

Abstract
Many cytokines exert their biological effect through members of the hemopoietin receptor family. Using degenerate oligonucleotides to the common WSXWS motif, we have cloned from human hemopoietic cell cDNA libraries various forms of the receptor that was recently shown to bind the obesity hormone, leptin. mRNAs encoding long and short forms of the human leptin receptor were found to be coexpressed in a range of human and murine hemopoietic organs, and a subset of cells from these tissues bound leptin at the cell surface. Ectopic expression in murine Ba/F3 and M1 cell lines revealed that the long, but not the short, form of the leptin receptor can signal proliferation and differentiation, respectively. In cultures of murine or human marrow cells, human leptin exhibited no capacity to stimulate cell survival or proliferation, but it enhanced cytokine production and phagocytosis of Leishmania parasites by murine peritoneal macrophages. Our data provide evidence that, in addition to its role in fat regulation, leptin may also be able to regulate aspects of hemopoiesis and macrophage function.
More than 20 cytokines exert their effects by binding to receptors that are members of the hemopoietin receptor family (1), a family defined by the presence of a 200-aa extracellular domain, containing four conserved cysteine residues and the five-aa motif WSXWS (2–4). Using degenerate oligonucleotides to this motif, it has been possible to clone new members of this family including the interleukin 11 (IL-11) receptor α-chain (5) and a subunit shared by the receptors for IL-4 and IL-13 (6). In this study, we also isolated from human and murine fetal liver, bone marrow, spleen, and peripheral blood leukocytes cDNAs that appeared to encode a novel member of this receptor family, with most similarity to the leukemia inhibitory factor (LIF) receptor α-chain and gp130. Recently, identical cDNAs have been cloned based on the ability of the protein they encode to bind the hormone leptin (7). Several alternatively spliced forms of the mRNA have been described that encode leptin receptors with different cytoplasmic tails (7–10). An inability to produce leptin causes obesity in the obese (ob) mouse strain (11), while the defect in the phenotypically related diabetic (db) mouse strain has been mapped to the leptin receptor locus (10, 12). It appears that in the db mouse strain, a single nucleotide substitution creates a new splice donor site, preventing production of the mRNA that encodes the longest form of the leptin receptor (8, 10). Similarly, the obese phenotype in fatty Zucker rats appears to result from a missense mutation in the rat leptin receptor (12, 13).
Although these studies have clearly demonstrated the pivotal role that the leptin receptor plays in regulating adipose tissue mass (7, 8, 10, 12, 13), our cloning of leptin receptor cDNAs from hemopoietic tissue together with the pleiotropic actions of many cytokines prompted us to investigate the possible role of leptin in the regulation of hemopoiesis. In the present study we have examined the expression of the long and short forms of the leptin receptor in hemopoietic cells by Northern blot analyses, reverse transcriptase–PCR (RT-PCR) and 125I-labeled leptin binding combined with autoradiography. In all cases examined, both the long and short forms of the leptin receptor were coexpressed in hemopoietic populations, but only the long form was capable of signaling proliferation or differentiation when expressed in hemopoietic cell lines. Leptin was also found to activate the function of mature macrophages.
MATERIALS AND METHODS
DNA Constructs.
Using PCR, derivatives of the human leptin cDNA and the long and short cytoplasmic forms of the human leptin receptors were generated, which encoded the IL-3 signal sequence (MVLASSTTSIHTMLLLLLMLFHLGLQASIS) and an N-terminal FLAG epitope-tag (DYKDDDDK) preceding the mature coding region. The PCR products were cloned into the mammalian expression vector pEF-BOS (14). Constructs were sequenced in their entirety prior to use.
Cytokines.
Recombinant murine forms of LIF and thrombopoietin were produced in Escherichia coli and CHO cells, respectively, and purified as described (15, 16). Stem cell factor and macrophage colony-stimulating factor (M-CSF) were produced by expression of the recombinant protein and subsequent purification in Pichia pastoris and Sacchromyces cerevisiae, respectively. Recombinant murine IL-6 was a kind gift from Richard Simpson (Joint Protein Structure Laboratory, Melbourne, Victoria, Australia). Recombinant murine Flk-ligand was produced in COS cells and purified using affinity chromatography. Purified recombinant murine IL-3 was purchased from Pepro Tech (Rocky Hill, NJ), recombinant murine granulocyte-macrophage colony-stimulating factor (GM-CSF) from Schering, and recombinant human granulocyte colony-stimulating factor (G-CSF) and human erythropoietin from Amgen Biologicals. Human leptin was produced as a FLAG-tagged fusion protein in CHO cells, and the monomeric form was purified to apparent homogeneity using an anti-FLAG (M2) affinity column according to the manufacturer’s instructions (Eastman Kodak), followed by gel filtration chromatography.
