

Abstract
The Gfi-1 protooncogene encodes a nuclear zinc-finger protein that carries a novel repressor domain, SNAG, and functions as a position- and orientation-independent active transcriptional repressor. The Gfi-1 repressor allows interleukin 2 (IL-2)-dependent T cells to escape G1 arrest induced by IL-2 withdrawal in culture and collaborates with c-myc and pim-1 for the induction of retrovirus-induced lymphomas in animals. Here we show that overexpression of Gfi-1 also inhibits cell death induced by cultivation of IL-2-dependent T-cell lines in IL-2-deficient media. Similarly, induction of Gfi-1 in primary thymocytes from mice carrying a metal-inducible Gfi-1 transgene inhibits cell death induced by cultivation in vitro. The protein and mRNA levels of the proapoptotic regulator Bax are down-regulated by Gfi-1 in both immortalized T-cell lines and primary transgenic thymocytes. The repression is direct and depends on several Gfi-1-binding sites in the p53-inducible Bax promoter. In addition to Bax, Gfi-1 also represses Bak, another apoptosis-promoting member of the Bcl-2 gene family. Therefore, Gfi-1 may inhibit apoptosis by means of its repression of multiple proapoptotic regulators. The antiapoptotic properties of Gfi-1 provide a potential explanation for its strong collaboration with c-myc during oncogenesis.
Stimulation of naive T cells by antigen and other costimulatory signals induces within 2–5 hr the expression of interleukin 2 (IL-2) and its receptor. Expression of these molecules commits the stimulated T cells to activation. During this process IL-2 and additional cytokines, expressed sequentially, inhibit programmed cell death (PCD), and induce cellular proliferation and differentiation (1).
To study the role of IL-2 in T-cell activation and oncogenesis we employed an insertional mutagenesis genetic strategy aimed at the identification of genes that promote the progression of IL-2-dependent T-cell lines to IL-2 independence. The Gfi-1 gene, which was cloned using this strategy, encodes a zinc-finger protein with six Cys2His2 type, tandem zinc-finger motifs located at the C-terminal half of the protein. Gfi-1 expression in adult animals is restricted to the thymus, spleen, and testis. In mitogen-stimulated splenocytes, Gfi-1 expression begins to rise at 12 hr after stimulation and reaches very high levels after 50 hr, suggesting that Gfi-1 may be functionally involved in events occurring after the interaction of IL-2 with its receptor. In agreement with this, Gfi-1 does not induce the expression of IL-2 (2). The Gfi-1 protein is localized in the nucleus, binds DNA in a sequence-specific manner, and functions as a position- and orientation-independent active transcriptional repressor (3, 4). The Gfi-1-induced transcriptional repression is mediated by a 20-amino acid, N-terminal, novel repressor domain (SNAG), which is coincident with a nuclear localization motif and defines a novel family of evolutionarily conserved transcriptional repression domains (4).
In addition to promoting progression of T-cell lines to IL-2 independence, Gfi-1 has also been associated with oncogenesis. Thus, Gfi-1 is a locus of common proviral integration in T-cell tumors induced by Moloney murine leukemia virus (MoMuLV) (2), mink-cell focus-forming (MCF) virus (5), and murine acquired immunodeficiency syndrome (MAIDS) virus (6). B- and T-cell lymphomas arising in MoMuLV-inoculated Eμ/myc and Eμ/pim1 transgenic mice carry provirus insertions in the pal-1 locus, which is synonymous with Gfi-1 (7). Moreover, due to its localization to the human chromosome 1p22, it has been suggested that Gfi-1 may contribute to the induction of several human neoplasms (8).
The promotion of IL-2 independence by Gfi-1 and the oncogenic properties of this gene depend, at least in part, on its effects on cell cycle progression. Overexpression of Gfi-1 in immortalized IL-2-dependent T cells allows them to escape G1 arrest induced by IL-2 withdrawal. A single amino acid substitution in the SNAG domain (P2A), which inhibits the Gfi-1-mediated transcriptional repression, also inhibits G1 arrest induced by removal of IL-2 from the culture medium, suggesting that the latter depends on the repressor activity of the SNAG domain (4). However, IL-2 withdrawal from these IL-2-dependent cultures also induces apoptosis in addition to G1 arrest. Here we show that overexpression of Gfi-1 in an immortalized IL-2-dependent T-cell line lowers the rate of cell death induced by IL-2 withdrawal. Moreover, we show that Gfi-1 prolongs the survival of primary thymocytes explanted from transgenic mice carrying a metallothionein promoter/Gfi-1 (MT-Gfi-1) transgene. Inhibition of cell death by Gfi-1 in both primary T cells and IL-2-dependent T-cell lines grown in the absence of IL-2 correlates with the direct inhibition of Bax expression and the direct or indirect inhibition of Bak. Therefore, the promotion of IL-2 independence by Gfi-1 and its oncogenic properties are due not only to the Gfi-1-mediated stimulation of cell cycle progression but also to its inhibitory effects on apoptosis. The antiapoptotic properties of Gfi-1 provide a potential explanation for its strong collaboration with c-myc during oncogenesis.
