

Abstract
Background:
Previous human clinical trials of insulin-like growth factor type I (IGF-1) in amyotrophic lateral sclerosis (ALS) have been inconsistent. This phase III, randomized, double-blind, placebo-controlled study was undertaken to address whether IGF-1 benefited patients with ALS.
Methods:
A total of 330 patients from 20 medical centers were randomized to receive 0.05 mg/kg body weight of human recombinant IGF-1 given subcutaneously twice daily or placebo for 2 years. The primary outcome measure was change in their manual muscle testing score. Secondary outcome measures included tracheostomy-free survival and rate of change in the revised ALS functional rating scale. Intention to treat analysis was used.
Results:
There was no difference between treatment groups in the primary or secondary outcome measures after the 2-year treatment period.
Conclusions:
Insulin-like growth factor type I does not provide benefit for patients with amyotrophic lateral sclerosis.
GLOSSARY
- ALS
= amyotrophic lateral sclerosis;
- ALSFRS-r
= revised ALS functional rating scale;
- AUC
= area under the curve;
- DVT
= deep venous thromboses;
- IGF-1
= insulin-like growth factor type I;
- MMT
= manual muscle testing;
- PE
= pulmonary embolisms.
Amyotrophic lateral sclerosis (ALS) is a progressive neurodegenerative disease resulting in death in the majority of those afflicted. Treatment options are disappointing and more effective treatments are necessary. Motor neuron tissue cultures have shown that IGF-I is effective in enhancing survival of embryonic motor neurons and reduce their susceptibility to glutamate induced neurotoxicity.1 Previous work with animal models has shown that IGF-I can delay motor neuron cell death.2,3 The results of two previous phase III clinical trials of insulin-like growth factor type I (IGF-1) in ALS were inconsistent.4,5 This study was undertaken to determine the benefit of IGF-1 for patients with ALS. We conducted a double-blind, placebo-controlled, randomized, phase III clinical trial using IGF-1 or placebo for 2 years. The objective was to determine if IGF-1 slowed the rate of progressive muscle weakness in ALS. The primary outcome measure was the rate of change in the averaged manual muscle testing score.6 Secondary outcome measures included overall survival and rate of change in the revised ALS functional rating scale (ALSFRS-r).7 Intention to treat analysis was used.
METHODS
A total of 330 patients from 20 medical centers were randomized to receive either IGF-1 or placebo. Inclusion criteria for participation included being aged 18 years or older with a history of progressive weakness of 30 months or less and fulfilling the probable or definite classification for ALS by the El Escorial criteria.8 Subjects were required to have a manual muscle testing (MMT) score of less than 9 (see below for the MMT score) and a forced vital capacity on pulmonary testing of 60% or greater. Exclusion criteria included history of malignancy (other than localized non-melanotic skin cancer); active renal disease or other systemic illness that would make participation unsafe; diabetes mellitus; history of tracheostomy; active depression (as measured by the Beck Depression Inventory9 with a scores greater than 16); or use of any investigational drugs for the treatment of ALS within the previous 30 days.
Intervention.
The intervention consisted of a subcutaneous injection of either recombinant human IGF-1 or placebo. All drug and placebo were provided, free of cost, by Cephalon, Inc. (Frazer, PA). A dose of 0.05 mg per kg body weight of IGF-1 was subcutaneously injected twice daily. This represented the highest tolerated dose in phase I human dose escalation studies, with hepatic toxicity being the dose limiting factor. The placebo was an acidic solution identical to the vehicle for IGF-1. This placebo was chosen to maintain blinding of the investigators and the study subjects. Localized site reactions are common because of the acidic vehicle used to deliver IGF-1.
Sample size.
The sample size was determined using preliminary data from another study6 where there was a mean change of 0.7 units in the MMT score over a 6-month period (SD 0.83). Assuming the rate of change remained constant over the 2-year period we predicted a decline of 2.8 points for the control group. The standard error of this 2.8-point decline would be somewhat higher than the 0.83 estimated for the 6-month change. We approximated the SD of the 2.8-point decline to be double that of the 6-month decline, or 1.66 points. Calculation of the sample size was based on a (two sided) t test comparing the mean decline in MMT for the two treatment groups. We assumed equal sample sizes for each treatment group. In order to detect a 25% difference between the two treatment groups with 90% power and a Type I error rate of 5%, assuming a death rate of 50% and a dropout rate of 5%, enrollment and randomization of 330 patients was performed.
