

SUMMARY
Motile dendritic filopodial processes are thought to be precursors of spine synapses, but how motility relates to cell-surface cues required for axon-dendrite recognition and synaptogenesis remains unclear. We demonstrate with dynamic imaging that loss of EphBs results in reduced motility of filopodia in cultured cortical neurons and brain slice. EphB knockdown and rescue experiments during different developmental time windows show that EphBs are required for synaptogenesis only when filopodia are most abundant and motile. In the context of EphB knockdown and reduced filopodia motility, independent rescue of either motility with PAK or of Ephephrin binding with an EphB2 kinase mutant is not sufficient to restore synapse formation. Strikingly, the combination of PAK and kinase-inactive EphB2 rescues synaptogenesis. Deletion of the ephrin-binding domain from EphB2 precludes rescue, indicating that both motility and trans-cellular interactions are required. Our findings provide a mechanistic link between dendritic filopodia motility and synapse differentiation.
INTRODUCTION
The establishment of precise synaptic connections between appropriate neurons is essential for the development of functional neural networks. In the mammalian CNS, formation of glutamatergic synaptic inputs is characterized by an early phase of slow addition followed by a burst of synaptogenesis, ending in maturation and pruning of contacts (Goda and Davis, 2003; Waites et al., 2005). Coinciding with this rapid phase of synapse addition is the presence of thin, elongated filopodia-like protrusions on dendrites. In vitro and in vivo studies have demonstrated that these dendritic filopodia are highly dynamic structures, capable of exploring their local cellular environment and possibly initiating contact with appropriate presynaptic partners (Dailey and Smith, 1996; Fiala et al., 1998; Lendvai et al., 2000; Zito et al., 2004; Ziv and Smith, 1996). Moreover, during the critical period in development, changes in sensory input in vivo cause alterations in the motility of filopodia, which suggests that this motility is involved in sensory map formation (Lendvai et al., 2000). As development proceeds, synapse and dendritic spine density increase while filopodia density and protrusion motility decrease (Dailey and Smith, 1996; Ziv and Smith, 1996). Thus, the motility of these filopodia is likely to play a significant role in establishment of contact between axons and dendrites and, ultimately, the formation of a synapse; however, it remains unclear whether motile filopodia are essential for synapse formation.
It is thought that if dendritic filopodia function as initial bridges between neurons during synaptogenesis, their motility must be paired with an ability to (1) recognize a presynaptic axonal partner, (2) provide for axon-dendrite adhesion, and (3) trigger differentiation of synaptic terminals (Dailey and Smith, 1996; Ziv and Smith, 1996). One attractive set of candidates for linking motility to these events are trans-synaptic molecules that not only function as cellular adhesion proteins but also control various components of pre- and/or postsynaptic organization (Dalva et al., 2007). Although these synaptogenic signals act as recognition and adhesion factors, it is not known whether any are also involved in dendritic filopodia motility or, therefore, how motility is coupled to the cell-surface molecules required for synapto-genesis.
The postsynaptic EphB receptor tyrosine kinase is part of the Eph-ephrin trans-synaptic signal that, through independent domain-specific functions, is able to regulate clustering of NMDA- and AMPA-type glutamate receptors (Dalva et al., 2000; Kayser et al., 2006). EphB-ephrinB reverse signaling into the presynaptic axon also leads to differentiation of presynaptic terminals (Kayser et al., 2006). Finally, EphB forward signaling induces the formation of dendritic spines. EphBs signal in a kinase-dependent manner to phosphorylate guanine exchange factors (GEFs) such as Tiam1, kalirin-7, and intersectin, that catalyze the Rho family GTPases Rac1 and Cdc42 into the active state (Irie and Yamaguchi, 2002; Penzes et al., 2003; Tolias et al., 2007). EphB2 also phosphorylates the transmembrane heparan sulfate proteoglycan syndecan-2 (Ethell et al., 2001). Each of these signaling pathways activates molecules that lead to reorganization of the actin cytoskeleton and spine morpho-genesis. For example, phosphorylation of kalirin-7 and activation of Rac1 cause phosphorylation of p21-activated kinase (PAK), while syndecan-2 appears to work coordinately with intersection/Cdc42 to activate N-WASP and the Arp2/3 complex (Irie and Yamaguchi, 2002; Penzes et al., 2003). With expression of dominant-negative EphB kinase mutants or in the absence of EphBs, mature neurons in culture not only have fewer spines and synapses but also more filopodia (Ethell et al., 2001; Henkemeyer et al., 2003; Penzes et al., 2003). Many of the same molecules through which EphBs signal, such as Rac1 and Cdc42, control the motility of filopodia in nonneuronal cells (Small et al., 2002) and have also been implicated in the modulation of dendritic filopodia motility in neurons (Tashiro and Yuste, 2004).
Here, we investigate the potential role for EphBs in the control of dendritic filopodia motility during neuronal synapse formation. Using time-lapse imaging, we find that loss of EphBs causes a reduction in motility of postsynaptic dendritic filopodia both in dissociated culture and in slice. Neurons lacking EphBs fail to undergo the rapid phase of synapse addition normally found when motile filopodia are abundant during the second week in culture and instead add synapses at a constant rate during the first 2 weeks in vitro. In addition, knockdown of EphB expression during multiple development time windows reveals that EphB is necessary for normal synapse formation only during the time when dynamic filopodia processes are numerous. Finally, we find that the temporally restricted synaptogenic role of EphB consists of two distinct actions, both of which are essential. First, EphB forward signaling controls dendritic filopodia motility, potentially allowing pre- and postsynaptic partners to initiate contact, and second, EphB trans-synaptic interactions stabilize nascent synaptic contacts. These two functions of EphB can be reconstituted by combining a form of EphB incapable of downstream signaling with expression of constitutively active PAK, which we find promotes motility. These results identify a molecular signal that directly couples the motility of postsynaptic dendritic filopodia to trans-cellular interactions and suggest that EphBs specifically direct formation of postsynaptically induced dendritic spine synapses.