Radioiodination of Leptin and Binding Studies.
Purified human leptin was radioiodinated using a modification of the iodine monochloride method (17), as described (18). Binding studies and autoradiography were carried out essentially as described (18, 19).
Northern Blot Analyses and RT-PCR.
Northern blot analyses were performed as described (20). RT-PCR was performed on cDNA generated from total RNA isolated using the TRIzol extraction method as instructed by the manufacturer (GIBCO/BRL). To amplify the long and short forms of the leptin receptor, a common forward primer (5′-GAAGGAGTGGGAAAACCAAAG-3′) was used in separate reactions with specific reverse primers: 5′-CATAGGTTACCTCAGTACCCTC-3′ for the long form of the leptin receptor, and 5′-CCACCATATGTTAACTCTCAG-3′ for the short form of the leptin receptor. Control reactions were performed using primers specific for actin. Thirty cycles of PCR were performed at 94°C for 30 sec, 60°C for 30 sec, and 72°C for 1 min. PCR products were resolved by agarose gel electrophoresis, blotted onto a nylon membrane (GeneScreen Plus, NEN), and hybridized with 32P-end-labeled oligonucleotides specific for the long form (5′-CTGTGGTCTCTCTACTTTCAAC-3′) or the short form (5′-CTAATCATGATCACTACAGATG-3′) of the leptin receptor. Autoradiographs were then exposed for 4 hr.
Transfection of Hemopoietic Cell Lines.
M1 and Ba/F3 cells (21, 22) were transfected by electroporation. Briefly, cells were washed twice in ice-cold PBS and resuspended in cold PBS at 5 × 106 per ml. Cells (4 × 106) were transferred into 0.4-mm electroporation cuvettes (Bio-Rad) with 2 μg of the linearized selectable marker plasmid pPGKpuro (a kind gift of Suzanne Cory, The Walter and Eliza Hall Institute for Medical Research, Victoria, Australia) and 20 μg of the linearized leptin receptor expression vectors. DNA and cells were then electroporated at 270 V and 960 μF in a Bio-Rad Gene-Pulser, mixed with 1 ml of culture medium, centrifuged through 3 ml of fetal calf serum (FCS), and resuspended in 100 ml of culture medium also containing, for Ba/F3 cells, 10 ng/ml IL-3. Cells were then dispensed into two 48-well dishes. After 2 days, selection was commenced by the addition of 20 μg/ml puromycin (Sigma). After 10–14 days, clones of proliferating cells were tested for receptor expression using flow cytometry with an antibody directed against the FLAG epitope tag.
Ba/F3 Cell Proliferation.
The proliferation of Ba/F3 cells in response to IL-3 or human leptin was measured in Lux 60 microwell HL-A plates (Nunc). Cells were washed three times in DMEM containing 20% (vol/vol) newborn calf serum and resuspended at a concentration of 2 × 104 cells per ml in the same medium. Aliquots of 10 μl of the cell suspension were placed in the culture wells with 5 μl of serial 2-fold dilutions of purified recombinant IL-3 (1 ng/ml) or human leptin (500 ng/ml). After 2 days of incubation at 37°C in a fully humidified incubator containing 10% CO2/90% air, viable cells were counted using an inverted microscope.
M1 Cell Differentiation.
To determine the responsiveness of M1 cells, 100 parental or transfected cells were cultured in 35-mm Petri dishes containing 1 ml of DMEM with 20% (v/v) FCS, 0.3% (w/v) agar, and 0.1 ml of serial dilutions of LIF (10 ng/ml) (23) or human leptin (500 ng/ml). After 7 days of culture at 37°C in a fully humidified atmosphere of 10% CO2/90% air, colonies of M1 cells were counted and classified as differentiated if they were composed of dispersed cells or had a corona of dispersed cells around a tightly packed center.