Randomization and blinding.
Randomization was performed using a dynamic allocation strategy, stratifying on five variables: site of onset (bulbar vs non-bulbar), age (55 years of age and less vs over the age of 55), riluzole use at randomization (yes vs no), MMT score (baseline score of 6 or less vs greater than 6), and medical center. The dynamic allocation strategy was employed to ensure balanced treatment groups with any of the strata. Treatment group assignment was performed by the Mayo Clinic research pharmacy. The study maintained a double blind approach. Only the Mayo Clinic research pharmacy and the Data Safety and Monitoring Board where unblinded to the treatment assignments. No formal assessment of the effectiveness of the blinding was performed.
Interim analyses.
One planned interim analysis was completed when half of the patients had completed 12 months of follow-up. Stopping rules were defined by the O’Brien-Flemming model. At the time of the planned interim analysis a p value of 0.0054 was required to halt the trial for efficacy. No formal futility analysis was included. In addition to the planned interim analysis, the DSMB called for two additional analyses for reasons of safety due to the frequency of vascular events noted in the study population. These three interim analyses dropped the level of significance to 0.0370 for the final analysis.
Statistical methods.
The primary outcome measure was the rate of change in the MMT score. The MMT examinations were performed by annually certified physical therapists. MMT involved the examination of 34 muscle groups with standard positioning. Muscle groups included were neck flexion and extension, bilateral shoulder abduction and external rotation, elbow flexion and extension, wrist flexion and extension, palmar thumb abduction, abduction of the fifth digit, hip flexion, extension and abduction, knee flexion and extension, ankle dorsiflexion and plantarflexion and flexion of the great toe. The final MMT score represented an average of the 34 muscles examined. The individual muscle score was based on the MRC grading scale (1–5) modified to a 10-point system to allow for MRC modifications of plus and minus (e.g., 4+, 3−). The MMT score was performed at randomization and then again at 3, 6, 12, 18, and 24 months post randomization. The final 24-month MMT score was performed at the subject’s home for all subjects living but incapacitated at the end of the study period. For all subjects who died during the follow-up period, a MMT score of 0 was imputed at the day of their death.
Analysis of MMT as the primary outcome was calculated as a ratio of change from baseline to last follow-up to time to duration until last follow-up. For the patients who died during the study period, the last follow-up time was considered as the time of death with a zero score for MMT measurement. Analysis was performed using intention to treat approach. Thus for the patients who had only baseline measurement and no follow-up information, a score of zero was assigned at the 24-month visit. Comparison of rate of change in MMT scores between the placebo and IGF-1 group was made using two-sample t test or Wilcoxon rank sum test as appropriate. Further analysis was also performed using analysis of covariance where MMT score and patient age at baseline were treated as covariates. Because these rate of change measurements require the assumption of linearity, the contingency primary analysis if linearity was violated was a nonlinear area under the curve analysis of MMT scores. In the analysis of the study the MMT scores violated linearity and the area under the curve (AUC) analysis was used as the primary analysis. In addition, various sensitivity analyses that include worst case scenario, randomization test, and rate of change or area under the curve of MMT score while patient was on drug were also performed.
Secondary outcome measures included tracheostomy-free survival. Patients who elected to proceed to tracheostomy were assessed on the day of their procedure. Subjects who continuously utilized NIPPV for greater than 10 days were assessed as being ventilator-dependent on the first day they began continuous NIPPV. All subjects were followed for the 24-month time period. Survival between groups was compared using the Cox proportional hazards model. Additionally, Kaplan-Meier survival curves for placebo and IGF-1 groups were generated for tracheostomy-free survival.
