

Abstract
A naturally-occurring β-(1→3)-d-rhamnotetraose has been constructed under conditions of sequential β -selective mannosylation controlled by the 4,6-O-[1-cyano-2-(2-iodophenyl)-ethylidene] protecting group. The route is concise, proceeding through a late stage radical deoxygenation that successfully uncovers all four deoxy subunits at once.
6-Deoxy-d-mannose, called d-rhamnose, has been found exclusively in antigenic lipopolysaccharides (LPSs) associated with the cell walls of microorganisms.1 Some examples include Xanthomonas campestris,2 Pseudomonas cepacia,1c P. syringae pv. Morspurunorum,3 P. aeruginosa IID 1008,4 P. maltophilia 555,5 Myxobacterium 402,6 and Escherichia hermanii.7 To date this novel subunit has not been encountered in humans, plants, or animals. Given the rise in antimicrobial resistance, enzymes involved in biosynthesis of the d-rhamnopyranosides would make promising targets for potentialy xenobiotic antiinfectives.
E. hermanii is a member of the family of enterobacteriaceae, related to E. coli. It has been isolated from human wounds and sputum and has demonstrated pathogenicity against humans in vivo.8 E. hermanii produces a β-lactamase and exhibits a distinctive antibiotic resistance, with resistance to penicillin, ampicillin, and carbenicillin.9 Degradation studies have resulted in characterization of a repeat (1→3)-β-d-rhamnan from the cell walls of E. hermanii strain ATCC 33651.10 The high content of the difficult β-d-rhamnosyl linkage, combined with its potential medicinal relevance make this LPS O-chain constituent an appropriate candidate for development of methods aimed at synthesis of the β-d-rhamnopyranosides.
The stereoeoselective synthesis of the 1,2-cis-equatorial glycosidic bond as is found in both the β-mannosides and the β-rhamnosides is of perennial difficulty in carbohydrate chemistry.11 Without the possibility of invoking neighboring group participation, the synthesis of such a linkage is rendered somewhat more sensitive than that of the trans-glycosidic bond. Captivated by this challenge, our group has found considerable success in employing the torsionally and electronically disarming 4,6-O-benzylidene protecting group.12 In our researches this group has been used to synthesize a variety of β-d-mannopyranosides, including the (1→2), (1→4), and alternating (1→3), (1→4)-mannans.13 However, even with this technology in hand, the challenge of the cis-glycosidic bond is considerably magnified in the biologically important rhamnopyranosides, which lack the functional arm for incorporation of a benzylidene-type directing effect.
In general, strategies for synthesis of polysaccharides containing deoxy-sugars proceed via prior synthesis of an appropriately protected deoxy subunit, followed by extensive optimization of conditions for stereoselective glycosidation; recent efforts in this vein have been frustrated by low selectivities.14 However, the reliability of the benzylidene-mediated mannosylations combined with their close structural relationship to the β-rhamnosides suggests inverting such a paradigm: synthesis of β-mannosyl linkages followed by a deoxygenation to provide the otherwise demanding subunits. This strategy is particularly attractive in the case of the β-d-rhamnopyranosides, where the starting material, d-mannose, is easily available in bulk. Thus, we have recently developed a protecting group that readily combines the stereoselectivity of benzylidene acetal with a latent radical fragmentation pathway, providing a high-yielding deoxygenation in the last stage of oligosaccharide synthesis.15 Herein we demonstrate the broadest capabilities of this method to date, with a concise total synthesis of a tetrameric fragment from the (1→3)-rhamnan of E. hermanii (ATCC 33651) via a one-pot quadruple radical fragmentation. To the best of our knowledge this is the first synthesis of a β-rhamnan (of either the d- or l-modification) and an unique example of such a multiple radical deoxygenation.16
Benzylidene protected hexopyranosides are known to undergo deoxygenation at C-6 via the NBS-mediated Hanessian-Hullar reaction.17 However, the initiation step of this reaction, radical abstraction of the benzylidene proton, has proven indiscriminate in consort with the standard host of non-participating protecting groups necessary for oligosaccharide synthesis.18 Similar incompatibilities are observed with Roberts’ thiol-catalyzed benzylidene fragmentation.19 Whereas the Hanessian-Hullar reaction likely occurs via a radical/polar crossover mechanism, Roberts’ sequence proceeds via a purely radical mechanism.20 This mechanism favors fragmentation to a primary radical at C-6 due to a conformationally less strained transition state arising from planarization at the incipient C-6 radical.21
To avoid the problematic hydrogen atom abstraction step we introduced the 4,6-O-[α-(2-(2-iodophenyl)-ethylthiocarbonyl)-benzylidene] group.22 This group enabled the synthesis of the tetrameric subunit from E hermanii (ATCC 33650 and 33652).23 However, the limited functional group compatibility of a key transesterification required to introduce the group minimized the overall scope. In subsequent work we have identified a second generation 4,6-O-[1-cyano-2-(2-iodophenyl)-ethylidene] acetal as a surrogate for the benzylidene fragmentation that is easily prepared, easily installed, and which is orthogonal to many protecting group manipulations.