MATERIALS AND METHODS
Cell Death Assays.
The IL-2-dependent 2780 cell line was infected with SRα (9) and SRα/Gfi-1 retroviruses, and G418-resistant cells were selected. Cells (107) from each of three SRα-infected and three SRα/Gfi-1-infected mass cultures were placed in 10 ml of IL-2 (100 units/ml)-containing RPMI 1640 medium supplemented with fetal bovine serum (10%) and penicillin (50 units/ml), streptomycin (50 μg/ml), and kanamycin (100 μg/ml). Twenty-four hours later, while the cells were growing logarithmically, they were harvested, washed twice, and subcultured in triplicate (106 cells per ml) in IL-2-deficient media. Live and dead cells from each subculture were counted daily in trypan blue. The numbers of live and dead cells were used to calculate the ratio dead/(dead + live) cells at sequential time points in each subculture. The mean value of these ratios and the SEM at 24 and 48 hr after IL-2 withdrawal were calculated. This experiment was repeated three times with similar results. RNA was isolated from aliquots of these cells at 24 hr after IL-2 withdrawal, and Northern blots were prepared as described previously (2). Finally, also at 24 hr after withdrawal of IL-2, aliquots of these cells were lysed in Nonidet P-40 (NP-40) lysis buffer (1% NP-40/20 mM Hepes, pH 7.5/440 mM NaCl/2 mM EDTA/50 μg/ml leupeptin/50 μg/ml aprotinin/1 mM phenylmethylsulfonyl fluoride) and total cell lysates were analyzed by Western blotting. The blots were probed with the Gfi-1 antiserum (4) or with the anti-Bax antiserum P-19 (Santa Cruz Biotechnology).
Single-cell suspensions of thymocytes from mice carrying a metal-inducible Gfi-1 transgene (MT-Gfi-1 transgenic mice), and age- and sex-matched nontransgenic control CD1 mice were washed three times and cultured at 37°C in RPMI 1640 medium supplemented with 10% fetal bovine serum and penicillin/streptomycin/kanamycin at 106 cells per ml. At regular intervals following exposure to ZnSO4 (50 μM), samples were harvested, resuspended in FACS buffer (3.4 mM sodium citrate/10 mM NaCl/0.1% Nonidet-P-40/75 μM ethidium bromide) (10) and analyzed by flow cytometry to determine their DNA content. The data were analyzed by using the cellquest program (Becton Dickinson).
Transgenic Mice.
The construct used to generate the transgenic mice consisted of the full-length rat Gfi-1 cDNA (2) placed under the control of the metal-inducible 1.7-kb mouse metallothionein-1 promoter (11). Following its release by restriction endonuclease digestion, the MT-Gfi-1 DNA insert was purified by gel electrophoresis followed by using a Geneclean kit (Bio 101). Fertilized mouse oocytes were obtained from superovulated BALB/c females crossed with CD-1 males and were cultured, injected, and implanted using standard procedures (12). Positive founders were identified by Southern blot or slot blot analysis of tail DNA samples.
Antibodies and Western Blot Analyses.
Gfi-1 antisera were raised in rabbits against a multiple antigen peptide (MAP), corresponding to Gfi-1 amino acid sequence 12–26 (synthesized by Research Genetics, Huntsville, AL). Reactive antisera were affinity purified by standard protocols (13) using columns of Sepharose-bound MAP peptides. Western blotting was carried out using Immobilon P membranes (Millipore). Membrane-bound proteins were detected by enhanced chemiluminescence (ECL) (Amersham).
Reporter and Expression Constructs and Chloramphenicol Acetyltransferase (CAT) Assays.