Bone Marrow Colony Formation.
Agar cultures of the above type were prepared containing 50,000 C57BL/6 murine or human bone marrow cells (the latter was obtained with the approval of the Institute Ethics Committee) with serial dilutions of 0.1 ml of human leptin (500 ng/ml) with or without the addition of 10 ng GM-CSF, G-CSF, M-CSF, or IL-3; 100 ng SCF; 500 ng IL-6 or Flk-ligand; or 50 ng thrombopoietin or 0.2 unit of erythropoietin. Colony formation was scored after 7 days of incubation at 37°C in a fully humidified atmosphere of 10% CO2/90% air. After scoring, all cultures were fixed with 1 ml of 2.5% glutaraldehyde, stained for acetylcholinesterase, and then stained with Luxol Fast Blue and haematoxylin.
CSF Production.
Peritoneal cell populations were harvested from 2-month-old wild-type C57BL/6 or GM-CSF transgenic mice (24), washed three times by centrifugation, then cultured at 5 × 106 cells/ml in 35-mm Petri dishes containing 1 ml DMEM [10% (vol/vol) FCS] and 0.1 ml of saline, 500 ng/ml human leptin, or saline containing 0.05 ng/ml endotoxin (Difco). After incubation for 3 hr the medium was collected, Millipore filtered, then assayed in microwell cultures for GM-CSF (using FDC-P1 cells) or for G-CSF (using Ba/F3GR cells) (25).
Phagocytosis of Leishmania.
Promastigotes of the virulent cloned line V121 of Leishmania major (26) were grown to stationary phase in Schneider’s Drosophila medium supplemented with 10% FCS at 26°C. Resident peritoneal macrophages from C57BL/6 or the lipopolysaccharide-unresponsive C3H/HeJ mice were plated on glass coverslips at a concentration of 2 × 105 cells in a volume of 500 μl mouse tonicity RPMI 1640 medium with 10% FCS and left to adhere for 1 hr at 37°C. The monolayers were then infected with L. major promastigotes at a ratio of 5 parasites per cell in the presence or absence of 50 ng/ml human leptin or 0.05 ng/ml endotoxin (Difco). Infection was allowed to proceed for 1 hr, free parasites were removed by vigorous washing, and infected cells were cultured in the presence or absence of leptin or endotoxin for an additional 18 hr. Cells were washed, stained with Giemsa, and the percentage of infected cells and number of intracellular amastigotes were counted. A minimum of 300 cells were counted in each duplicate sample.
RESULTS
During screens of cDNA libraries from human fetal liver, bone marrow, spleen, and peripheral blood using degenerate oligonucleotides to the WSXWS motif, we isolated clones encoding several forms of a receptor-like polypeptide that appeared most similar to the LIF receptor α-chain and gp130. The cDNAs were identical in sequence to the recently cloned long (Ob-Rb) and short (Ob-Ra) forms of the leptin receptor (7, 10).
Northern blot analyses of mRNA from a variety of murine tissues using probe sequences present in both the long and short leptin receptor forms revealed a 5.8-kb transcript in most tissues, as well as variable expression of smaller mRNA species of heterogeneous size. Notably, a transcript of ≈3.5 kb was particularly prominent in the placenta (Fig. 1A). In hemopoietic tissues, expression was observed in the fetal liver, bone marrow, and spleen (Fig. 1A). To examine specifically expression of the long (Ob-Rb) and short (Ob-Ra) leptin receptor transcripts in human hemopoietic cells, we used a RT-PCR strategy that distinguished these mRNAs. Transcripts encoding the long and short forms were found to be coexpressed in human fetal liver, bone marrow, spleen, and CD34+ progenitor cells and in two human hemopoietic cell lines (Meg01 and HEL) (Fig. 1B).
Figure 1.