The mechanism for the cyano-group transfer/fragmentation (Scheme 1) is based upon chemistry first articulated by Beckwith and later expanded by Rychnovsky.24 As is frequently the case with radical reactions propagated by tin hydrides several competing reactions are possible, including premature reduction of radicals 2, 3, and 4, making the rapid rate of cyano group migration essential for our synthesis. The challenge of synthesizing a polymeric rhamnan by this methodology can be seen as one of minimizing a possible three different by-products per monomer subunit or, for a tetramer, promoting one product in 34 = 81. With this in mind we commenced our synthesis.
Scheme 1.

Radical fragmentation mechanism.
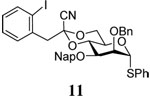
The synthesis of the (1→3)-tetrasaccharide required preparation of only one suitably protected monomer, 10. This was achieved from diol 9, which was prepared from 4,6-O-benzylidene protected thiomannoside 825 in 72% yield over 4 steps using standard reactions (Scheme 2). The 4,6-O-[1-cyano-2-(2-iodophenyl)-ethylidene] acetal was cleanly introduced as a single diastereomer via Lewis-acid promoted transcyanation chemistry developed by Utimoto and co-workers.26 Monomer 10 was then employed in sequential, linear synthesis of a β-mannotetraose. After each coupling deprotection was achieved prior to chromatographic purification of the newly synthesized polymer. Thus, a single purification provided the acceptor for each subsequent step in the elaboration of the growing polymer. All couplings resulted in high yields and high β-selectivities, as is consistent with a benzylidene-type directing effect (Table 1). The β-anomers could be readily assigned by their characteristic H-5 multiplets in the 1H NMR spectrum, with the reducing end H-5 resonance at δ ~3.3, and subsequent residues further upfield still at δ 2.6–2.9.27
Scheme 2.

Synthesis of donor 11.
Table 1.
Preparation of Fragmentation Precursor 18 by Iterative Glycosidation.a
| donor | acceptor | product | yield (selectivity) |
|---|---|---|---|
 |
C6H11OH | 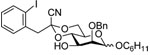 |
12 β; 13 α 94% (β: α = 6:1) |
| 11 | 12 | 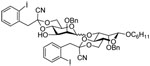 |
14 β; 15 α 78% (β: α = 10:1) |
| 11 | 14 | 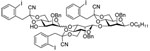 |
16 β; 17 α 79% (β: α = 8:1) |
| 11 | 16 | 18 β; 19 α 86% (β: α = 10:1) |
Reagents: 1) Ph2SO (1.5 equiv.), TTBP (3.0 equiv.), Tf2O (1.7 equiv.), CH2Cl2, −70/−20/−70 °C. 2) DDQ, CH2Cl2H2O (17:1).
The fully protected tetramer was subjected to conditions of radical fragmentation in refluxing xylenes. In the development of the protecting group, it was found that the higher boiling point of xylenes favored the fragmentation pathway over the reduction of the benzylidene radical 4. Adapting the conditions from the developmental work, a 4 h addition of tin hydride to a 0.0015M solution of substrate in xylenes at reflux, followed by NaBH4 reduction to facilitate removal of the tin residues, and then saponification allowed initial separation of the desired product cleanly on silica gel from traces of by-products arising from incomplete fragmentation. The pure tetraol, 20, was isolated in 22% yield from 18. Subsequent global deprotection with palladium hydroxide proceeded to give the tetrasaccharide in 90% yield (Scheme 3).
Scheme 3.

Radical fragmentation and deprotection of tetrasaccharide 18.
In both the 1H and 13C NMR spectra of the synthetic tetrasaccharide, resonances from the two end units can be distinguished from the compounded peaks of the internal subunits. Despite these differences, there is excellent agreement between shifts of the internal residues of the synthetic polymer and those of the natural polymer, as is illustrated in Table 2.
Table 2.
