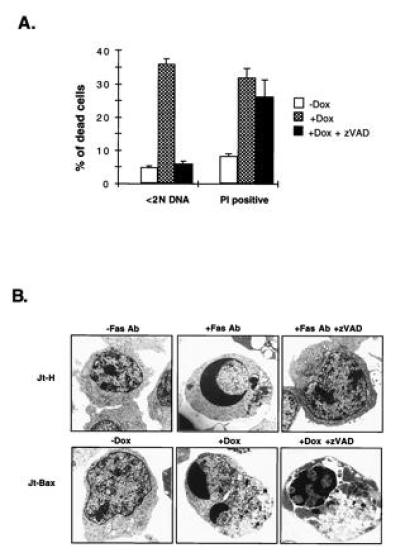
zVAD-fmk blocks DNA fragmentation but not cell death after BAX induction. (A) Jt-Bax-21 cells were incubated with doxycycline (1 μg/ml) for 24 hr in the presence or absence of zVAD-fmk (50 μM). DNA fragmentation (<2N DNA) and cell viability (propidium iodide exclusion) were measured as described (1). (B) Jt-H and Jt-Bax-21 cells were treated with either anti-FAS antibody (100 ng/ml) for 4 hr or doxycycline (1 μg/ml) for 20 hr, respectively, in the presence or absence of zVAD-fmk. The ultrastructure of the cells was analyzed by electron microscopy.
Several cell-free systems have implicated mitochondria as being necessary for apoptosis (21, 22). A fall in mitochondrial membrane potential (ΔΨm) and the production of ROS have been noted to be early events in several apoptotic systems (23). Since BAX is predominantly localized to mitochondria, we asked whether BAX might induce alteration of mitochondrial functions. A reduction in ΔΨm was noted within 6 hr of BAX induction (Fig. 4A). Production of ROS, measured by hydroethidine, which principally detects superoxide anion, followed closely (Fig. 4A). However, other ROS including hydrogen peroxide and hydroxyl radical measured by DCFH-AD were not affected by BAX (Fig. 4A) (24, 25). Moreover, no extracellular release of ROS was detected by electron paramagnetic resonance spectroscopy (EPR) using 5,5-dimethyl-pyrraline-N-oxide as a trapping reagent (14, 15). In FAS-induced apoptosis, the fall in ΔΨm and ROS production are relatively late events and are blocked by zVAD-fmk, indicating that they lie downstream of ICE-like protease activation (Fig. 4B). In contrast, the reduction in ΔΨm and production of ROS induced by BAX continued in the absence of ICE-like protease activity when zVAD-fmk was present (Fig. 4B).
Figure 4.
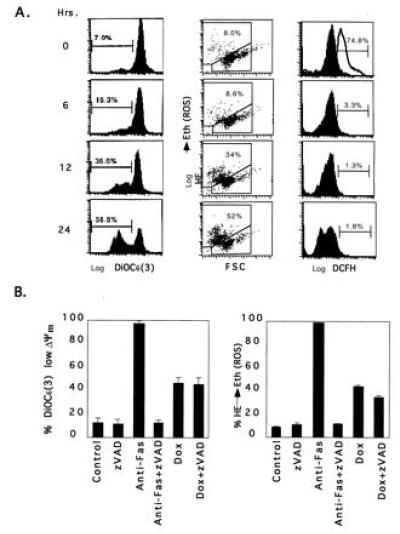
Reduction of mitochondrial membrane potential (ΔΨm) and production of ROS after BAX induction. (A) Jt-Bax-21 cells were treated with doxycycline for indicated periods of time. Aliquots of 1 × 106 cells were incubated with 50 nM 3,3′-dihexyloxacarbocynine iodide [DiOC6(3)], 2 μM hydroethidine, or 5 μM DCFH-DA and analyzed by cytofluorometry. The percentages reflect the reduction of ΔΨm [DiOC6 (3)] (Left), ROS production (hydroethidine converted to ethidum) (Middle), and production of peroxides (DCFH) (Right). Data are representative of three experiments. The open peak in the DCFH panel of 0 hour was a positive control after 10 mM H202 treatment. (B) Jt-Bax cells were induced with Dox (1 μg/ml) or anti-FAS antibody (100 ng/ml) for 24 hr in the presence or absence of 50 μM zVAD-fmk. The reduction of ΔΨm (Left) and ROS production (Right) were measured as described above.
DISCUSSION
Available evidence indicates that the BCL-2/BAX decisional point is proximal to the irreversible damage of cellular constituents. Genetic ordering in the nematode C. elegans argues that the ICE-like gene ced-3 is downstream of its BCL-2 homolog, ced-9 (26). In mammalian cells, the expression of BCL-2 blocks the activation of CPP32 (19, 27). Introduction of the baculovirus p35 gene into C. elegans inhibited CED-3 and blocked developmental cell death also arguing for an essential role of this protease in the nematode (28). However, mammalian apoptosis could prove more complex, varying with different death stimuli within different lineages. For example, FAS-induced apoptosis requires the activation of ICE-like proteases. However, this may reflect a more proximal role for the cysteine protease FLICE/MACH that is recruited into a death-inducing complex with the cytoplasmic tail of CD95 (FAS, APO-1) (29, 30). In contrast, induction of the common apoptotic pathway by the distal death agonist, BAX, activates an ICE-like protease pathway, but also a pathway apparently independent of known ICE-like proteases (Fig. 5). Elimination of ICE-like protease activity prevented DNA degradation, but this path was not required for actual cell death.