Reporter constructs were generated in the modified pCAT Basic vector (Promega) C.2 (4). To generate the C.2 vector we first mutated the plasmid pCAT Basic backbone by USE mutagenesis (14) to eliminate potential Gfi-1-binding sites. We then introduced a HindIII restriction site 3′ of the CAT gene by replacing the EcoRI fragment of pCAT Basic with the equivalent fragment from the TK-CAT plasmid pBLCAT2 (15). Subsequently, a double-stranded oligonucleotide containing an EcoRI, a BglII, and a BamHI site was cloned 5′ of the CAT gene. The Bax promoter (Fig. 3A) was amplified from clone TM-604 (kindly provided by J. Reed, Burnham Institute, La Jolla, CA) using primers generating BamHI sites at its ends, and was cloned in C.2. Overlap extension PCR was used to eliminate five Gfi-1-binding sites in the Bax promoter (AATC cores were mutated to TTTC and AAGC cores were mutated to TTGC). All cloned PCR products were sequenced to verify that additional mutations were not inadvertently introduced. Oligonucleotides required for these manipulations were synthesized by the Fox Chase DNA synthesis facility. The Gfi-1 expression construct was generated by inserting the C2B cDNA (2) into the cytomegalovirus (CMV)-based CMV5 vector (16) (CMV5-Gfi-1). The β-galactosidase expression construct [CMV(mutant B)-β-galactosidase] was generated by inserting the β-galactosidase gene into a CMV5 vector in which the two Gfi-1-binding sites in the CMV MIE promoter were mutated so that they no longer bind Gfi-1. As expected, the mutant B CMV MIE promoter is no longer responsive to Gfi-1 (3).
Figure 3.
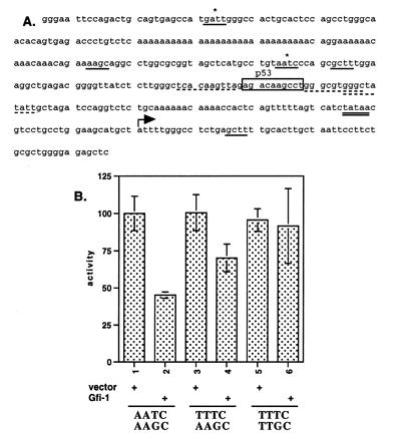
The repression of Bax by Gfi-1 is direct. (A) The Bax promoter contains several Gfi-1-binding sites with AATC (underlined and marked by asterisks) or AAGC (underlined) cores. Four overlapping p53-binding sites are shown by broken underlines (low-affinity sites) and a box (high-affinity site). The TATA box is marked by a double underline and the transcription start site is marked by an arrow (20). (B) Cotransfection of the CMV5-Gfi-1 expression construct represses the Bax promoter/CAT construct (bars 1 and 2). Mutation of the Gfi-1-binding sites containing an AATC core into TTTC reduces the Gfi-1 repression (bars 3 and 4), while mutation of the Gfi-1-binding sites containing AATC and AAGC cores into TTTC and TTGC, respectively, abrogates the Gfi-1-mediated repression (bars 5 and 6).
The Bax promoter/CAT reporter constructs (300 ng), a CMV5-Gfi-1 expression construct (10 ng), and a CMV(mutant B)-β-galactosidase expression construct (50 ng) (3) were cotransfected into NIH 3T3 cells by using Lipofectamine (GIBCO/BRL) according to the manufacturer’s protocol. Following lysis by several consecutive freeze–thaw cycles at 38 hr after transfection, the lysates were normalized for transfection efficiency according to the results of a microtiter-plate-based β-galactosidase assay (17). Lysates were analyzed for CAT activity by measuring diffusion of the acetylated form of chloramphenicol in scintillation fluid as described (3). All transfections were performed in triplicate and have been repeated at least twice with different Qiagen column preparations of plasmid DNA.
RESULTS
To determine whether forced expression of Gfi-1 affects cell viability after IL-2 withdrawal, a subline of the IL-2-dependent T-cell lymphoma line 2780 (2780a) from which Gfi-1 was originally cloned was infected with the SRα retrovirus vector or with an SRα/Gfi-1 construct. Three independent SRα/Gfi-1 infections gave rise to three independently maintained cell lines, all overexpressing Gfi-1. Cultures in logarithmic-phase growth from each of the three independent SRα- and SRα/Gfi-1-infected sublines of 2780a cells were subcultured in triplicate in IL-2-deficient media, as described in Materials and Methods. Live and dead cells were counted daily, and their numbers were used to calculate the ratio dead/(dead + live) cells in each culture. The mean value of these ratios and the SEM were calculated for the 24- and 48-hr time points (Table 1). As analyzed by a a two-sided t test, the differences in the fraction of dead cells between the SRα and SRα/Gfi-1 cultures were 12.7% versus 8.0% at 24 hr and 34.9% versus 25.6% at 48 hr, and these were shown to be statistically significant at both the 24-hr (P ≤ 0.0052) and the 48-hr (P ≤ 0.0192) time points. Therefore, the rate of cell death induced by IL-2 withdrawal was significantly lower in the cells overexpressing Gfi-1.
Table 1.