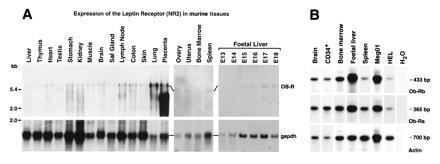
Expression of mRNA for long and short forms of the leptin receptor in hemopoietic cells. (A) Northern blot analysis of leptin receptor (Ob-R) and glyceraldehyde-3-phosphate dehydrogenase (gapdh) expression in mRNA from various murine tissues. (B) RT-PCR analysis of leptin receptor RNA from various human tissues. PCR was performed on cDNA generated from the indicated tissues using primers specific for the long form of the leptin receptor, Ob-Rb (Top), the short form of the leptin receptor, Ob-Ra (Middle), or actin (Bottom).
To determine which hemopoietic cells express cell-surface leptin receptors, autoradiography was carried out after incubation with 125I-human leptin. With murine bone marrow cells, low levels of labeling of blast cells, immature granulocytes, and monocytes and mature monocytes were observed. Some lymphocyte-like cells were labeled, and a subset of these (comprising <1% of marrow cells) exhibited intense labeling. Selective labeling of C57BL/6 peritoneal macrophages and high levels of labeling of peritoneal macrophages from GM-CSF transgenic mice were observed (Table 1). No labeled cells were observed in the thymus, spleen, or lymph nodes. Labeling of marrow cells and transgenic macrophages was blocked by an excess of unlabeled leptin. However, the significance of the labeling of normal peritoneal macrophages is unclear because competition by unlabeled leptin was incomplete.
Table 1.
Labeling of murine hemopoietic cells by 125I-labeled human leptin
| Cell type | % labeled | Grain count of labeled cells |
|---|---|---|
| C57BL/6 bone marrow | ||
| Blasts | 63 | 9.1 ± 6.6 |
| Promyelocytes/myelocytes | 72 | 10.8 ± 7.9 |
| Metamyelocytes/polymorphs | 15 | 1.6 ± 0.8 |
| Promonocytes | 66 | 5.7 ± 4.2 |
| Monocytes | 34 | 5.9 ± 6.1 |
| Eosinophils | 0 | — |
| Lymphocytes | 7 | 14.9 ± 30.9 |
| Nucleated erythroid cells | 5 | 1.5 ± 0.7 |
| C57BL/6 peritoneal cells | ||
| Macrophages | 40 | 7.3 ± 6.4 |
| Lymphocytes | 0 | — |
| Eosinophils | 0 | — |
| Mast cells | 0 | — |
| GM-CSF transgenic cells | ||
| Macrophages | 87 | 26.0 ± 28.0 |
Mean grain counts ± SD after subtraction of background grain counts. Labeling of cells was blocked by coincubation with a 100-fold excess of unlabeled leptin except that of C57BL/6 peritoneal macrophages where labeling was reduced by only 30%.
To assess the capacity of the long and short forms of the leptin receptors to signal in hemopoietic cells, cDNAs encoding these human molecules were transfected separately into the murine cytokine-responsive cell lines Ba/F3 and M1. Cell surface expression of the receptor was confirmed by fluorescence-activated cell sorter analysis of the epitope-tag incorporated into the N terminus of each form of the leptin receptor. Ba/F3 cells are dependent on IL-3 for their survival and proliferation, while M1 cells are autonomous myeloid leukemic cells that can differentiate into macrophages when stimulated by LIF or IL-6. Neither parental cell line responded to leptin. However, expression of the long form, but not the short form, of the leptin receptor conferred responsiveness on Ba/F3 cells to proliferative stimulation by human leptin (Fig. 2A). Similarly, expression of the long form of the leptin receptor in M1 cells allowed human leptin to induce morphological differentiation and clonal suppression in cultures of M1 cells (Fig. 2B). M1 cells that expressed the short receptor form failed to respond to leptin. For both cell types, leptin exerted a half-maximal effect at between 1 and 10 ng/ml, similar to the concentration found in human serum (27, 28).
Figure 2.
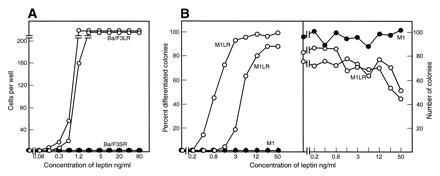
Capacity of the long and short forms of the leptin receptor to signal proliferation and differentiation. (A) Two hundred Ba/F3 cells expressing either the long (○) or short (•) form of the human leptin receptor were stimulated by the indicated concentrations of human leptin and cell counts performed after 48 hr of incubation. (B) Human leptin had no action on parental M1 cells (•), but in cultures of two clones of M1 cells expressing long leptin receptors (○), human leptin induced differentiation and suppressed the numbers of colonies developing.