Figure 5.
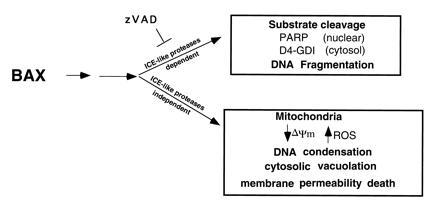
Schematic model of BAX-induced apoptosis. The ICE-like protease-dependent and -independent pathways may be parallel or sequential.
In contrast, another path apparently independent of the known ICE-like proteases was essential for cell death after BAX induction (Fig. 5). While we cannot formally exclude a role for an as yet uncharacterized member of the ICE-like family; this may not be an adequate explanation for the residual death activity. (i) BAX did induce a classic ICE-like protease pathway that was blocked by zVAD-fmk preventing cleavage of both endogenous substrates and a sensitive fluorogenic peptide reporter. Inhibition of ICE-like proteases did alter the process by interfering with the nuclear events of chromatin condensation and DNA degradation. (ii) Any undefined protease after BAX induction would vary from the known ICE-like proteases. The IC50 value for zVAD-fmk is within a reasonable range for the divergent members of this family (19), and t-butoxycarbonyl-Asp-fluoromethyl ketone also failed to block death. The substrate specificity would also be much different not recognizing either the DEVD or YVAD reporters or the endogenous PARP or D4-GDI substrates tested.
Instead, the requisite portion of the BAX death pathway that appears independent of ICE-like proteases may reside in the ongoing alterations in mitochondrial function (Figs. 4B and 5). A fall in mitochondrial membrane potential occurs early in BAX-induced death and is followed by the generation of selected ROS (Figs. 4A and 5). Whether these parameters directly participate in cell death or occur concomitant with other mitochondrial dysfunction remains to be determined. These findings suggest that the localization of BAX at mitochondrial membranes may initiate a death program involving this organelle.
Acknowledgments
We thank Dr. H. Bujard for the reverse rtTA-inducible system, Dr. K. A. Roth for assistance with TUNEL, and Dr. Tom Lin’s lab for EPR. We are also grateful to M. Pichler for secretarial assistance.
Footnotes
Abbreviations: ICE, interleukin 1β-converting enzyme; PARP, poly(ADP ribose) polymerase; zVAD-fmk, benzyloxycarbonyl-Val-Ala-Asp-fluoromethyl ketone; ΔΨm, mitochondrial membrane potential; ROS, reactive oxygen species; DEVD, acetyl-Asp-Glu-Val-Asp; YVAD, acetyl-Tyr-Val-Ala-Asp; AFC, aminotrifluoromethylcoumarin; D4-GDI, GDP dissociation inhibitor for the rho family; TUNEL, TdT-mediated dUTP-cyanine-3 nick end-labeling; DCFH-AD, 2′,7′-dichlorofluorescin diacetate;
References
- 1.Oltvai Z N, Milliman C L, Korsmeyer S J. Cell. 1993;74:609–619. doi: 10.1016/0092-8674(93)90509-o. [DOI] [PubMed] [Google Scholar]
- 2.Sedlak T W, Oltvai Z N, Yang E, Wang K, Boise L H, Thompson C B, Korsmeyer S J. Proc Natl Acad Sci USA. 1995;92:7834–7838. doi: 10.1073/pnas.92.17.7834. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Oltvai Z N, Korsmeyer S J. Cell. 1994;79:189–192. doi: 10.1016/0092-8674(94)90188-0. [DOI] [PubMed] [Google Scholar]
- 4.Knudson C M, Tung K S, Tourtellotte W G, Brown G A, Korsmeyer S J. Science. 1995;270:96–99. doi: 10.1126/science.270.5233.96. [DOI] [PubMed] [Google Scholar]
- 5.Deckwerth T L, Ellitott J L, Knudson C M, Johnson E M, Snider W D, Korsmeyer S J. Neuron. 1996;17:401–411. doi: 10.1016/s0896-6273(00)80173-7. [DOI] [PubMed] [Google Scholar]
- 6.Chinnaiyan A M, Dixit V M. Curr Biol. 1996;6:555–562. doi: 10.1016/s0960-9822(02)00541-9. [DOI] [PubMed] [Google Scholar]
- 7.Henkart P A. Immunity. 1996;4:195–201. doi: 10.1016/s1074-7613(00)80428-8. [DOI] [PubMed] [Google Scholar]