The rate of cell death induced by IL-2 withdrawal is lower in 2780a cells infected with the SRα/Gfi-1 retrovirus
| Retrovirus | % cell death
|
|
|---|---|---|
| At 24 hr | At 48 hr | |
| SRα | 12.7 ± 0.93 | 34.9 ± 1.34 |
| SRα/Gfi-1 | 8.0 ± 0.08 | 25.6 ± 1.54 |
Live and dead cells were counted at 24 and 48 hr after IL-2 withdrawal. The percentage of dead cells was calculated from the ratios dead/(dead + live) cells. The numbers show the mean (±SEM) values of nine independent measurements for each time point (three subcultures of three independently infected cultures of 2780a cells). When a two-sided t test was used, the differences in the percentage of dead cells between the SRα- and SRα/Gfi-1-infected cultures were found to be statistically significant (at 24 hr P ≤ 0.0052 and at 48 hr P ≤ 0.00192).
To determine whether the Gfi-1-mediated inhibition of cell death was limited to immortalized cell lines, we examined the survival of explanted thymocytes from a transgenic mouse line carrying a metal-inducible MT-Gfi-1 transgene. Explanted primary thymocytes from MT-Gfi-1 transgenic and nontransgenic control mice were cultured at a concentration of 106 cells per ml. Cells were harvested at regular intervals after exposure to zinc (50 μM ZnSO4) and were analyzed for DNA content by flow cytometry. Nuclei with less than diploid DNA content, detected by this analysis, identify cells undergoing apoptosis (18). DNA histograms of thymocytes from MT-Gfi-1 (mtgfi) and control (tgneg) mice were identical at the start of the experiment (Fig. 1A, 0 hr). The addition of zinc in the MT-Gfi-1 but not the control cultures, however, reduced the number of subdiploid nuclei relative to the number of nuclei with ≥2N DNA content (Fig. 1A, 72 hr + Zn). Graphic display of a representative experiment in which nuclei with ≥2N DNA content were quantitated demonstrated that transgenic zinc-induced thymocytes survived longer in culture (Fig. 1B, compare mtgfi to tgneg, + Zn at 72 hr). Therefore, explanted thymocytes forced to express Gfi-1 exhibit prolonged survival in culture.
Figure 1.
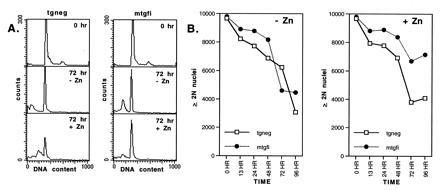
Gfi-1 induction inhibits apoptosis of explanted thymocytes from MT-Gfi-1 transgenic mice. (A) Single-cell suspensions of thymocytes from MT-Gfi-1 transgenic and control mice, cultured at a concentration of 1 × 106 per ml, were harvested at the indicated times, resuspended in FACS buffer (10), and analyzed by flow cytometry to determine their DNA content. (Top) DNA histograms of MT-Gfi-1 transgenic (mtgfi) and nontransgenic (tgneg) thymocytes demonstrate identical profiles at the start of the experiment (O hr). (Middle and Bottom) At 72 hr all samples contain nuclei with a subdiploid DNA content, a diagnostic feature of apoptosis (18). However, the subdiploid nuclei peak is reduced in the MT-Gfi-1 transgenic thymocytes cultured in the presence of zinc (72 hr, + Zn). Similar results were obtained in three separate experiments. (B) The number of nuclei with a ≥2N DNA content, detected in 10,000 flow events, was quantitated by using the cellquest program (Becton Dickinson) and plotted against time. The results show that the number of these nuclei is higher in cultures of MT-Gfi-1 thymocytes maintained in zinc-containing media. When the numbers of subdiploid nuclei were plotted against time the opposite plots were generated, demonstrating that the number of subdiploid nuclei is lower in MT-Gfi-1 thymocytes maintained in zinc-containing media. Similar results were obtained in three separate experiments.
Since Gfi-1 is a transcriptional repressor, we questioned whether it represses the expression of genes that induce apoptosis. One of these proapoptotic genes, Bax, contains multiple Gfi-1-binding sites in its promoter and therefore represents an excellent candidate for direct Gfi-1-mediated repression. To address this hypothesis, we first examined the expression of Bax mRNA and protein in IL-2-dependent 2780a cells infected with SRα (9) or SRα/Gfi-1 retroviruses, cultivated in the presence or absence of IL-2. The results showed that IL-2 withdrawal enhances Bax expression, and this enhancement is inhibited by Gfi-1 (Fig. 2A). The Gfi-1-mediated repression of the activation of Bax translates into lower levels of Bax protein in SRα/Gfi-1- as opposed to SRα-infected cells grown in the absence of IL-2 (Fig. 2C). The effects of Gfi-1 on the expression of Bax were more pronounced in subline B, which expresses the highest Gfi-1 levels (Fig. 2 A and C). In addition to its effects on Bax expression, overexpression of Gfi-1 in 2780 cells correlates with the appearance of a novel protein band (Fig. 2C) that is detected with the Bax rabbit antiserum and exhibits slower mobility than Bax. This band most likely represents post-translationally modified Bax protein, although we cannot exclude that it may represent a novel protein crossreacting with the Bax antibody.