Human leptin was unable to stimulate clonal proliferation in cultures of C57BL/6 or GM-CSF transgenic murine marrow or peritoneal cells or human bone marrow cells and did not enhance colony formation stimulated by GM-CSF, G-CSF, M-CSF, IL-3, IL-6, Flk-ligand, erythropoietin, stem cell factor, or thrombopoietin. Similarly, in cultures of C57BL/6 marrow cells, human leptin did not support the survival of progenitor cells when the addition of IL-3 was progressively delayed (data not shown).
To determine whether leptin may influence function of mature hemopoietic cells, its effect on cytokine production and phagocytosis by macrophages was investigated. Although some variability was encountered, human leptin appeared able to stimulate peritoneal cell populations from C57BL/6 or GM-CSF transgenic mice to produce higher levels of GM-CSF and G-CSF (Fig. 3). The leptin preparation was found to contain 0.05 ng/ml endotoxin, but the addition of endotoxin at this concentration did not elevate CSF production above that in control cultures.
Figure 3.
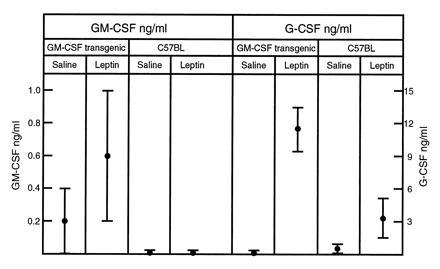
Capacity of human leptin to enhance cytokine production by primary macrophages. In 3-hr cultures of 5 × 106 cells/ml of C57BL/6 or GM-CSF transgenic peritoneal cells, addition of 50 ng of human leptin stimulated the production of GM-CSF and G-CSF. Mean data from three experiments using triplicate cultures ± SD (note different scales).
We further addressed the biological effect of leptin on macrophage function by examining phagocytosis of L. major, an obligatory intracellular pathogen of mononuclear phagocytes. In this system, leptin-treated peritoneal macrophages displayed higher numbers of attached promastigotes after 1 hr of infection compared with untreated controls (Fig. 4). Similarly, a significantly higher number of intracellular amastigotes were observed in the infected leptin-treated macrophages after overnight incubation compared with controls (Fig. 4). The effect of leptin on parasite phagocytosis was not due to the presence of endotoxin, since the addition of endotoxin at a concentration similar to that detected in the leptin preparation had no effect on parasite uptake. Moreover, a similar level of increased phagocytosis was observed in macrophages from C3H/HeJ mice that do not respond to lipopolysaccharide (29) (Fig. 4). Similar results were obtained in three additional experiments, although the level of phagocytosis varied between experiments from spectacular enhancement to levels similar to that described here.
Figure 4.
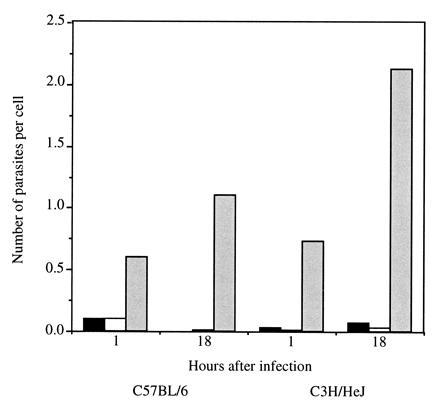
Phagocytosis of L. major promastigotes by leptin-treated macrophages. Resident peritoneal macrophages from C57BL/6 or C3H/HeJ mice were infected with promastigotes for 1 hr or 18 hr in the presence of leptin (░⃞), endotoxin (□), or medium alone (▪). The number of parasites associated with the cells were counted in Giemsa-stained preparations.