The final secondary outcome measure was the rate of change in the ALSFRS-r score. The ALSFRS-r was completed at each visit (randomization and then at 3, 6, 12, 18, and 24 months post-randomization). As with the MMT scores a score of 0 was imputed on the day of death. Analysis of the ALSFRS-r scores as a secondary outcome was performed in similar manner as MMT score.
Finally, summary statistics by treatment group were reported comparing baseline demographics, subject deaths, subjects who terminated their assignment treatment prior to death, and common and serious adverse events. Comparisons were made using two-sample t test, Wilcoxon rank sum test, or χ2 test as appropriate. The final analyses were performed by Mayo Department of Biostatistics.
RESULTS
Between July 2003 and August 2005, 330 subjects were randomized from 20 medical centers throughout the United States. The final patient follow-up was August 2007. There was a mean age of 55.4 years with 210 men (63.6%) and 120 women (36.4%). Ethnicity and demographics of the study population appear in table 1. The flow of subjects after randomization is illustrated in figure 1.
Table 1 Distribution of baseline demographics by treatment group
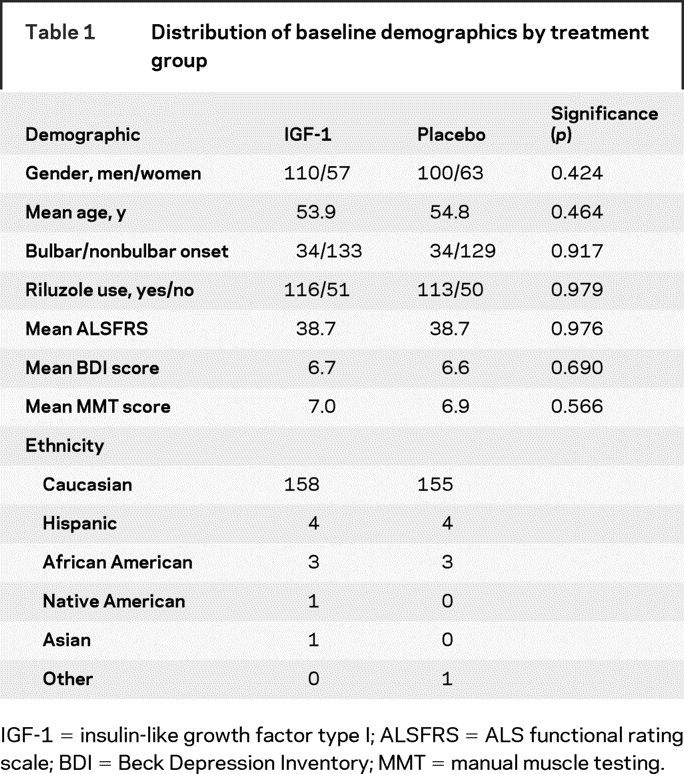
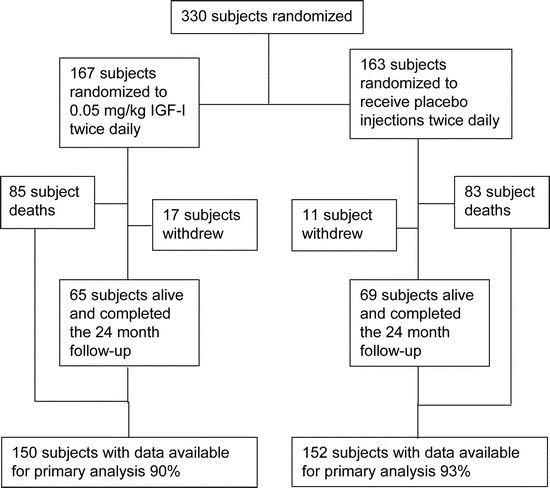
Figure 1 Patient flow chart
There was an overall mean rate of change in the MMT score of 0.41 units per month. The IGF-1 treatment group changed at a rate of 0.44 units per month and the placebo group changed at a rate of 0.39 units per month (p = 0.529). The overall mean rate of change for the ALSFRS-r was 2.35 units per month. The IGF-I treatment group changed at a rate of 2.5 units per month and the placebo group changed at a rate of 2.2 units per month (p = 0.321). Because the rates of change for MMT and ALSFRS were not linear, the nonlinear area under the AUC analysis was used as the primary analysis. When comparing two groups using AUC, a group with higher average AUC is considered to be progressing at a slower rate than the other group. There was an overall mean AUC in the MMT scores of 2,756 MMT units/days followed. The IGF-1 treatment group had a mean AUC of 2,726 MMT units/days followed and the placebo group had a mean AUC of 2,786 MMT units/days followed (p = 0.455). There was an overall mean AUC in the ALSFRS-r scores of 15,308 ALSFRS-r units/days followed. The IGF-1 treatment group had a mean AUC of 14,908 ALSFRS-r units/days followed and the placebo group had a mean AUC of 15,718 ALSFRS-r units/days followed (p = 0.314). Furthermore, as a number of subjects discontinued drug during the course of the trial, an additional sensitivity analysis was completed for subjects while on drug. All analyses yielded the same results for both MMT and ALSFRS-r.