Figure 2.
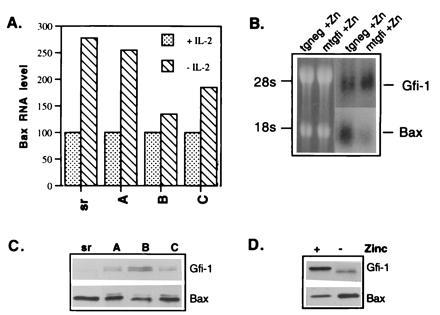
Gfi-1 represses Bax expression. (A) Gfi-1 represses the induction of Bax following IL-2 withdrawal from cultures of 2780a cells. RNA extracted from empty vector (SRα, labeled sr in the figure) or SRα/Gfi-1-infected cells (sublines A, B, and C) grown in the presence of IL-2, or 24 hr after IL-2 withdrawal, was hybridized to a murine Bax cDNA probe. The level of Bax mRNA was quantitated by using an AMBIS densitometer. The Bax mRNA in cells grown in the presence of IL-2 was normalized to 100. (B) MT-Gfi-1 (mtgfi) and nontransgenic (tgneg) control mice were provided with drinking water containing 25 mM ZnSO4 (19) for 8 days prior to sacrifice. The addition of 25 mM ZnSO4 in the drinking water is not toxic to mice (19). Total RNA extracted from the thymi of these mice was subjected to formaldehyde/gel electrophoresis and, following transfer to a nylon membrane, it was hybridized to Gfi-1 and Bax probes as shown. Photography of the ethidium bromide-stained gel was used to demonstrate equal loading of the two lanes. Quantitation of the autoradiograms by AMBIS densitometry revealed that in zinc-treated transgenic mice the level of Gfi-1 RNA is 1.32-fold higher, while Bax RNA levels are 2.1-fold lower than controls. (C) Western blots of lysates made from cells in A (24 hr after IL-2 withdrawal) were probed with antisera to Gfi-1 (4) or Bax (P-19 from Santa Cruz Biotechnology). The differences in Gfi-1 and Bax protein expression were quantitated by AMBIS densitometry, and sublines A, B, and C were shown to contain 8-, 20-, and 8.8-fold greater Gfi-1, but 0.8-, 0.65-, and 0.74-fold lower Bax levels than empty vector controls (sr). (D) Western blots of lysates from MT-Gfi-1 transgenic littermates, supplied with drinking water without added zinc (− zinc) or containing 25 mM ZnSO4 (+ zinc) for 8 days were probed with anti-Gfi-1 or anti-Bax antisera as in C.
Bax mRNA and protein expression were also examined in explanted thymocytes from MT-Gfi-1 transgenic and control mice given control or zinc-containing water for 8 days prior to sacrifice (19). The results showed that the induction of Gfi-1 in transgenic mice given the zinc-containing water correlates with Bax repression (Fig. 2 B and D). In nontransgenic mice, zinc failed to induce Gfi-1 and to repress Bax. Therefore, Bax repression was causally linked to the induction of Gfi-1.
The preceding results showed that Gfi-1 represses Bax expression. To determine whether the observed repression was direct, we examined the effects of Gfi-1 on the activity of the Bax promoter in NIH 3T3 cells. The results showed that the activity of a Bax promoter/CAT construct was down-regulated when a Gfi-1 expression construct was cotransfected (Fig. 3B, bars 1 and 2). The Bax promoter (20) contains two Gfi-1-binding sites with an AATC and three sites with an AAGC core (Fig. 3A). Mutation of the AATC- and AAGC-containing binding sites to TTTC and TTGC, respectively, abrogated the Gfi-1-mediated repression (Fig. 3B, bars 3–6). Therefore, the repression of Bax by Gfi-1 is direct.
Recent studies by others have shown that explanted thymocytes from Bax “knock-out” mice fail to survive in culture longer than control thymocytes (21). This suggests that the repression of Bax by Gfi-1 contributes to, but may not be sufficient for, the Gfi-1-induced delay in apoptosis of explanted thymocytes. This prompted us to examine the effects of Gfi-1 on the expression of other members of the Bcl-2 family of proteins. The results showed that induction of Gfi-1 down-regulates the expression of not only Bax but also Bak, while it does not alter the expression of Bcl-2 (Fig. 4). Therefore, Gfi-1 may contribute to the delay in apoptosis by altering the balance between proteins critical to the regulation of cell death.