DISCUSSION
Many cytokines, originally isolated on the basis of particular biological actions, have subsequently been shown to be capable of stimulating a variety of biological responses in a wide spectrum of cell types. Examples of cytokines with extreme functional pleiotropy are LIF and IL-6 (30, 31). Leptin was discovered on the basis of a very specific biological action: the ability to regulate adipose tissue mass (11). Consistent with such an action, receptors for leptin have been found in the hypothalamus (7), the brain center responsible for satiety. Receptors for leptin have, however, also been found elsewhere—notably at high levels in the choroid plexus, where they have been postulated to aid transport of leptin from the serum to the cerebrospinal fluid (7), and in the lungs and kidneys (7, 9), where, some have suggested, they act to clear excess leptin (32). Because the leptin receptor gene is also transcribed by tissues that are not classically involved in cytokine clearance, such as the bone marrow, ovaries, and prostate (7, 9, 10) (Fig. 1), and leptin receptors were found to be abundant on the surface of macrophages (Table 1), we have explored whether leptin might act also to influence various aspects of the biology of hemopoietic cells.
mRNA encoding several forms of the leptin receptor, which differ in their predicted cytoplasmic domains, have been described (7–10). Previous studies have demonstrated that alternative splicing results in distinct leptin receptor transcripts encoding an identical extracellular domain and a variety of cytoplasmic domains (7–10). The receptor with the longest cytoplasmic domain (OB-Rb), which is absent in db mice, shares sequence similarity with the corresponding regions of the LIF receptor α-chain and gp130 and has been postulated to be the form necessary for transmitting the satiety signal in the hypothalamus (7, 8, 10). We have compared the expression and signaling capacity of long and short forms of the leptin receptor in hemopoietic cells and tissue. In all tissues examined, whether of adult or fetal origin, and including primitive CD34+ positive progenitor cells, both forms of the leptin receptor were coexpressed. These experiments do not, however, allow us to determine whether both forms of the receptor are expressed in a single cell.
In transfection experiments, the long form of the receptor, which has a cytoplasmic tail similar in sequence to the LIF receptor α-chain and gp130, was capable of signaling for cell survival and proliferation in Ba/F3 cells and the differentiation of M1 cells into macrophages. The short form of the receptor, which contains the conserved box 1, but not box 2, motifs of the hemopoietin receptor family, was inactive in these assays. These experiments indicate that leptin has the ability to initiate responses in hemopoietic cells engineered to express inserted full-length leptin receptors, and raise the possibility that hemopoietic cells naturally expressing such leptin receptors might similarly respond to leptin. Because the long form of the receptor (Ob-Rb) is not produced in db mice (8, 10), our data concerning functional differences in the signal transduction capacity of long and short forms of the leptin receptor add experimental support to the view that defects observed in db mice arise because of the absence of leptin receptor signal transduction (7, 8, 10, 33). In preliminary experiments we have also found that both the long and short forms of the leptin receptor were capable of internalizing cell surface-bound leptin and targeting it for intracellular degradation (N.A.N., unpublished data) consistent with the possibility that the short receptor form, which is expressed in the liver and kidney, may play a primary role in the clearance of leptin from the circulation.
Although low numbers of leptin receptors were demonstrated on early granulocytes and monocytes and higher numbers on a subset of lymphocyte-like cells that could represent stem or progenitor cells, leptin was not able to stimulate the clonal proliferation of such cells in semisolid cultures or to enhance responses to other cytokines. With mature peritoneal macrophages, however, leptin did appear to be able to enhance the production of cytokines and to increase the attachment and subsequent receptor-mediated process of phagocytosis of the obligatory intracellular parasite L. major (34). This activity could be mediated by an up-regulation of macrophage receptors for the parasite, among them CR1 and CR3, or by increased phagocytic activity. A major remaining question is whether leptin promotes parasite survival or death in treated macrophages.
These functional experiments suggest that leptin may have actions on tissues other than the hypothalamus and suggest that further exploration of the actions of leptin on various hemopoietic populations is warranted.