There were 168 deaths (51%) in the course of the study: 104 of these occurred in patients while still receiving their assigned treatment, and 53 occurred in patients who had discontinued their assigned treatment prior to death. There were an additional 12 patients who died within 1 month of completing their 24 months of treatment and were identified at their scheduled 1-month follow-up visit. In addition to subject deaths, there were an additional 19 subjects who underwent tracheostomy and 21 subjects who began continuous NIPPV. Kaplan-Meier survival curves by treatment assignment appear in figure 2.
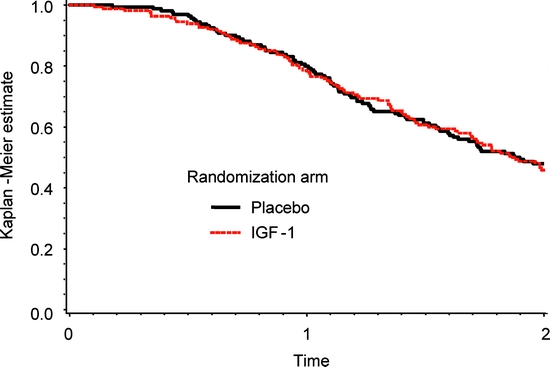
Figure 2 Kaplan-Meier survival curves, p = 0.415
A total of 128 subjects discontinued their assigned treatment for a reason other than death. Of these, 15 did so because of an adverse event and 21 more withdrew their consent for any further contact. The mean duration of treatment for subjects who discontinued their assigned treatment was 183 days, with a range from 0 to 542 days. Of the 122 subjects, 55 subsequently died prior to their 24-month visit. Of the remaining 67 subjects, 36 completed their 24-month assessment in their own home, and 28 declined to provide the final assessment. An additional three subjects agreed to complete the assessment but died prior to its scheduled completion within the 1-month follow-up period. There were no differences by treatment assignment.
The most commonly encountered adverse event was localized site reactions to the injections which occurred in 180 subjects. Headache occurred in 44, change in visual acuity in 43, hepatotoxicity in 32, other abnormal blood chemistries in 23, and deep venous thromboses in 15. There were 30 patients with presumed hypoglycemia including 2 cases of documented hypoglycemia. Twenty-one of these subjects were in the IGF-1 treated arm and 9 were in the placebo arm (p = 0.034). Disease attributed adverse events commonly occurred. There were 195 subjects with a deterioration of the BDI score suggestive of depression, 105 subjects with aspiration pneumonia or respiratory failure, and 61 hospitalizations for PEG tube placement. Table 2 reports the distribution of these common adverse events by treatment assignment.