Comparison of 1H NMR chemical shifts and coupling constants of internal residues of 21 with those of the natural rhamnan.
| chemical shift (ppm) | coupling constant (Hz) | ||||
|---|---|---|---|---|---|
| H | isolateda | syntheticb | isolateda | syntheticb | Δδ |
| H-1 | 4.81 | 4.66 | s | s | −0.15 |
| H-2 | 4.26 | 4.11 | 2.7 | 3.0 | −0.15 |
| H-3 | 3.87 | 3.72 | 9.7 | 9.5 | −0.15 |
| H-4 | 3.54 | 3.38 | 9.2 | 9.5 | −0.16 |
| H-5 | 3.46 | complex | 5.9 | complex | -- |
| H-6 | 1.34 | 1.19 | -- | 6.0 | −0.15 |
Data for isolated polysaccharide from ref. 10.
Values for synthetic polysaccharide recorded in D2O at room temperature at 500 MHz
In conclusion, we have developed a concise synthetic route to a β-(1→3)-d-rhamnotetraose, in which four challenging β-glycosidic linkages are installed with a high degree of stereoselectivity due to the disarming effect of the 4,6-O-[1-cyano-2-(2-iodophenyl)-ethylidene] protecting group. The key step is a late stage radical deoxygenation, which occurs simultaneously on all four residues of the tetramer.
Supplementary Material
Full experimental details and characterization data. This material is available free of charge via the Internet at http://pubs.acs.org.
Acknowledgment
We thank the NIH (GM 57335) for financial support.
References
- 1.(a) Hirooka M, Yoshimura A, Saito I, Ikawa F, Uemoto Y, Koto S, Takabatake A, Taniguchi A, Shinoda Y, Morinaga A. Bull.Chem. Soc. Jpn. 2003;76:1409. [Google Scholar]; (b) Spitali M, Smith ARW. J. Phytopath. 2000;148:563. [Google Scholar]; (c) Knirel YA, Shashkov AS, Senchenkova SYN, Ajiki Y, Fukuoka S. Carbohydr. Res. 2002;337:1589. doi: 10.1016/s0008-6215(02)00216-1. [DOI] [PubMed] [Google Scholar]; (d) Cerantola S, Montrozier H. Eur. J. Biochem. 1997;246:360. doi: 10.1111/j.1432-1033.1997.00360.x. [DOI] [PubMed] [Google Scholar]; (e) Winn AM, Wilkinson SG. Carbohydr. Res. 1996;294:109. doi: 10.1016/s0008-6215(96)90623-0. [DOI] [PubMed] [Google Scholar]; (f) Senchenkova SN, Shashkov AS, Kecskes ML, Ahohuendo BC, Knirel YA, Rudolph K. Carbohydr. Res. 2000;329:831. doi: 10.1016/s0008-6215(00)00250-0. [DOI] [PubMed] [Google Scholar]; (g) Vinogradov EV, Campos-Portuguez S, Yokota A, Mayer H. Carbohydr. Res. 1994;261:103. doi: 10.1016/0008-6215(94)80009-x. [DOI] [PubMed] [Google Scholar]; (h) Vinogradov EV, Shashkov AS, Knirel YA, Zdorovenko GM, Solyanik LP, Gubanova NY, Yakovleva LM. Carbohydr. Res. 1991;212:307. doi: 10.1016/0008-6215(91)84071-l. [DOI] [PubMed] [Google Scholar]; (i) Molinaro A, Silipo A, Lanzetta R, Newman M-A, Dow JM, Parrilli M. Carbohydr. Res. 2003;338:277. doi: 10.1016/s0008-6215(02)00433-0. [DOI] [PubMed] [Google Scholar]; (j) Beynon LM, Bundle DR, Perry MB. Can. J. Chem. 1990;68:1456. [Google Scholar]
- 2.Hickman J, Ashwell G. J. Biol Chem. 1966;241:1424. [PubMed] [Google Scholar]
- 3.Smith ARW, Zamze SE, Munro SM, Carter KJ, Hignett RC. Eur. J. Biochem. 1985;149:73. doi: 10.1111/j.1432-1033.1985.tb08895.x. [DOI] [PubMed] [Google Scholar]
- 4.Yokota S, Kaya S, Sawada S, Kawamura T, Araki Y, Ito E. Eur. J. Biochem. 1987;167:203. doi: 10.1111/j.1432-1033.1987.tb13324.x. [DOI] [PubMed] [Google Scholar]
- 5.Di Fabio JL, Perry MB, Bundle DR. Biochem. Cell Biol. 1987;65:968. doi: 10.1139/o87-126. [DOI] [PubMed] [Google Scholar]
- 6.Morrison IM, Young R, Perry MB, Adams GA. Can. J. Chem. 1967;45:1987. [Google Scholar]