RESULTS
EphBs Control Dendritic Filopodia Motility
Mature (21 days in vitro [DIV]) neurons cultured from EphB1•/•, EphB1•/•, EphB2•/•, EphB3•/• triple-knockout (TKO) mice have fewer excitatory synapses and spines than EphB1•/•, EphB3•/• double-knockout (DKO) or wild-type controls, as well as more filopodia (Henkemeyer et al., 2003) (the presence of only EphB2 in DKO animals results in synapse formation similar to wild-type by all examined measures; Henkemeyer et al., 2003; Kayser et al., 2006). We investigated whether the absence of EphBs affects filopodia motility during week 2 in vitro, when dendritic protrusion motility is at its peak (Ziv and Smith, 1996). Cortical neurons were cultured from P1–3 TKO or DKO littermate controls. At 3–5DIV, neurons were transfected with GFP, and images of live cells were captured every 3 min for 30 min at 9–10DIV. We analyzed movement of dendritic filopodia over time by tracking the location of the protrusion tip throughout the series and calculating the total distance moved by an individual filopodium during the imaging period. There was no difference in density or average length of filopodia on dendrites of TKO and DKO neurons at this time in development (data not shown). Surprisingly, although 10DIV neurons cultured from TKO animals have abundant lengthy dendritic protrusions, we found that the absence of EphBs resulted in a 55% reduction of dendritic filo-podia motility (Figures 1A and 1B). While dendritic filopodia on DKO control cells often appeared to be actively searching out their local environment with multiple extensions, retractions, and sweeping motions, the filopodia on TKO cells were often stationary (Figure 1A and Movie S1 available online). These findings indicate that EphB signaling is required for the normal dynamic motility of postsynaptic dendritic filopodia during the second week of synapse formation.
Figure 1. Reduced Expression of EphB Results in Decreased Motility of Postsynaptic Dendritic Filopodia during Synapse Formation In Vitro.

(A) Representative frames from time-lapse images of 9–10DIV neurons cultured from EphB1•/•, EphB3•/•(DKO) or EphB1•/•, EphB2•/•, EphB3•/•(TKO) and transfected at 3–5DIV with GFP. Colored arrowheads track motile protrusions through the series. Open arrowheads indicate less motile protrusions.
(B) Total distance individual protrusions move throughout 30 min of imaging of DKO (n = 150 protrusions, 9 cells) or TKO cells (n = 120 protrusions, 8 cells).
(C) Total distance individual protrusions move throughout 30 min of imaging of control (n = 235 protrusions, 12 cells) or EphB2 shRNA transfected cells (n = 281 protrusions, 13 cells).
(D) Representative images of initial frame in time-lapse series (grayscale) overlayed on 30 min time projection showing increased area of coverage by motile filopodia (magenta) (control: n = 102 protrusions, 7 cells; EphB2 shRNA: n = 121 protrusions, 8 cells). *p < 0.0001; t test. Scale bars, 2 mm. Error bars indicate SEM.
Neurons cultured from TKO animals lack EphBs in both axons and dendrites, and despite evidence that EphB functions cell-autonomously to organize postsynaptic specializations (Kayser et al., 2006), the possibility remains that synapse formation defects arise from early developmental abnormalities in axon guidance. To determine whether reduced filopodia motility in the absence of EphBs results from a postsynaptic function of EphB receptors, we used a previously described shRNA that specifically targets EphB2 (Kayser et al., 2006; Figure S1). Work from our laboratory has demonstrated that knockdown of EphB2 at 3DIV and examination of neurons at 9–10DIV reveals a reduced density of excitatory synaptic inputs (Kayser et al., 2006), and we have found that knockdown of EphB2 alone in cultured neurons results in a similar synapse-loss phenotype to neurons cultured from animals lacking all three EphBs (see Figures 3A and 4A). Thus, using this shRNA we investigated whether filopodia motility is also reduced during the second week in vitro following acute knockdown of postsynaptic EphB2 expression. Wild-type cortical neurons (3DIV) were transfected with GFP and either vector control or EphB2 shRNA and imaged live at 9–10DIV. Because we achieve a relatively low transfection efficiency (• 0.1%), transfected neurons receive an overwhelming majority of inputs from wild-type axons, ensuring that our observed effects are due to changes in postsynaptic dendritic motility and not changes in the axon. Filopodia on dendrites of control neurons were highly dynamic, with motility comparable to DKO controls. In contrast, EphB2 knockdown neurons displayed a reduction in protrusion movement similar to that found in TKO cells (Figure 1C and Movie S2), demonstrating that reduced expression of postsynaptic EphB2 results in decreased filopodia motility. EphBs appear to play a similar function in other cell types, as knockdown of EphB2 in cultured hippocampal neurons also results in reduced filopodia motility and synapse number (Motility: Control, total distance moved per filopodium over 30 min = 7.06 ± 0.389 mm, n = 124 filopodia, 8 cells; EphB2 shRNA, total distance = 3.10 ± 0.251 mm, n = 109 filopodia, 8 cells; p < 0.0001; t test. Synapse density: Control, 13.2 ± 1.49 synapses/100 mm, n = 21 cells; EphB2 shRNA, 6.40 ± 0.76 synapses/100 mm, n = 18 cells; p % 0.0005; t test.).
Figure 3. Synapse Formation Is Abnormal during the Second Week In Vitro in the Absence of EphBs.

(A) Average synapse density on cortical neurons cultured from EphB1•/•, EphB3•/•(DKO) or EphB1•/•, EphB2•/•, EphB3•/•(TKO) mice, fixed between 5– 21DIV, and immunostained with antibodies recognizing PSD-95 and vGlut1 (n = 18, 18, 16, 23, 22, 39, 16, 32, 20, and 31 cells from left to right; *p < 0.0001; ANOVA). Error bars indicate SEM.
(B) Representative images of dendrites from neurons in (A) at 7, 14, and 21DIV. Arrows indicate colocalized PSD-95 (red) and vGlut1 (blue) on GFP-transfected cells.
(C) Average density of spines and filopodia on dendrites of 21DIV DKO and TKO neurons, and representative images of each (n = 387 protrusions from 8 DKO cells, and 408 protrusions from 9 TKO cells; *p < 0.003; t test). Error bars indicate SEM.
(D) Plot of number of synapses per cell at each developmental time point, derived from synapse density and total dendritic arbor length measured from each neuron (n = 6 cells for both conditions at every time point). Scale bars, 2 mm. Error bars indicate SEM.
Figure 4. Distinct Early and Late Developmental Roles for EphB2 in Synaptogenesis.

(A) Cortical neurons cultured from E18 rats were transfected at the indicated time with GFP and either control or EphB2 shRNA, fixed at the indicated time, and immunostained with antibodies recognizing GFP, PSD-95 (red), and vGlut1 (blue; n = 21, 19, 35, 27, 24, 25, 30, 31, 16, and 34 cells from left to right; *p < 0.0001; t test).