Figure 4.
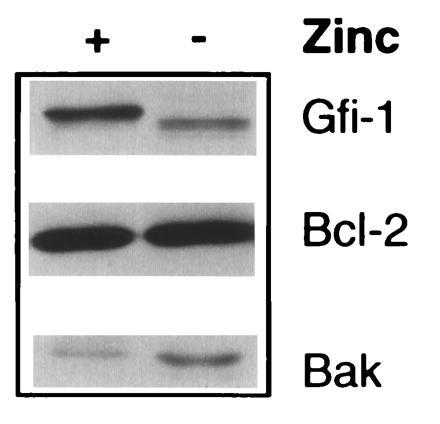
Gfi-1 also represses the expression of Bak but does not affect the expression of Bcl-2 in MT-Gfi-1 transgenic thymocytes. Western blots of lysates from thymocytes of MT-Gfi-1 transgenic mice supplied with drinking water without added zinc (−) or containing 25 mM ZnSO4 (+) were probed with antisera to Gfi-1, Bcl-2 (Santa Cruz Biotechnology N-19), or Bak (Santa Cruz Biotechnology G23).
DISCUSSION
Our earlier studies had shown that Gfi-1 allows IL-2-dependent cells to escape from G1 arrest induced by IL-2 withdrawal (4). However, cultivation of IL-2-dependent T cells in IL-2-deficient media also induces apoptotic cell death. In this report we have presented evidence showing that Gfi-1 also delays T-cell death induced by IL-2 withdrawal. In addition, we have shown that Gfi-1 not only protects T-cell lines from death induced by cultivation in IL-2-free media but also delays apoptosis of in vitro cultivated primary murine thymocytes. The effects of Gfi-1 on cell survival appear to depend on its ability to modulate the expression of genes involved in the regulation of PCD.
The regulation of PCD is a process of fundamental biological significance. Decisions underlying embryonic development, the renewal of differentiating tissues in adult animals, and the response of the immune system to antigenic signals depend in part on the execution of programs regulating cell death. In lymphoid cell populations PCD is a highly regulated process which selects cells with functional antigen receptors and eliminates cells that are autoreactive or no longer needed (reviewed in ref. 22). One family of proteins that plays a critical role in the regulation of PCD is the Bcl-2 family, which includes Bcl-2, Bcl-xL, Bcl-xS, Bax, Bak, and Bad (reviewed in ref. 23). Overexpression of the first two of these proteins inhibits, whereas overexpression of the remainder promotes, PCD. The inhibition of PCD by the former proteins is hypothesized to be regulated by means of the formation of heterodimers between these negative and the positive regulators of apoptosis, a process that sets up a PCD-regulating rheostat. When a death signal is received by the cell, if Bcl-2 or Bcl-X is in excess the cell is protected, while if Bax or Bak is in excess, PCD is activated and the cell dies by apoptosis (24). Since the role of IL-2 in protecting cells from PCD has been linked to the expression of several members of the Bcl-2 family of proteins (25), we examined whether the expression of Bax, a member of this family and a major proapoptotic regulator, is repressed in Gfi-1-expressing cells. Our results showed that the expression of Bax in thymocytes and its induction in IL-2-dependent T-cell lines after IL-2 withdrawal are indeed repressed and that the repression is due to the direct action of Gfi-1. Given that Bax induces apoptosis, its repression is expected to inhibit cell death.
The repression of Bax may be sufficient to inhibit apoptosis in IL-2-dependent T-cell lines after IL-2 withdrawal. However, since explanted thymocytes from Bax “knock-out” mice fail to survive longer than control thymocytes in culture (21), the prolonged survival of MT-Gfi-1 transgenic thymocytes is unlikely to be solely due to Bax repression. Our data confirmed that overexpression of Gfi-1 in murine thymocytes also represses the expression of Bak, another member of the Bcl-2 family of proteins, but does not affect the expression of Bcl-2. The Gfi-1-mediated inhibition of cell death in explanted murine thymocytes, therefore, is likely to be the result of readjustment of the dynamic equilibrium between the various members of this family of proteins. The lower levels of Bax and Bak shift the balance toward an excess of Bcl-2. Since Bcl-2 overexpression in thymocytes confers resistance to apoptosis and predisposes these mice to T-cell lymphomas (26, 27), Gfi-1 overexpression is likely to be equivalent to Bcl-2 overexpression.