Table 2 Common and serious adverse events by treatment assignment
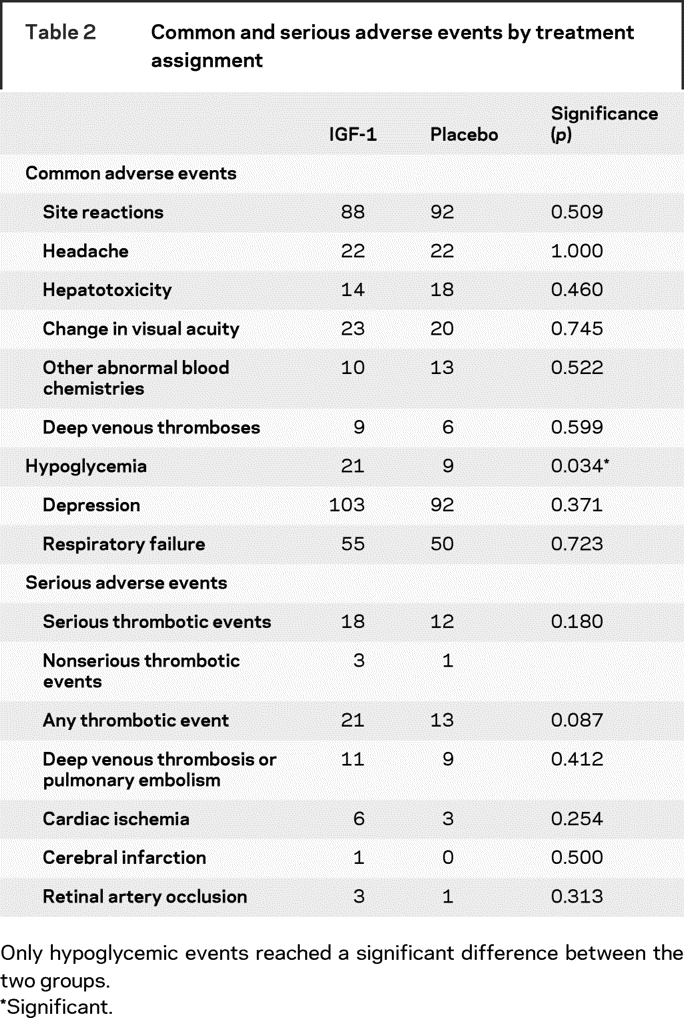
The most commonly occurring serious adverse event not attributable to ALS was thromboembolic. During the course of the study, there were 34 thromboembolic events noted. These included 20 subjects with deep venous thromboses (DVT) or pulmonary embolisms (PE). There were nine cases of cardiac ischemia and one case of cerebral infarction. Interestingly, there were four cases of retinal artery occlusions. The distribution of the thromboembolic events by treatment assignment appears in table 2. The occurrence of these was noted and monitored early by the DSMB.
DISCUSSION
The use of IGF-1 in ALS has previously produced conflicting results. Animal studies have demonstrated neurotrophic properties of IGF-110–12 and its importance in the development of the nervous system.13 Experience in transgenic SOD mice has been mixed9 but direct therapeutic studies in this model have been positive.14–17 Human clinical trials have also generated conflicting results. One large trial demonstrated a benefit that the other failed to confirm.4,5
The primary outcome measure and secondary outcome measures for this study were negative. A variety of alternative analyses were performed to determine whether the use of different statistical models would affect the outcome. These included dropping the intent to treat design, dropping the imputations, and analysis of data only while receiving treatment. All confirmed the negative result with any difference favoring placebo. The sensitivity analyses demonstrated that the result was robust, even in the presence of random perturbation of the data or systematic errors in measurement.
The results of our study most resemble those of the previous European study4 with no benefit in either survival or functionals scales. Unlike the European study, our study groups were balanced for the known prognostic variables, addressing that previous criticism of the European results. It is disappointing that we were unable to confirm the benefit that was noted in the previous North American study.5
The strengths of the present study include a number of novel methodologic features. We were able to minimize missing data by performing the strength examinations in the homes of patients. Additionally, we utilized trained physical therapists with annual recertifications in performing the strength examinations. By doing so we were successful in obtaining our primary outcome measure in 92% of all randomized patients after 2 years of follow-up. This 2-year duration is the longest to date in an ALS clinical trial, improving upon previous short duration studies where the long-term impact of the intervention remains unknown.