(B–E) Representative images of immunostained dendrites from control cells (B) and those expressing the EphB2 knockdown construct from 3–21DIV (C), 10–21DIV (D), and 14–21DIV (E). Panels on the right show only the colocalized PSD-95 and vGlut1 puncta on the transfected cell, with other puncta subtracted from the image. Arrows in (B) indicate synapses occurring in spines. Arrowheads in (E) indicate synapses occurring on filopodia-like processes with spine-like heads. Scale bars, 2 mm.
(F) Average density of spines and filopodia on dendrites of 21DIV neurons transfected with control or EphB2 shRNA at either 3DIV or 10DIV (n = 829 protrusions from 18 control cells, and n = 680 protrusions from 22 EphB2 shRNA cells for 3–21DIV; n = 1361 protrusions from 19 control cells, and n = 1025 protrusions from 18 EphB2 shRNA cells for 10–21DIV; *p < 0.0001; t test).
(G) Neurons cultured from TKO mice transfected with GFP or EphB2-YFP at 3DIV, and DKO neurons transfected with GFP, fixed at 10DIV, and immunostained with antibodies recognizing GFP or EphB2, PSD-95, and vGlut1 (n = 6, 14, and 11 cells from left to right; *p < 0.01; ANOVA).
(H) Neurons cultured from TKO mice transfected with GFP or EphB2-YFP at 3DIV or 10DIV, and DKO neurons transfected with GFP, fixed at 21DIV, and immunostained with antibodies recognizing GFP or EphB2, PSD-95, and vGlut1 (n = 20, 23, 8, and 33 cells from left to right; *p < 0.0001; ANOVA). Error bars indicate SEM.
We also investigated whether reduced motility with EphB2 knockdown is generalized to all types of dendritic protrusions at this time. We examined dendritic spines and—although there are few spines at this stage in development (Ethell et al., 2001; Papa et al., 1995; Ziv and Smith, 1996)—found no difference in their motility in knockdown compared to control cortical cells (EphB2 shRNA: total distance moved per spine over 30 min = 0.63 ± 0.076 mm, n = 65 spines, 11 cells; control: total distance = 0.55 ± 0.071 mm, n = 60 spines, 9 cells; p = 0.5; t test). In addition, to control for the possibility that the reduced motility of filo-podia is due nonspecifically to the shRNA, we cotransfected an EphB2 rescue construct (B2-R) insensitive to our knockdown construct along with EphB2 shRNA, which rescued the motility deficit (see Figure 5A). Finally, we measured the area explored by dendritic filopodia in control or EphB2 knockdown neurons. For filopodia on control cells, we found on average that motility in a 30 min period enables a filopodium to explore 458.8% ± 82.7% more territory than the area covered by that filopodium in the first frame of the series (Figure 1D). In contrast, knockdown of EphB2 reduced this area explored to less than twice the size of the filopodium (179.3% ± 13.4%; p < 0.003; t test). These findings support the idea that reduced EphB2 expression results in substantial defects in the ability of filopodia to sample their local environment.
Figure 5. Normal Filopodia Motility in the Absence of EphB Signaling Is Not Sufficient to Direct Normal Synapse Formation.

(A) Total distance individual protrusions move throughout 30 min of imaging of 9–10DIV cells transfected with the indicated constructs + GFP at 3DIV (control: n = 31 protrusions [p], 2 cells [c]; B2 shRNA: 50 p, 3 c; B2 rescue (B2-R): 135 p, 12 c; wild-type (WT) PAK: 179 p, 12 c; constitutively active (CA) PAK: 215 p, 16 c; **p < 0.03, *p < 0.0001 compared to B2 shRNA; ANOVA).
(B) Synapse density on 10DIV cells transfected with the indicated constructs + GFP at 3DIV (control, n = 18 cells; B2 shRNA, 7; B2-R, 13; WT PAK, 23; CA PAK, 22; *p < 0.0001 compared to B2 shRNA; ANOVA). Error bars indicate SEM.
(C) Representative images of dendrites from 10DIV cells quantified in (B). Arrows indicate synapses (colocalization of PSD-95 [red] and vGlut1 [blue]). Scale bars, 2 mm. Error bars indicate SEM.
To examine whether EphBs control filopodia motility in a preparation that more closely mimics that found in vivo, we performed motility assays in organotypic brain slice cultures. We showed previously that cortical brain sections from TKO mice have an • 40% reduction in synapse density, with a specific loss of postsynaptic specializations from dendritic protrusions in cultured brain slices (Kayser et al., 2006). Here, cortical slices were made from P3–4 TKO, DKO, and wild-type mice, and after 2 days in culture, neurons were transfected with GFP. Using two-photon microscopy, images of live cells were captured every 3 min for 30 min at 4–5 days following transfection (Figure 2A), and we analyzed movement of dendritic filopodia over time as described above. Filopodia on dendrites of wild-type and DKO neurons did not exhibit different amounts of motility, demonstrating that the presence of EphB2 alone is sufficient to direct normal filopodial exploration. Interestingly, filopodia motility in brain slice appeared different than that in dissociated neuronal culture, with a predominance of extensions and retractions and few side-to-side sweeping motions (Movie S5). Similar to results in dissociated culture, however, filopodia on neurons in TKO slices showed reduced motility in comparison to those observed in the DKO control neurons over 30 min (Figures 2A and 2B and Movie S5). Thus, EphBs are required for normal filopodia motility in brain slices. To confirm as we did in dissociated neurons that these results are due specifically to the loss of EphB2 function in the postsynaptic neuron, we again used a knockdown approach. DKO neurons were transfected with GFP and either vector control or EphB2 shRNA. We found that DKO neurons in which EphB2 was knocked down exhibited reduced motility compared to DKO controls (Figure 2C and Movie S6). Additionally, EphB2 knockdown in neurons of brain slices cultured from wild-type mice resulted in a similar motility defect (Figure 2D). These data validate our in vitro approach and demonstrate that EphBs are required for normal filopodial motility in the more intact brain slice preparation.
Figure 2. Filopodia Motility Is Reduced in the Absence of EphBs and following Knockdown of EphB2 in Cultured Brain Slices.