The tumor suppressor gene p53 has been shown to down-regulate Bcl-2 and up-regulate Bax expression (28). Therefore, the effects of Gfi-1 on the expression of the members of the Bcl-2 family of proteins appear to be, at least in part, opposite to the effects of p53. Mutations in p53 represent the most commonly detected genetic change in neoplasia (reviewed in refs. 29 and 30). While such mutations are frequent in Eμ-Myc-induced lymphomas, they are rare in tumors from Eμ-Bcl-2/Eμ-Myc double transgenic mice (31), suggesting that the biological effects of Bcl-2 overexpression and of p53 mutations may overlap. Since our results suggest that the overexpression of Gfi-1 and Bcl-2 may be equivalent, we conclude that the biological outcomes of Gfi-1 overexpression and of p53 mutations may also overlap.
Oncogenicity studies using transgenic mice indicate a strong collaboration between Gfi-1 and c-myc or pim-1 (7). The collaboration of c-myc with both Gfi-1 (7) and Bcl-2 (31) and the functional relationship between Gfi-1 and Bcl-2 (this report) suggest that c-myc may collaborate with both of these genes on a similar basis. Given that c-myc promotes G1 progression but also induces apoptosis, the collaboration of Bcl-2 with c-myc appears to be due to the Bcl-2-mediated inhibition of c-myc-induced apoptosis (32). Therefore, Gfi-1 may also collaborate with c-myc by inhibiting apoptosis induced by c-myc. Thus, the antiapoptotic properties of Gfi-1 provide a potential explanation for its strong collaboration with c-myc during oncogenesis.
The collaboration between Gfi-1 and c-myc and the potential opposition of Gfi-1 to the biological effects of p53 reinforce the importance of the antiapoptotic function of Gfi-1 and suggest that Gfi-1 overexpression may contribute to the induction and progression of multiple types of neoplasms.
Acknowledgments
We thank Dr. C. McMahon for help in detecting endogenous Gfi-1, Dr. S. Litwin (Fox Chase Cancer Center) for the statistical analysis of the rate of cell death in IL-2-depleted cultures of SRα- and SRα/Gfi-1-infected 2780 cells, Dr. J. Reed (Burnham Institute) for the Bax promoter construct and for the murine Bax cDNA, and Pat Bateman for secretarial assistance. This work was supported by Public Health Service Grant CA56110 and National Cancer Institute of Canada Grant 4805. Additional support was provided by Public Health Service Grant CA06927 and by an appropriation from the Commonwealth of Pennsylvania to the Fox Chase Cancer Center. H.L.G. is funded by National Research Service Award CA59302.
Footnotes
Abbreviations: IL-2, interleukin 2; PCD, programmed cell death; CAT, chloramphenicol acetyltransferase; CMV, cytomegalovirus.
References
- 1.Crabtree G R. Science. 1989;243:355–361. doi: 10.1126/science.2783497. [DOI] [PubMed] [Google Scholar]
- 2.Gilks C B, Bear S E, Grimes H L, Tsichlis P N. Mol Cell Biol. 1993;13:1759–1768. doi: 10.1128/mcb.13.3.1759. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Zweidler-McKay P A, Grimes H L, Flubacher M M, Tsichlis P N. Mol Cell Biol. 1996;16:4024–4034. doi: 10.1128/mcb.16.8.4024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Grimes H L, Chan T O, Zweidler-McKay P A, Tong B, Tsichlis P N. Mol Cell Biol. 1996;16:6263–6272. doi: 10.1128/mcb.16.11.6263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Liao X, Buchberg A M, Jenkins N A, Copeland N G. J Virol. 1995;69:7132–7137. doi: 10.1128/jvi.69.11.7132-7137.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Morse H C, III, Hartley J W, Tang Y, Chattopadhyay S K, Giese N, Fredrickson T N. In: Viruses and Cancer. Minson A C, McRae M A, editors. Cambridge, U.K.: Cambridge Univ. Press; 1994. pp. 265–291. [Google Scholar]