Acknowledgments
The excellent technical assistance of Bronwyn Roberts, Maria Harrison-Smith, Naomi Sprigg, Sandra Mifsud, and Ladina DiRago is gratefully acknowledged. We would like to thank Drs. M. Hasegawa and H. Nomura of Chugai Pharamceutical Company’s Institute of Molecular Medicine in Tsukuba, Japan, and Dr. James Goding and Murrat Erciyas of the Department of Pathology and Immunology, Monash Medical School, Prahran, Victoria, Australia, for their kind gift of leptin cDNAs. Dr. Goding is also thanked for his helpful discussions and for critically reading this manuscript. Lou Fabri, Aanastasia Tsigos, Rosa De Fazio, Julianna Chang, and Xu Ping of AMRAD Operations Pty. Ltd., Richmond, Victoria, Australia, are thanked for their help with production and purification of leptin. This work was supported by the Anti-Cancer Council of Victoria, Melbourne, Australia; the National Health and Medical Research Council, Canberra, Australia; the J. D. and L. Harris Trust; National Institutes of Health Grant CA-22556; and the Australian Federal Government Cooperative Research Centres Program. D.J.H. was supported by a Queen Elizabeth II Postdoctoral Fellowship from the Australian Research Council.
Footnotes
Abbreviations: IL, interleukin; LIF, leukemia inhibitory factor; ob, obese; db, diabetic; M-CSF, macrophage colony-stimulating factor; GM-CSF, granulocyte-macrophage colony-stimulating factor; G-CSF, granulocyte colony-stimulating factor; FCS, fetal calf serum.
References
- 1.Kishimoto T, Taga T, Akira S. Cell. 1994;76:253–262. doi: 10.1016/0092-8674(94)90333-6. [DOI] [PubMed] [Google Scholar]
- 2.Gearing D P, King J A, Gough N M, Nicola N A. EMBO J. 1989;8:3667–3676. doi: 10.1002/j.1460-2075.1989.tb08541.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Bazan J F. Proc Natl Acad Sci USA. 1990;87:6934–6938. doi: 10.1073/pnas.87.18.6934. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Cosman D, Lyman S D, Idzerda R L, Beckmann M P, Park L S, Goodwin R G, March C J. Trends Biochem Sci. 1990;15:265–270. doi: 10.1016/0968-0004(90)90051-c. [DOI] [PubMed] [Google Scholar]
- 5.Hilton D J, Hilton A A, Raicevic A A, Rakar S, Harrison-Smith M, Gough N M, Metcalf D, Nicola N A, Willson T A. EMBO J. 1994;13:4765–4775. doi: 10.1002/j.1460-2075.1994.tb06802.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Hilton D J, Zhang J G, Metcalf D, Alexander W S, Nicola N A, Willson T A. Proc Natl Acad Sci USA. 1996;93:497–501. doi: 10.1073/pnas.93.1.497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Tartaglia L A, Dembski M, Weng X, Deng N, Culpepper J, Devos R, Richards G J, Campfield L A, Clark F T, Deeds J, Muir C, Sanker S, Moriarty A, Moore K J, Smutko J S, Mays G G, Woolf E A, Monroe C A, Tepper R I. Cell. 1994;83:1263–1271. doi: 10.1016/0092-8674(95)90151-5. [DOI] [PubMed] [Google Scholar]
- 8.Chen H, Charlat O, Tartaglia L A, Woolf E A, Weng X, Ellis S J, Lakey N D, Culpepper J, Moore K J, Breitbart R E, Duyk G M, Tepper R I, Morgenstern J P. Cell. 1996;84:491–495. doi: 10.1016/s0092-8674(00)81294-5. [DOI] [PubMed] [Google Scholar]
- 9.Cioffi J A, Shafer A W, Zupancic T J, Smith-Grur J, Mikhail A, Platika D, Snodgrass R. Nat Med. 1996;2:585–589. doi: 10.1038/nm0596-585. [DOI] [PubMed] [Google Scholar]
- 10.Lee G-H, Proenca R, Montez J M, Carroll K M, Darvishzadeh J G, Lee J I, Friedman J M. Nature (London) 1996;379:632–635. doi: 10.1038/379632a0. [DOI] [PubMed] [Google Scholar]