(A) Representative frames from time-lapse images of neurons in slices made from EphB1•/•, EphB3•/•(DKO) and EphB1•/•, EphB2•/•, EphB3•/•(TKO) mice and transfected with GFP. Arrows as in Figure 1.
(B–D) Total distance individual filopodia move throughout 30 min of imaging neurons in slice culture from (B) DKO (n = 67 filopodia, 8 cells) or TKO cells (n = 172 filopodia, 23 cells), (C) DKO cells transfected with control (n = 53 filopodia, 9 cells) or EphB2 shRNA (n = 63 filopodia, 9 cells), or (D) WT cells transfected with control (n = 98 filopodia, 9 cells) or EphB2 shRNA (n = 52 filopodia, 8 cells). *p < 0.005; t test. Scale bars, 4 mm. Error bars indicate SEM.
Temporal Specificity of EphB Synaptogenic Activity
Throughout the first week in vitro, cultured neurons have few dendritic protrusions of any kind, while by week three and beyond, dendritic spines predominate. During the second week in vitro, dendritic filopodia are at their highest density, and protrusions are most motile (Papa et al., 1995; Ziv and Smith, 1996). Because our results indicate that EphBs regulate both filopodia motility and synapse number (Figure 1; Kayser et al., 2006), we conducted a series of experiments to determine whether EphBs are required for excitatory synapse development only during the second week in vitro. Cortical neurons were cultured from TKO mice, DKO littermate controls, or wild-type mice. We transfected these cultures with GFP at 3DIV and fixed and immunostained the neurons with antibodies recognizing preand postsynaptic marker proteins at 5, 7, 10, 14, and 21DIV. During the first week in vitro, we found no difference in excitatory synapse density (defined as colocalization of PSD-95 and vGlut1 puncta) (Figures 3A and 3B). However, a significant reduction in synapse density emerged in TKO cells compared to DKO and wild-type by 10DIV that persisted through 21DIV (Figures 3A and 3B; there was no difference between DKO and wild-type neurons at any time point [data not shown]). The decrease in density was not due to an obvious reduction in the stability of pre- and postsynaptic contacts that occur (Supplemental Data and Figure S2), suggesting that EphB is an inductive synaptogenic signal without which fewer contacts are established. In addition, because there is normally a shift from immature filopodia-like structures to mature spines during dendritic development, we examined dendritic protrusion morphology at each time point. There was no difference in density of dendritic protrusions at 7DIV, but—consistent with results in hippocampal cells (Henkemeyer et al., 2003)—a 56% reduction at 21DIV in density of dendritic spines and 180% increase in density of filopodia-like protrusions in TKO cortical neurons compared to controls (Figures 3B and 3C). Thus, while TKO and DKO neurons initially form similar numbers of synapses and dendritic protrusions, TKO neurons fail to elaborate as many contacts as DKO neurons, beginning at the time that filopodia are normally most abundant and motile.
Although synapse density remains constant in TKO neurons, dendritic arborization continues from 10 to 21DIV. Therefore, even in the absence of EphBs, some synapses must continue to be added throughout development to keep overall density constant despite increasing dendritic length. To investigate how the rate of synaptogenesis differs with and without EphBs, we calculated the total number of synapses on TKO versus DKO neurons from 0 to 21DIV. Consistent with previous work in wild-type neurons (Papa et al., 1995; Rao et al., 1998; Ziv and Smith, 1996), the rate of synapse addition increases markedly between the first week (• 24 synapses added/day) and second week (• 71 synapses/day) in vitro in DKO cells (Figure 3D). In contrast, the rate of addition remains virtually unchanged during the first 2 weeks in vitro in TKO cells (Figure 3D; first week: • 24 synapses/day; second week: • 19 synapses/day). The rate of synaptogenesis then tapers off in both TKO and DKO cells during the third week in vitro (• 9 synapses/day for each condition), suggesting that the signals mediating a decreased rate of synapse addition at this later stage of development are still intact in the absence of EphBs. Thus, cortical neurons lacking EphBs undergo an initial phase of synapse formation during early development (1–7DIV) that continues through the second week in vitro, but fail to undergo a rapid phase of synaptogenesis between 7–14DIV. These results demonstrate that, coincident with the time when filopodia motility is hypothesized to be most important for synaptogenesis (Dailey and Smith, 1996; Ziv and Smith, 1996), EphBs direct formation of a rapidly added subpopulation of excitatory synapses. The rate of synapse addition appears unaffected in the absence of EphBs at developmental times when fewer filopodia are normally present.
To further refine when EphBs function in synaptogenesis and examine the potential for a later developmental role of EphB in synapse maturation, we took a comprehensive knockdown and rescue approach in wild-type and TKO neurons, respectively. First, we cultured cortical neurons from E18–19 animals, knocked down EphB2 expression during multiple time periods, and assessed synapse density at each point. This approach is advantageous because it enables us to study the role of EphB exclusively in the postsynaptic neuron. We found that acute knockdown of EphB2 alone early in development phenocopies TKO neurons at each time tested: cotransfection of neurons with EphB2 shRNA and GFP at 0DIV did not reduce synapse number at 7DIV (Figure 4A), while knockdown at 3DIV and fixation at either 10DIV or 21DIV resulted in fewer synapses than control (Figures 4A–4C). Moreover, much like that observed in TKO neurons, knockdown from 3 to 21DIV resulted in a shift from spines to filopodia (Figures 4C and 4F). We conducted a number of additional controls to ensure that the observed effects of our shRNA constructs on synapse formation are specific to the loss of EphB2 (Figure S1), including showing that EphB2 shRNA has no effect in TKO neurons. shRNAs targeting EphB2, therefore, generate not only the same motility defect (see Figure 1) but also the same reduction in synapse density and shift in protrusion morphology throughout development in vitro as that found in neurons cultured from animals lacking EphB1-3.
Taking advantage of the temporal control offered by the RNAi approach, we tested whether the presence of EphB only early in development—when filopodia first become abundant—is sufficient to drive normal synapse formation or whether EphB signaling is also required later in synaptogenesis. Ten DIV neurons were transfected with EphB2 shRNA or control along with GFP, and we examined synapse density at 21DIV. We found that reducing EphB2 expression even at this later time point results in a 54% decrease in synapse density (Figures 4A and 4D). However, as opposed to the shift from spines to filopodia found with early knockdown (3–21DIV) or in TKO neurons, later knockdown (10–21DIV) caused a 55% reduction of spine density but no concurrent increase in filopodia density (Figures 4D and 4F). These data show that EphB2 is required throughout the second week in vitro either for the formation or stabilization of synaptic contacts and that late knockdown results in spine and synapse loss without the increase in filopodia density seen when EphB expression is reduced earlier in development (Figures 4C and 4F).