- 7.Perry MB, Bundle DR. Infect. Immun. 1990;58:1391. doi: 10.1128/iai.58.5.1391-1395.1990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Dahl KM, Barry J, DeBiasi RL. Clin. Infect. Dis. 2002;35:96. doi: 10.1086/342304. [DOI] [PubMed] [Google Scholar]
- 9.(a) Fitoussi F, Arlet G, Grimont PAD. J. Antimicrob. Chemother. 1995;36:537. doi: 10.1093/jac/36.3.537. [DOI] [PubMed] [Google Scholar]; (b) Chaudhury A, Nath G, Tikoo A, Sanyal SC. J. Diarr. Dis. Res. 1999;17:85. [PubMed] [Google Scholar]
- 10.Perry MB, Richards JC. Carbohydr. Res. 1990;205:371. doi: 10.1016/0008-6215(90)80154-u. [DOI] [PubMed] [Google Scholar]
- 11.(a) Barresi F, Hindsgaul O. In: Modern Methods in Carbohydrate Synthesis. Khan SH, O’Neill RA, editors. Amsterdam: Harwood Academic Publishers; 1996. p. 251. [Google Scholar]; b) Demchenko AV. Synlett. 2003:1225. [Google Scholar]; c) Pozsgay V. In: Carbohydrates in Chemistry and Biology. Ernst B, Hart GW, Sinay P, editors. Vol. 1. Weinheim, Germany: Wiley-VCH; 2000. p. 319. [Google Scholar]; d) Gridley JJ, Osborn HMI. J. Chem. Soc., Perkin Trans. 2000;1:1471. [Google Scholar]
- 12.(a) Crich D, Li H. J. Org. Chem. 2002;67:4640. doi: 10.1021/jo0108818. [DOI] [PubMed] [Google Scholar]; (b) Crich D, Dai Z. Tetrahedron. 1999;55:1569. [Google Scholar]; (c) Crich D, Barba GR. Tetrahedron Lett. 1998;39:9339. [Google Scholar]; (d) Crich D, de la Mora MA, Cruz R. Tetrahedron. 2002;58:35. [Google Scholar]; (e) Crich D, Banerjee A. Org. Lett. 2005;7:1395. doi: 10.1021/ol050224s. [DOI] [PubMed] [Google Scholar]; (f) Crich D, Dudkin V. J. Am. Chem. Soc. 2002;124:2263. doi: 10.1021/ja0123958. [DOI] [PubMed] [Google Scholar]; (g) Dudkin VY, Crich D. Tetrahedron Lett. 2003;44:1787. [Google Scholar]; (h) Dudkin VY, Miller JS, Danishefsky SJ. J. Am. Chem. Soc. 2004;126:736. doi: 10.1021/ja037988s. [DOI] [PubMed] [Google Scholar]; (i) Miller JS, Dudkin VY, Lyon GJ, Muir TW, Danishefsky SJ. Angew. Chem. Int. Ed. 2003;42:431. doi: 10.1002/anie.200390131. [DOI] [PubMed] [Google Scholar]; (j) Nicolaou KC, Mitchell HJ, Rodriguez RM, Fylaktakidou KC, Suzuki H, Conley SR. Chem. Eur. J. 2000;6:3149. doi: 10.1002/1521-3765(20000901)6:17<3149::aid-chem3149>3.0.co;2-l. [DOI] [PubMed] [Google Scholar]; (k) Kim KS, Kang SS, Seo YS, Kim HJ, Lee YJ, Jeong K-S. Synlett. 2003:1311. [Google Scholar]; (l) Wu X, Schmidt RR. J. Org. Chem. 2004;69:1853. doi: 10.1021/jo0354239. [DOI] [PubMed] [Google Scholar]; (m) Kwon YT, Lee YJ, Lee K, Kim KS. Org. Lett. 2004;6:3901. doi: 10.1021/ol048648u. [DOI] [PubMed] [Google Scholar]; (n) Tanaka S-I, Takashina M, Tokimoto H, Fujimoto Y, Tanaka K, Fukase K. Synlett. 2005:2325. [Google Scholar]
- 13.(a) Crich D, Banerjee A, Yao Q. J. Am. Chem. Soc. 2004;126:14930. doi: 10.1021/ja047194t. [DOI] [PubMed] [Google Scholar]; (b) Crich D, Li W, Li H. J. Am. Chem. Soc. 2004;126:15081. doi: 10.1021/ja0471931. [DOI] [PubMed] [Google Scholar]; (c) Crich D, Li H, Yao Q, Wink DJ, Sommer RD, Rheingold AL. J. Am. Chem. Soc. 2001;123:5826. doi: 10.1021/ja015985e. [DOI] [PubMed] [Google Scholar]
- 14.Bedini E, Carabellese A, Barone G, Parrilli M. J. Org. Chem. 2005;70:8064. doi: 10.1021/jo051153d. [DOI] [PubMed] [Google Scholar]
- 15.Crich D, Bowers AA. J. Org. Chem. 2006;71:3452. doi: 10.1021/jo0526688. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.For a previous example of a multiple radical fragmentation in oligosaccharide synthesis see: Crich D, Hermann F. Tetrahedron Lett. 1993;34:3385.