- 11.Zhang Y, Proenca R, Maffei M, Barone M, Leopold L, Friedman J M. Nature (London) 1994;372:425–431. doi: 10.1038/372425a0. [DOI] [PubMed] [Google Scholar]
- 12.Chua S C, Chung W K, Wu-Peng S X, Zhang Y, Liu S-M, Tartaglia L, Leibel R L. Science. 1996;271:994–996. doi: 10.1126/science.271.5251.994. [DOI] [PubMed] [Google Scholar]
- 13.Phillips M S, Liu Q, Hammond H A, Dugan V, Hey P, Caskey C T, Hess J F. Nat Genet. 1996;13:18–19. doi: 10.1038/ng0596-18. [DOI] [PubMed] [Google Scholar]
- 14.Mizushima S, Nagata S. Nucleic Acids Res. 1990;18:5322. doi: 10.1093/nar/18.17.5322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Gearing D P, Nicola N A, Metcalf D, Foote S, Willson T A, Gough N M, Williams R L. Bio/Technology. 1989;7:1157–1161. [Google Scholar]
- 16.Alexander W S, Roberts A W, Nicola N A, Li R, Metcalf D. Blood. 1996;87:2162–2170. [PubMed] [Google Scholar]
- 17.Contreras M A, Bale W F, Spar I L. Methods Enzymol. 1983;92:277–292. [PubMed] [Google Scholar]
- 18.Hilton D J, Nicola N A. J Biol Chem. 1992;267:10238–10247. [PubMed] [Google Scholar]
- 19.Hilton D J, Nicola N A, Metcalf D. Proc Natl Acad Sci USA. 1988;85:5971–5975. doi: 10.1073/pnas.85.16.5971. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Alexander W S, Metcalf D, Dunn A R. EMBO J. 1995;14:5569–5578. doi: 10.1002/j.1460-2075.1995.tb00244.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Ichikawa Y. J Cell Physiol. 1969;74:223–234. doi: 10.1002/jcp.1040740303. [DOI] [PubMed] [Google Scholar]
- 22.Palacios R, Steinmetz M. Cell. 1985;41:727–734. doi: 10.1016/s0092-8674(85)80053-2. [DOI] [PubMed] [Google Scholar]
- 23.Hilton D J, Nicola N A, Metcalf D. Anal Biochem. 1988;173:359–367. doi: 10.1016/0003-2697(88)90200-x. [DOI] [PubMed] [Google Scholar]
- 24.Lang R A, Metcalf D, Cuthbertson R A, Lyons I, Stanley E, Kelso A, Kannourakis G, Williamson D J, Klintworth G K, Gonda T J, Dunn A R. Cell. 1987;51:675–686. doi: 10.1016/0092-8674(87)90136-x. [DOI] [PubMed] [Google Scholar]
- 25.Metcalf D, Willson T A, Hilton D J, DiRago L, Mifsud S. Leukemia (Baltimore) 1995;9:1556–1564. [PubMed] [Google Scholar]
- 26.Handman E, Hocking R E, Mitchell G F, Spithill T W. Mol Biochem Parasitol. 1983;7:111–126. doi: 10.1016/0166-6851(83)90039-7. [DOI] [PubMed] [Google Scholar]
- 27.Maffei M, Halaas J, Ravussin E, Pratley R E, Lee G H, Zhang Y, Fei H, Kim S, Lallone R, Ranganathan S, Kern P A, Friedman J M. Nat Med. 1995;1:1155–1161. doi: 10.1038/nm1195-1155. [DOI] [PubMed] [Google Scholar]
- 28.Schwartz M W, Peskind E, Raskind M, Boyko E J, Porte D., Jr Nat Med. 1996;2:589–593. doi: 10.1038/nm0596-589. [DOI] [PubMed] [Google Scholar]
- 29.Sultzer B M, Nilsson B S. Nat New Biol. 1972;240:198–200. doi: 10.1038/newbio240198a0. [DOI] [PubMed] [Google Scholar]
- 30.Hilton D J. Trends Biochem Sci. 1992;17:72–76. doi: 10.1016/0968-0004(92)90505-4. [DOI] [PubMed] [Google Scholar]
- 31.Hirano T, Akira S, Taga T, Kishimoto T. Immunol Today. 1991;11:443–449. doi: 10.1016/0167-5699(90)90173-7. [DOI] [PubMed] [Google Scholar]
- 32.Scott J. Nature (London) 1996;379:113–114. doi: 10.1038/379113a0. [DOI] [PubMed] [Google Scholar]
- 33.Barinaga M. Science. 1996;271:913. doi: 10.1126/science.271.5251.913. [DOI] [PubMed] [Google Scholar]
- 34.Mauel J. Adv Parasitol. 1996;38:1–51. doi: 10.1016/s0065-308x(08)60032-9. [DOI] [PubMed] [Google Scholar]