We next knocked down EphB2 after the robust phase of synapse addition that occurs between 7 and 14DIV and at a time when filopodia have become sparse to investigate whether EphB2 is required for late-forming synapses or maintenance of pre-existing contacts. Fourteen DIV cortical neurons were transfected with EphB2 shRNA or control and assessed for synapse density at 21DIV. Remarkably, knockdown at 14DIV did not alter synapse density (Figures 4A and 4E), suggesting that EphB2 is no longer necessary to maintain or form synaptic inputs later in development when filopodia density and motility are low (Ziv and Smith, 1996). EphB2 knockdown at 14DIV did, however, induce an abnormal morphological phenotype, with the overlapped pre- and postsynaptic puncta now found on long, filpodia-like protrusions with spine-like heads rather than short mushroom-shaped spines (Figure 4E). To test whether this dendritic protrusion phenotype could be a precursor to synapse loss that might occur with longer-term knockdown, we examined effects of EphB2 shRNA from 14 to 25DIV. However, even with expression of the shRNA for 11 days, there was no change in synapse density and a similar extension of spine-like protrusions to that found with knockdown from 14 to 21DIV (Figure S3). To examine whether the difference in the role of EphB2 between the second week in vitro and the third/fourth weeks might be a reflection of alterations in EphB2 expression levels, we lysed cultured cortical neurons at different developmental time points and assayed western blots with an antibody recognizing EphB2. Consistent with work in vivo (Henderson et al., 2001), EphB2 expression was initially low but then greatly increased by 7DIV (Figure S3). Levels remained elevated as development proceeded, though EphB2 expression appeared reduced by 14DIV and beyond in comparison to week 2 in vitro (Figure S3). Together, this work suggests that EphB2 is required for the maintenance of dendritic spines late in neuronal development but not necessary for the continued adhesion of pre- and postsynaptic terminals, which remain intact even when EphB2 expression is knocked down. In addition, our results suggest that the temporal change in EphB2 function might be due in part to changes in levels of EphB2 expression during development.
Having addressed the necessity of EphB signaling early in synaptogenesis, we next used a rescue approach at multiple time points in TKO neurons to investigate whether EphB is sufficient to direct normal synapse formation when present only later in development. Consistent with work in cultured brain slices (Kayser et al., 2006), we were able to rescue defects in synapse formation with transfection of YFP-tagged EphB2 (EphB2-YFP) in TKO neurons at 3DIV and fixation at 10DIV (Figure 4G). To confirm that this rescue persists, we also transfected 3DIV TKO neurons and examined synapse density at 21DIV, which again resulted in successful rescue (Figure 4H). We then asked whether late expression of EphB2 would overcome early defects or whether older neurons become unable to respond to EphB if it is absent early. When we expressed EphB2-YFP in TKO neurons from 10 to 21DIV, synapse density was not rescued and remained at TKO levels (Figure 4H). Taken together, these data indicate that the presence of EphB early in development is required for its function as a synaptogenic signal and raise the possibility that EphB acts as a molecular switch enabling the rapid phase of synapse formation and filopodia motility beginning at 7DIV.
Filopodia-Based Synapse Formation Requires Motility and Trans-Synaptic Interactions
The fact that EphB’s synaptogenic function is restricted to a specific developmental time period, along with its role in regulating filopodia motility, suggests a model in which EphBs control motility-based synaptogenesis during the second week in vitro. However, this hypothesis raises the question of the extent to which filopodia motility alone underlies EphB’s role as a synaptogenic signal. To address this issue, we sought to independently increase filopodia motility downstream of EphB in the context of EphB2 knockdown and determine how synapse formation is affected. Recent work has shown that overexpression of PAK, a serine/threonine kinase that signals to reorganize the actin cytoskeleton and upon which a number of Rho family GTPases downstream of EphB2 converge (Penzes et al., 2003; Tolias et al., 2007), leads to an increase in spine, filopodia, and synapse density (Zhang et al., 2005). We examined whether PAK overexpression might increase dendritic protrusion motility in EphB2 knockdown cells. Three DIV wild-type cortical neurons were cotransfected with GFP and either vector control, EphB2 shRNA, or EphB2 shRNA + myc-tagged wild-type (WT) or constitutively active (CA) PAK, and we performed time-lapse imaging of live cells at 9–10DIV. While coexpression of WT PAK along with EphB2 shRNA resulted in a small but significant increase in motility of dendritic filopodia, CA PAK cotransfected with EphB2 shRNA rescued motility to control levels (Figure 5A and Movie S3). To rule out the possibility that expression of any constitutively active serine/threonine kinase, such as PAK, increases filopodia motility in the context of EphB2 knockdown, we cotransfected 3DIV neurons with GFP, EphB2 shRNA, and constitutively active aCaMKII. Unlike expression of CA PAK with EphB2 shRNA, however, aCaMKII did not rescue the motility of dendritic filopodia in 9DIV cortical neurons in the presence of EphB2 knockdown (aCaMKII + EphB2 shRNA: total distance moved per filopodium over 30 min = 2.02 ± 0.23 mm, n = 102 filopodia, 6 cells; p = 0.87 compared to EphB2 shRNA alone; ANOVA).
We next examined whether increasing dendritic filopodia motility in EphB2 knockdown neurons is also sufficient to direct normal synapse formation. We transfected neurons at 3DIV with GFP and either vector control, EphB2 shRNA, or EphB2 shRNA + WT PAK or CA PAK. Neurons were fixed at 10DIV and immuno-stained with antibodies recognizing PSD-95 and vGlut1 to determine synapse density. Despite previous results showing that PAK expression can induce synapse formation (Zhang et al., 2005), neither WT nor CA PAK were able to rescue the reduced synapse density found with knockdown of EphB2, although cotransfection with B2-R did restore motility and synapse density to control levels (Figures 5A–5C). Thus, motility of postsynaptic dendritic filopodia is not alone sufficient to drive normal synaptogenesis.