- 17.(a) Hanessian S, Plessas NR. J. Org. Chem. 1969;34:1035. [Google Scholar]; (b) Hanessian S, Plessas NR. J. Org. Chem. 1969;34:1045. [Google Scholar]; (c) Hanessian S, Plessas NR. J. Org. Chem. 1969;34:1053. [Google Scholar]; (d) Chana JS, Collins PM, Farnia F, Peacock DJ. J. Chem. Soc., Chem. Commun. 1988;2:94. [Google Scholar]; (e) Binkley RW, Goewey GS, Johnston JC. J. Org. Chem. 1984;49:992. [Google Scholar]; (f) Hanessian S. Org. Synth. 1987;65:243. [Google Scholar]; (g) Hullar TL, Siskin SB. J. Org. Chem. 1970;35:225. doi: 10.1021/jo00826a046. [DOI] [PubMed] [Google Scholar]
- 18.Liotta LJ, Dombi KL, Kelley SA, Targontsidis S, Morin AM. Tetrahedron Lett. 1997;38:7833. [Google Scholar]
- 19.(a) Roberts BP, Smits TM. Tetrahedron Lett. 2001;42:3663. [Google Scholar]; (b) Dang H-S, Roberts BP, Sekhon J, Smits TM. Org. Biomol. Chem. 2003;1:1330. doi: 10.1039/b212303g. [DOI] [PubMed] [Google Scholar]; (c) Fielding AJ, Franchi P, Roberts BP, Smits TM. J. Chem. Soc., Perkin Trans. 2002;2:155. [Google Scholar]; (d) Cai Y, Dang H-S, Roberts BP. J. Chem. Soc., Perkin Trans. 2002;1:2449. [Google Scholar]; (e) Jeppesen LM, Lundt I, Pedersen C. Acta Chem. Scand. 1973;27:3579. [Google Scholar]
- 20.(a) McNulty J, Wilson J, Rochon AC. J. Org. Chem. 2004;69:563. doi: 10.1021/jo035223x. [DOI] [PubMed] [Google Scholar]; (b) Crich D, Bowers AA, Yao Q. Carbohydr. Res. 2006;341:1748. doi: 10.1016/j.carres.2006.02.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Roberts BP, Dang H-S, Cai Y. J. Chem. Soc., Perkin Trans. 2002;1:2449. [Google Scholar]
- 22.Crich D, Yao Q. Org. Lett. 2003;5:2189. doi: 10.1021/ol034741r. [DOI] [PubMed] [Google Scholar]
- 23.Crich D, Yao Q. J. Am. Chem. Soc. 2004;126:8232. doi: 10.1021/ja048070j. [DOI] [PubMed] [Google Scholar]
- 24.(a) Beckwith ALJ, Easton CJ. J. Am. Chem. Soc. 1981;103:615. [Google Scholar]; (b) Rychnovsky SD, Swenson SS. Tetrahedron. 1997;53:16489. [Google Scholar]
- 25.Crich D, Li W, Li H. J. Am. Chem. Soc. 2004;126:15081. doi: 10.1021/ja0471931. [DOI] [PubMed] [Google Scholar]
- 26.Utimoto K, Wakabayashi Y, Horiie T, Inoue M, Shishiyama Y, Obayashi M, Nozaki H. Tetrahedron. 1983;39:967. [Google Scholar]
- 27.That it was the residues of the non-reducing end with upfield resonances was confirmed by NOE experiments on the β-dimer 13. In the NOE spectrum a clear correlation was observed between the cyclohexyl proton and the reducing end anomeric proton, which was further correlated to the downfield H-5 signal. The upfield H-5 multiplet correlated with the remaining anomeric peak.
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Full experimental details and characterization data. This material is available free of charge via the Internet at http://pubs.acs.org.