Despite these advantages, we did encounter limitations. The long duration of the study did create subject “fatigue.” Many subjects became dissatisfied with the ongoing rigors of the clinical trial as their disease progressed with many stopping their drug. As a result any apparent impact IGF-1 may have on ALS may be diminished as a result. Our sensitivity analysis comparing rate of change of MMT and ALSFRS-r while on drug demonstrated that this did not have an impact on our results but potentially could be an issue in subsequent studies.
The rate of progression in our study population exceeded that seen in our preliminary studies. This is most likely reflective of differences in the sample populations. This accelerated rate of progression led to improved power and our study retained a 90% power to detect a difference of less than 20% for the analyses.
An important statistical limitation of our methods is the variable follow-up time for our patients. This occurred because approximately half of the subjects died at varying time points during the 2-year duration of the study. This is an inherent problem in any progressive fatal disorder. It introduces a potential bias if the overall follow-up time differs between the study groups or if the rates of change measurements are not linear. To minimize the impact of this limitation we included the nonlinear sensitivity analysis AUC. Additionally, our overall follow-up time between the IGF-1 arm and the placebo arm was similar. A time to event analysis would eliminate this issue; however, this requires continuous observation, which is only possible for survival. After consultation with numerous statistical sources, there is no current consensus on a statistical methodology that adequately addresses this limitation.
IGF-1 appears to be safe and well tolerated in the vast majority of subjects. The most common serious adverse event that is not clearly disease-related was thromboembolic events. There were a total of 20 DVTs or PE. Our incidence rate for DVT was 3.0% per 12 months follow-up. This is comparable to previously published incidence rates. One published report of aggregated clinical trial data indicated an incidence rate of DVT in ALS of 2.7% per year follow-up.18 Another retrospective study indicated an incidence rate of 2.97% per year follow-up.19 Finally, the clinical trial studying topiramate in ALS reported an annual incidence rate of DVT or PE of 4.4% compared to 4.2% in our population.20 Given the similarities of these rates to those seen in our study, it is likely that this reflects an underlying increased risk of DVT and thromboembolic events in those with ALS. Although there was an imbalance in our population, this did not reach statistical significance at the end of the trial. As expected with the insulin-like properties of IGF-1, 20 subjects did encounter episodes of hypoglycemia. Interestingly, we also encountered four cases of retinal artery occlusion, an adverse event not previously reported in ALS clinical trials.
ACKNOWLEDGMENT
All study drug and placebo was provided by Cephalon, Inc.
Address correspondence and reprint requests to Dr. Eric J. Sorenson, Department of Neurology, Mayo Clinic, 200 1st St. SW, Rochester, MN 55905 sorenson.eric@mayo.edu
Supported by NIH grant # RO1 NS 42759 and by a grant from the ALS Association.
Disclosure: The authors report no disclosures.
Received June 3, 2008. Accepted in final form August 25, 2008.
REFERENCES
- 1.Corse AM, Bilak MM, Bilak SR, Lehar M, Rothstein JD, Kuncl RW. Preclinical testing of neuroprotective neurotrophic factors in a model of chronic motor neuron degeneration. Neurobiol Dis 1999;6:335–346. [DOI] [PubMed] [Google Scholar]
- 2.Vergani L, Losa M, Lesma E, et al. Glycosaminoglycans boost insulin-like growth factor-I-promoted neuroprotection: blockade of motor neuron death in the wobbler mouse. Neuroscience 1999;93:565–572. [DOI] [PubMed] [Google Scholar]