In addition to its kinase-domain-dependent signaling to the actin cytoskeleton (Ethell et al., 2001; Irie and Yamaguchi, 2002; Penzes et al., 2003; Tolias et al., 2007), EphB2 has a number of identified domain-specific activities at the synapse, including local protein-protein interactions such as ephrin-binding, an extracellular domain interaction with NMDARs, and PDZ binding domain-dependent clustering of AMPARs (Dalva et al., 2000; Kayser et al., 2006). We asked whether normal levels of dendritic protrusion motility combined with an EphB kinase mutant capable of local protein interactions but not competent to signal downstream to the actin cytoskeleton might be sufficient to direct normal synapse formation. To accomplish this, we coexpressed EphB2 shRNA with CA PAK and a kinase-inactive EphB2 mutant (B2KI-R) insensitive to knockdown by EphB2 shRNA. B2KI-R transfected with EphB2 shRNA alone in 3DIV neurons failed to rescue either the defective motility or synapse density at 9–10DIV, indicating that, without normal filopodial motility, the ability of EphB to interact with ephrinB and other synaptic proteins is not sufficient to induce wild-type levels of synapse formation (Figures 6A–6C). However, coexpression of B2KI-R + CA PAK in the context of EphB2 knockdown rescued both the motility of dendritic filopodia as well as synapse density (Figures 6A–6C andMovie S4), demonstrating that EphB-mediated local protein interactions combined with independent activation of PAK is sufficient to drive normal synaptogenesis.
Figure 6. Motility of Postsynaptic Filopodia Coupled with EphB-ephrinB Trans-Cellular Interactions Is Required for EphB-Dependent Synaptogenesis.

(A) Total distance individual protrusions move throughout 30 min of imaging of 9–10DIV cells transfected with the indicated constructs + GFP at 3DIV (B2 kinase inactive rescue (B2KI-R): 113 p, 9 c; B2KI-R + CA PAK: 82 p, 7 c; B2 kinase inactive ephrin binding domain deletion rescue (B2DebKI-R): 89 p, 6 c; B2DebKI-R + CA PAK: 101 p, 6 c; B2 ephrin-binding domain deletion rescue (B2Deb-R): 138 p, 9 c; B2Deb-R + CA PAK: 100 p, 9 c; *p < 0.0001 compared to B2 shRNA; ANOVA).
(B) Synapse density on 10DIV cells transfected with the indicated constructs + GFP at 3DIV (B2KI-R, 18 Cells; B2KI-R + CA PAK, 24; B2DebKI-R, 24; B2DebKI-R + CA PAK, 25; B2Deb-R, 18; B2Deb-R + CA PAK, 20; *p < 0.0001 compared to B2 shRNA; ANOVA). Error bars indicate SEM.
(C) Representative images of dendrites from 10DIV cells quantified in (B). Arrows indicate synapses (colocalization of PSD-95 [red] and vGlut1 [blue]). Scale bars, 2 mm. Error bars indicate SEM.
If filopodia motility leads to dendrite-axon contact initiation and synapse induction, we would predict that a cell-surface molecule involved in this process must be able to bind its trans-cellular partner in order for synapse formation to occur. Thus, we wanted to test specifically whether a trans-cellular interaction with the presynaptic axon is required for EphB motility-based synaptogenesis. We generated a kinase-inactive EphB2 mutant from which the ephrin-binding globular domain was deleted, also with silent mutations rendering the construct insensitive to EphB2 shRNA knockdown (B2DebKI-R). Deletion of the ephrin-binding domain from EphB prevents the recruitment of ephrinBs and recycling presynaptic vesicles in a heterologous cell coculture assay (Kayser et al., 2006). We cotransfected B2DebKI-R with EphB2 shRNA in 3DIV cortical neurons, either with or without CA PAK, and examined filopodia motility and synapse density at 9–10DIV. Expression of B2DebKI-R failed to rescue filopodia motility and synapse formation when cotransfected only with EphB2 shRNA (Figures 6A–6C). Moreover, while coexpression of CA PAK with B2DebKI-R increased filopodia motility, these constructs together also failed to restore synapse density (Figures 6A–6C). Thus, without EphB-ephrinB interactions, normal levels of filopodia motility are not sufficient to drive normal synapse development. Finally, to examine the possibility that EphB-dependent filopodia motility might be triggered independently of the ephrin-binding domain, we generated an EphB2 rescue mutant expression construct lacking this domain but with a functional kinase domain (B2Deb-R). Using an anti-phospho-EphB2 antibody, we found that B2Deb-R expressed in neurons is not constitutively active (data not shown), allowing us to test whether it becomes activated in an ephrin-binding-domain-independent manner in our motility and synapse formation assays. Coexpression of B2Deb-R with EphB2shRNA failed to rescue motility and, as expected, synapse formation (Figures 6A-6C). Taken together, this series of rescue experiments indicates that in 9–10DIV neurons, EphB-dependent synapse formation requires forward signaling that leads to PAK activation and increased motility of dendritic filopodia, combined with trans-cellular ephrin binding.
DISCUSSION
A number of studies have characterized the dynamics of filopo-dial protrusions that precede the formation of synapses and spines on dendritic branches (Dailey and Smith, 1996; Lendvai et al., 2000; Zito et al., 2004; Ziv and Smith, 1996). However, in contrast to the extensive understanding of molecular cues controlling spinogenesis (Ethell and Pasquale, 2005; Tada and Sheng, 2006), less is known regarding what signals regulate the motility of dendritic filopodia and how this motility is linked to cell-cell interactions required for the establishment of a synapse. We find that loss of EphB signaling causes reduced motility of filopodia and, in turn, a reduction in the cellular area explored by these filopodia. In addition, the synaptogenic activity of EphB is restricted to the time in neuronal development that filopodia are most prevalent. Knockdown and rescue experiments demonstrate that EphB-dependent motility-based synaptogenesis occurs at a rapid rate in comparison to synapses added via EphB-independent mechanisms. Finally, we show that neither normal filopodia motility nor the ability for EphB and ephrinB to interact is alone sufficient to drive normal synaptogenesis. Rather, both motility and a trans-cellular Ephephrin interaction are required. Thus, in EphB receptor signaling we find a molecular mechanism able to regulate filopodia motility, provide a trans-cellular interaction between the dendrite and axon, and induce synapse differentiation and spine morphogenesis.