- 8.Thornberry N A, Bull H G, Calaycay J R, Chapman K T, Howard A D, et al. Nature (London) 1992;356:768–774. doi: 10.1038/356768a0. [DOI] [PubMed] [Google Scholar]
- 9.Lazebnik Y A, Kaufmann S H, Desnoyers S, Poirier G G, Earnshaw W C. Nature (London) 1994;371:346–347. doi: 10.1038/371346a0. [DOI] [PubMed] [Google Scholar]
- 10.Whyte M. Trends Cell Biol. 1996;6:245–248. doi: 10.1016/0962-8924(96)20025-x. [DOI] [PubMed] [Google Scholar]
- 11.Enari M, Hug H, Nagata S. Nature (London) 1995;375:78–81. doi: 10.1038/375078a0. [DOI] [PubMed] [Google Scholar]
- 12.Gossen M, Freundlieb S, Bender G, Muller G, Hillen W, Bujard H. Science. 1995;268:1766–1769. doi: 10.1126/science.7792603. [DOI] [PubMed] [Google Scholar]
- 13.Tornusciolo D R Z, Schmidt R E, Roth K A. BioTechnology. 1995;19:800–805. [PubMed] [Google Scholar]
- 14.Carmichael A J, Steel-Goodwin L, Gray B, Arroyo C M. Free Radical Res Commun. 1993;19:s1–s16. doi: 10.3109/10715769309056s1. [DOI] [PubMed] [Google Scholar]
- 15.Dugan L L, Sensi S L, Canzoniero L M T, Handran S D, Rothman S M, Lin T-S, Goldberg M P. J Neurosci. 1995;15:6377–6388. doi: 10.1523/JNEUROSCI.15-10-06377.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Darmon A J, Nicholson D W, Bleackley R C. Nature (London) 1995;377:446–448. doi: 10.1038/377446a0. [DOI] [PubMed] [Google Scholar]
- 17.Enari M, Talanian R V, Wong W W, Nagata S. Nature (London) 1996;380:723–726. doi: 10.1038/380723a0. [DOI] [PubMed] [Google Scholar]
- 18.Na S, Chuang T H, Cunningham A, Turi T G, Hanke J H, Bokoch G M, Danley D E. J Biol Chem. 1996;271:11209–11213. doi: 10.1074/jbc.271.19.11209. [DOI] [PubMed] [Google Scholar]
- 19.Armstrong R C, Aja T, Xiang J, Gaur S, Krebs J F, Hoang K, Bai X, Korsmeyer S J, Karanewsky D S, Fritz L C, Tomaselli K J. J Biol Chem. 1996;271:16850–16855. doi: 10.1074/jbc.271.28.16850. [DOI] [PubMed] [Google Scholar]
- 20.Fraser A, Evan G. Cell. 1996;85:781–784. doi: 10.1016/s0092-8674(00)81005-3. [DOI] [PubMed] [Google Scholar]
- 21.Zamzami N, Susin S A, Marchetti P, Hirsch T, Gomez-Monterrey I, Castedo M, Kroemer G. J Exp Med. 1996;183:1533–1544. doi: 10.1084/jem.183.4.1533. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Newmeyer D D, Farschon D M, Reed J C. Cell. 1994;79:353–364. doi: 10.1016/0092-8674(94)90203-8. [DOI] [PubMed] [Google Scholar]
- 23.Zamzami N, Marchetti P, Castedo M, Zanin C, Vayssiere J L, Petit P X, Kroemer G. J Exp Med. 1995;181:1661–1672. doi: 10.1084/jem.181.5.1661. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Rosenkranz A R, Schmaldienst S, Stuhlmeier K M, Chen W, Knapp W, Zlabinger G J. J Immunol Methods. 1992;156:39–45. doi: 10.1016/0022-1759(92)90008-h. [DOI] [PubMed] [Google Scholar]
- 25.Hockenbery D M, Oltvai Z N, Yin X M, Milliman C L, Korsmeyer S J. Cell. 1993;75:241–251. doi: 10.1016/0092-8674(93)80066-n. [DOI] [PubMed] [Google Scholar]
- 26.Shaham S, Horvitz H R. Genes Dev. 1996;10:578–591. doi: 10.1101/gad.10.5.578. [DOI] [PubMed] [Google Scholar]
- 27.Boulakia C A, Chen G, Ng F W H, Teodoro J G, Branton P E, Nicholson D W, Poirier G G, Shore G C. Oncogene. 1996;12:529–535. [PubMed] [Google Scholar]
- 28.Xue D, Horvitz H R. Nature (London) 1995;377:248–251. doi: 10.1038/377248a0. [DOI] [PubMed] [Google Scholar]
- 29.Boldin M P, Goncharov T M, Goltsev Y V, Wallach D. Cell. 1996;85:803–815. doi: 10.1016/s0092-8674(00)81265-9. [DOI] [PubMed] [Google Scholar]
- 30.Muzio M, Chinnaiyan A M, Kischkel F C, O’Rourke K, Shevchenko A, Ni J, Scaffidi C, Bretz J D, Zhang M, Gentz R, Mann M, Krammer P H, Peter M E, Dixit V M. Cell. 1996;85:817–827. doi: 10.1016/s0092-8674(00)81266-0. [DOI] [PubMed] [Google Scholar]