- 3.Vergani L, Finco C, Di Giulio AM, Muller EE, Gorio A. Effects of low doses of glycosaminoglycans and insulin-like growth factor-I on motor neuron disease in wobbler mouse. Neurosci Lett 1997;228:41–44. [DOI] [PubMed] [Google Scholar]
- 4.Borasio GD, Robberecht W, Leigh PN, et al. A placebo-controlled trial of insulin-like growth factor-I in amyotrophic lateral sclerosis. European ALS/IGF-I Study Group. Neurology 1998;51:583–586. [DOI] [PubMed] [Google Scholar]
- 5.Lai EC, Felice KJ, Festoff BW, et al. Effect of recombinant human insulin-like growth factor-I on progression of ALS: a placebo-controlled study. The North America ALS/IGF-I Study Group.Neurology 1997;49:1621–1630. [DOI] [PubMed] [Google Scholar]
- 6.Great Lakes ALSSG. A comparison of muscle strength testing techniques in amyotrophic lateral sclerosis [see comment]. Neurology 2003;61:1503–1507. [DOI] [PubMed]
- 7.Cedarbaum JM, Stambler N, Malta E, et al. The ALSFRS-R: a revised ALS functional rating scale that incorporates assessments of respiratory function. BDNF ALS Study Group (Phase III). J Neurol Sci 1999;169:13–21. [DOI] [PubMed] [Google Scholar]
- 8.Brooks BR. El Escorial World Federation of Neurology criteria for the diagnosis of amyotrophic lateral sclerosis. Subcommittee on Motor Neuron Diseases/Amyotrophic Lateral Sclerosis of the World Federation of Neurology Research Group on Neuromuscular Diseases and the El Escorial “Clinical limits of amyotrophic lateral sclerosis” workshop contributors [see comment]. J Neurol Sci 1994;124 suppl:96–107. [DOI] [PubMed] [Google Scholar]
- 9.Beck A, Steer R, Garbin M. An inventory for measuring depression. Arch Gen Psychiatry 1962;4:561–571. [DOI] [PubMed] [Google Scholar]
- 10.Li L, Oppenheim RW, Lei M, Houenou LJ. Neurotrophic agents prevent motoneuron death following sciatic nerve section in the neonatal mouse. J Neurobiol 1994;25:759–766. [DOI] [PubMed] [Google Scholar]
- 11.Lewis ME, Neff NT, Contreras PC, et al. Insulin-like growth factor-I: potential for treatment of motor neuronal disorders. Exp Neurol 1993;124:73–88. [DOI] [PubMed] [Google Scholar]
- 12.Caroni P, Grandes P. Nerve sprouting in innervated adult skeletal muscle induced by exposure to elevated levels of insulin-like growth factors. J Cell Biol 1990;110:1307–1317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Russo VC, Gluckman PD, Feldman EL, Werther GA. The insulin-like growth factor system and its pleiotropic functions in brain. Endocrine Rev 2005;26:916–943. [DOI] [PubMed] [Google Scholar]
- 14.Messi ML, Clark HM, Prevette DM, Oppenheim RW, Delbono O. The lack of effect of specific overexpression of IGF-1 in the central nervous system or skeletal muscle on pathophysiology in the G93A SOD-1 mouse model of ALS. Exp Neurol 2007;207:52–63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Nagano I, Ilieva H, Shiote M, et al. Therapeutic benefit of intrathecal injection of insulin-like growth factor-1 in a mouse model of amyotrophic lateral sclerosis. J Neurol Sci 2005;235:61–68. [DOI] [PubMed] [Google Scholar]
- 16.Kaspar BK, Frost LM, Christian L, Umapathi P, Gage FH. Synergy of insulin-like growth factor-1 and exercise in amyotrophic lateral sclerosis. Ann Neurol 2005;57:649–655. [DOI] [PubMed] [Google Scholar]
- 17.Dobrowolny G, Giacinti C, Pelosi L, et al. Muscle expression of a local Igf-1 isoform protects motor neurons in an ALS mouse model. J Cell Biol 2005;168:193–199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Qureshi MM, Cudkowicz ME, Zhang H, Raynor E. Increased incidence of deep venous thrombosis in ALS [see comment]. Neurology 2007;68:76–77. [DOI] [PubMed] [Google Scholar]
- 19.Elman LB, Siderowf A, Houseman G, Kelley M, McCluskey LF. Venous thrombosis in an ALS population over four years. Amyotroph Lateral Scler Other Motor Neuron Disord 2005;6:246–249. [DOI] [PubMed] [Google Scholar]
- 20.Cudkowicz ME, Shefner JM, Schoenfeld DA, et al. A randomized, placebo-controlled trial of topiramate in amyotrophic lateral sclerosis [see comment]. Neurology 2003;61:456–464. [DOI] [PubMed] [Google Scholar]