Dendritic Filopodia Motility and Synaptogenesis
Motile filopodia are hypothesized to provide an extended volume within which a dendrite can establish increased numbers of transient axonal contacts (Ziv and Smith, 1996). In addition, work in rat hippocampal slices has found that induction of actin polymerization leads to hypermotility of dendritic protrusions and the formation of more spines and synapses (Zito et al., 2004). Increasing the chance of encounter between axons and dendrites, however, is not thought to be sufficient for synapse formation without a mechanism for stabilizing cell-cell interactions (Dailey and Smith, 1996). Here, we probed not only the role of filopodia motility but also how cell-cell interactions and a synaptogenic factor are related to motility during synapse formation in both dissociated cultures and brain slices.
Our data provide evidence to support the long-standing proposition that filopodia motility is only one aspect of establishing a synapse; in addition, a trans-cellular interaction between axon and dendrite is required. The molecular cue mediating this interaction appears to be surprisingly specific: even when motility is restored, the absence of only EphB prevents neurons from adding normal numbers of synapses, despite the presence of many other factors that enable cell-cell signaling and adhesion. It will be important to explore whether loss of other adhesion signals also results in reduced filopodia motility and synapse number or whether this effect is exclusive to EphB. Regardless, these findings suggest a model in which EphB downstream signaling leads to PAK activation and increased filopodia motility, resulting in enhanced short-range dendritic exploration for axonal partners (Figure 7A). Dendritic EphB would then be able to recognize and stabilize contacts with appropriate presynaptic axons bearing ephrinB. Following this initial axon-dendrite contact, both pre- and postsynaptic components can be assembled (Dalva et al., 2000; Kayser et al., 2006), and the filopodium matures into a dendritic spine either directly or following retraction to the dendritic shaft (Fiala et al., 1998; Ziv and Smith, 1996).
Figure 7. Model of EphB Functions during Synaptogenesis.
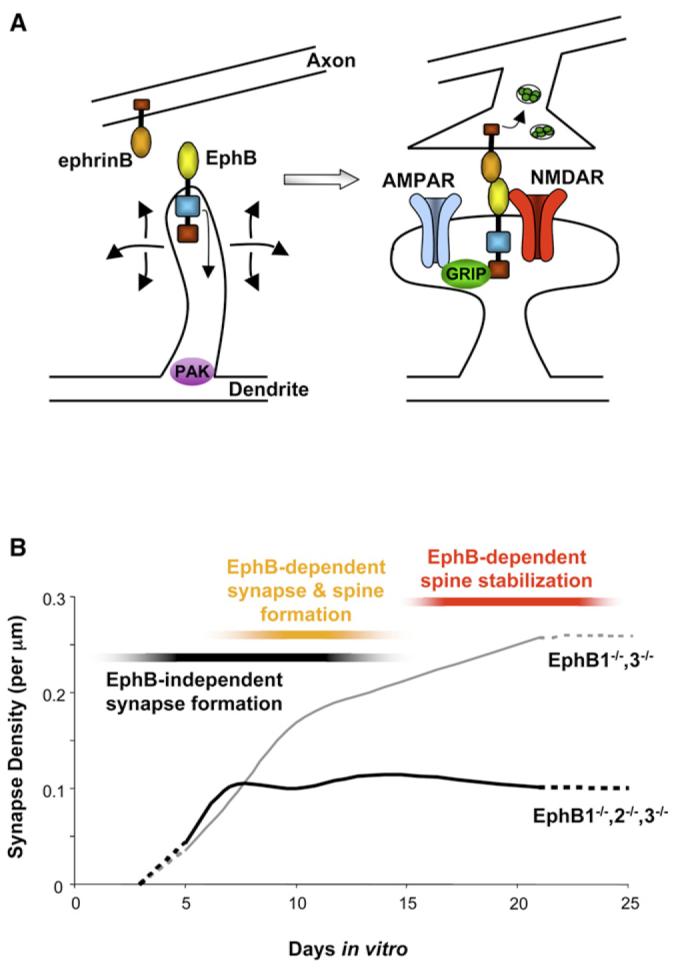
(A) Mechanism for EphB-dependent synapse formation: EphB directs synapse development by modulating filopodia motility via PAK and providing a trans-cellular interaction via ephrinB. Upon stable binding of axonal ephrinB, EphB is able to direct presynaptic terminal differentiation, NMDAR and AMPAR clustering, and formation of dendritic spines.
(B) EphB is active during a discrete time period to guide the formation of a subgroup of synapses. The earliest forming contacts do not require EphB. During the second week in vitro, these mechanisms continue, but a rapidly forming group of synapses that are EphB dependent are also added. By the third week in vitro, EphB is no longer involved in the formation or maintenance of synaptic contacts but is required for the stabilization of dendritic spines (hashed lines indicate extrapolated data from collected data points).
How do EphBs and PAK work together to coordinate the formation of a synaptic input? PAK has previously been demonstrated to regulate synapse number (Zhang et al., 2005), but we find that while expression of constitutively active PAK (CA PAK) in the context of EphB knockdown rescues filopodia motility, normal synaptogenesis is not restored. Likewise, expression of a kinase-inactive EphB2 rescue construct (B2KI-R) competent to bind ephrinB, NMDARs, and PDZ domain-containing proteins (Dalva et al., 2000; Kayser et al., 2006) is not sufficient to drive normal synapse formation when motility is reduced by EphB knockdown. Remarkably, independent reconstitution of motility with CA PAK and of local protein interactions with B2KI-R does restore synaptogenesis. Deletion of the ephrin-binding domain, with or without the kinase-inactivating mutation, prevents rescue of synapse formation, demonstrating that the ability of EphB to bind ephrinB is specifically required. Future work will be needed to understand how the EphB-ephrinB interaction functions both to induce forward signaling that leads to filopodia motility, as well as cell-cell adhesion and synapse formation. One possibility is that transient Eph-ephrin interactions between axons and dendrites induce EphB kinase-dependent motility, while stable interactions mediate synapse development. In this model, other factors such as metalloproteases or cell adhesion molecules could regulate the stability of the Eph-ephrin interactions. Alternatively, postsynaptic EphB might become activated through an interaction with postsynaptic ephrinB ligands in the cis configuration, resulting in enhanced filopodia motility, culminating in establishment of a synaptic contact through trans interactions with pre-synaptic ephrinBs. Although EphB signaling is already known to be important throughout neural development, with roles in border formation, axon guidance, synapse formation, and synaptic plasticity (Dalva et al., 2007; Flanagan and Vanderhaeghen, 1998), our work extends a defined function for EphB in the earliest stages of filopodia exploration and initial dendrite-axon contact formation.