- 7.Berns A, Van der Lugt N, Alkema M, Van Lohuizen M, Douen J, Acton D, Allen J, Laird P W, Jonkers J. Cold Spring Harbor Symp Quant Biol. 1994;59:435–447. doi: 10.1101/sqb.1994.059.01.049. [DOI] [PubMed] [Google Scholar]
- 8.Bell D W, Taguchi T, Jenkins N A, Gilbert D J, Copeland N G, Gilks C B, Zweidler-McKay P, Grimes H L, Tsichlis P N, Testa J R. Cytogenet Cell Genet. 1995;70:263–267. doi: 10.1159/000134048. [DOI] [PubMed] [Google Scholar]
- 9.Landau N R, Littman D R. J Virol. 1992;66:5110–5113. doi: 10.1128/jvi.66.8.5110-5113.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Sherley J L, Kelly T J. J Biol Chem. 1988;263:8350–8358. [PubMed] [Google Scholar]
- 11.Stuart G W, Searle P F, Chen H W, Brinster R L, Palmiter R D. Proc Natl Acad Sci USA. 1984;81:7318–7322. doi: 10.1073/pnas.81.23.7318. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Hogan B, Costantini F, Lacey E, editors. Manipulating the Mouse Embryo: A Laboratory Manual. Plainview, NY: Cold Spring Harbor Lab. Press; 1986. [Google Scholar]
- 13.Harlow E, Lane D, editors. Antibodies: A Laboratory Manual. Plainview, NY: Cold Spring Harbor Lab. Press; 1988. [Google Scholar]
- 14.Deng W P, Nickoloff J A. Anal Biochem. 1992;200:81–88. doi: 10.1016/0003-2697(92)90280-k. [DOI] [PubMed] [Google Scholar]
- 15.Luckow B, Schutz G. Nucleic Acids Res. 1987;15:5490. doi: 10.1093/nar/15.13.5490. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Andersson S, Davis D L, Dahlback H, Jornvall H, Russell D W. J Biol Chem. 1989;264:8222–8229. [PubMed] [Google Scholar]
- 17.Bignon C, Daniel D, Djiane J. BioTechniques. 1993;15:243–245. [PubMed] [Google Scholar]
- 18.Telford W G, King L E, Fraker P J. J Immunol Methods. 1994;172:1–16. doi: 10.1016/0022-1759(94)90373-5. [DOI] [PubMed] [Google Scholar]
- 19.Palmiter R D, Brinster R L, Hammer R E, Trumbauer M E, Rosenfeld M G, Birnberg N C, Evans R M. Nature (London) 1982;300:611–615. doi: 10.1038/300611a0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Miyashita T, Reed J C. Cell. 1995;80:293–299. doi: 10.1016/0092-8674(95)90412-3. [DOI] [PubMed] [Google Scholar]
- 21.Knudson C M, Tung K S K, Tourtellotte W G, Brown G A J, Korsmeyer S J. Science. 1995;270:96–99. doi: 10.1126/science.270.5233.96. [DOI] [PubMed] [Google Scholar]
- 22.Nuñez G, Merino R, Grillot D, Gonzalez-Garcia M. Immunol Today. 1994;15:582–588. doi: 10.1016/0167-5699(94)90221-6. [DOI] [PubMed] [Google Scholar]
- 23.White E. Genes Dev. 1996;10:1–15. doi: 10.1101/gad.10.1.1. [DOI] [PubMed] [Google Scholar]
- 24.Korsmeyer S J. Trends Genet. 1995;11:101–105. doi: 10.1016/S0168-9525(00)89010-1. [DOI] [PubMed] [Google Scholar]
- 25.Miyazaki T, Liu Z, Kawahara A, Minami Y, Yamada K, Barsoumian E L, Perlmutter R M, Taniguchi T. Cell. 1995;81:223–231. doi: 10.1016/0092-8674(95)90332-1. [DOI] [PubMed] [Google Scholar]
- 26.Linette G P, Hess J L, Sentman C L, Korsmeyer S J. Blood. 1995;86:1255–1260. [PubMed] [Google Scholar]
- 27.Sentman C L, Shutter J R, Hockenbery D, Kanagawa O, Korsmeyer S J. Cell. 1991;67:879–888. doi: 10.1016/0092-8674(91)90361-2. [DOI] [PubMed] [Google Scholar]
- 28.Miyashita T, Krajewski S, Krajewska M, Wang H G, Lin H K, Lieberman D A, Hoffman B, Reed J C. Oncogene. 1994;9:1799–1805. [PubMed] [Google Scholar]
- 29.Lee J M, Bernstein A. Cancer Metastasis Rev. 1995;14:149–161. doi: 10.1007/BF00665797. [DOI] [PubMed] [Google Scholar]
- 30.Chang F, Syrjanen S, Syrjanen K. J Clin Oncol. 1995;13:1009–1022. doi: 10.1200/JCO.1995.13.4.1009. [DOI] [PubMed] [Google Scholar]
- 31.Hsu B, Marin M C, McDonnell T J. Cancer Lett. 1995;94:17–23. doi: 10.1016/0304-3835(95)03836-l. [DOI] [PubMed] [Google Scholar]
- 32.Evan G I, Wyllie A H, Gilbert C S, Littlewood T D, Land H, Brooks M, Waters C M, Penn L Z, Hancock D C. Cell. 1992;69:119–128. doi: 10.1016/0092-8674(92)90123-t. [DOI] [PubMed] [Google Scholar]