The Role of EphB2 Signaling in Cultured Brain Slices
Our earlier work demonstrated that brain sections from animals lacking EphB1-3 show a 40% reduction in excitatory synapse density in cortex (Kayser et al., 2006). We report here that filopodia on neuronal dendrites in cultured brain slices from TKO animals are significantly less motile than filopodia in DKO or wild-type controls. In comparison to filopodia of control neurons in dissociated culture, the predominant motion of filopodia in brain slice is extension and retraction, with few observed sweeping motions. A simple explanation for this difference is that the more compact and complex cellular environment in brain slice might form a significant restraint to filopodia motility. Further work will be needed to determine whether these differences reflect changes in signaling in addition to the density of the extra-cellular milieu. Somewhat surprisingly, we also find that knockdown specifically of EphB2 in wild-type brain slices results in reduced motility, though knockout of EphB2 alone has no reported effect on synapse structure or density (Henderson et al., 2001). Thus, our data suggest that the acute knockdown of EphB2 is different than the embryonic knockout of the same molecule, possibly because knockdown does not provide adequate time for compensatory mechanisms. In sum, our data demonstrate that the effects of EphBs—and specifically EphB2—on dendritic filopodia motility are conserved in cortical brain slices, which more closely mimic the intact tissue. Even in this more complex environment where filopodia have increased potential axonal partners, we find that EphB plays a significant role in dendritic filopodia motility.
Temporally Restricted Role for EphBs in Synapse Formation
The development of synaptic inputs in vitro is a stereotyped and tightly regulated process. Although a relatively large number of molecules with roles in synaptogenesis have been identified (Dalva et al., 2007; Scheiffele, 2003), there has been little work examining whether particular synaptogenic signals are important at certain times during development. Our results demonstrate that EphB is only required as a synaptogenic signal during the period of neuronal development when filopodia are the predominant protrusion type and motility is high. Based on the distinct rates of synapse addition at different times in the presence and absence of EphB, we propose that there are at least three independent but overlapping phases of synaptogenesis (Figure 7B).
The first phase begins early and is EphB independent but stretches from before 5DIV through the second week in vitro. Synapses are added at a constant rate throughout this phase, which is obscured by the normal rapid rate of synaptogenesis during week 2 and thus only revealed in the absence of EphBs. Further work will be needed to determine the molecular mechanisms guiding the establishment of the earliest-forming EphB-independent populations of inputs. A second phase of synapto-genesis requires EphB and occurs during the second week in culture. It is within this EphB-dependent phase that synapses are added at a particularly rapid rate, requiring motility of dendritic filopodia. EphB-dependent and -independent mechanisms, therefore, function concurrently within single neurons during the second week in vitro. Synapses (and spines) are thought to form early in development either from motile dendritic filopodia contacting axons (Dailey and Smith, 1996; Fiala et al., 1998; Friedman et al., 2000; Ziv and Smith, 1996) or from pre-formed axonal packets contacting dendrites (Ahmari et al., 2000; Gerrow et al., 2006; Harris, 1999; Miller and Peters, 1981). Our results raise the possibility that EphB-dependent synapse formation is motility based and postsynaptically induced. Consistent with this hypothesis, synapse number scales linearly with dendritic arbor growth in the absence of EphBs, suggesting that these dendrites are only able to passively add new synapses.
The last phase of synaptogenesis is also EphB independent and begins around 14DIV. Neurons either with or without EphBs reach a similar plateau in the rate of synapse addition during this third week in culture. The EphB-independent mechanisms responsible for setting the low rate of addition at this time could be signals promoting contact formation, signals predominately involved in contact maintenance, and/or signals that increase the rate of synapse elimination. For example, it appears that two synaptogenic factors, SALM2 and neuroligin, are particularly important later in neuronal maturation for synapse maintenance (Ko et al., 2006; Varoqueaux et al., 2006), and recent work has begun to identify specific molecular pathways regulating synapse elimination (Ding et al., 2007). Alternatively, what we characterize as a final distinct phase of addition based on the low rate of synapse formation might be a continuation of the earliest EphB-independent period of synaptogenesis. Regardless, because each phase of synapse development is characterized by a unique rate of addition, our findings suggest that distinct molecular mechanisms are likely to guide synapse formation during different time periods. As discussed above, it will be interesting to examine whether other known synaptogenic factors also have temporally restricted functions like EphBs. In addition, more work will be required to determine whether synapses added at particular times in development differ in terms of molecular composition and/or function in the neuron.
Early and Late Functions for EphBs in Synapse/ Spine Development
Expression of EphBs is required both at the onset and throughout the second week in vitro to direct normal motility-based synapse addition. Re-expression of EphB2 in TKO neurons in the middle of the second week in vitro—after the normal time of initiation of the rapid phase of synaptogenesis at • 7DIV—does not rescue synapse number. In addition, knockdown of EphB2 in wild-type neurons in the middle of the second week in vitro (10DIV) results in fewer synapses at 21DIV, while knockdown from 0–7DIV or from 14–21DIV does not affect synapse number. Thus, the period in which EphBs act as regulators of synapse formation has a defined beginning (• 7DIV) and end point (• 14DIV), before which they are not required and after which they play no apparent role. Furthermore, EphB expression must be maintained throughout this window of rapid synapse addition for it to progress normally. Our results suggest a model in which EphB acts as a molecular switch to induce the formation of a discrete class of synapses defined by the timing and rate of their addition, as well as a reliance on filopodia motility. Consistent with this model, EphB expression in vitro is upregulated at the onset of this phase and remains high during the period of EphB-dependent synapse development. In vivo, the rapid phase of synaptogenesis occurs as neurons are forming the specific functional contacts found in adults (Blue and Parnavelas, 1983; Dalva and Katz, 1994). Interestingly, the expression of EphB2 in hippocampus and cortex is also upregulated in vivo during this period of synapse development (Henderson et al., 2001), further supporting a temporally specific role. Thus, defining the mechanisms that control EphB expression during neuronal maturation should provide insights into mechanisms that regulate synapse development.
Although EphBs are only essential for synapse addition during the second week in vitro, we define a broader temporal role for EphB in dendritic spine morphogenesis. In fact, we find that the functions of EphB as a synaptogenic and spinogenic signal are temporally separable. During the second week in vitro, EphBs appear to control formation of numerous synapses and spines. Throughout the late phase of synaptogenesis (after 14DIV), however, EphBs are not required for maintaining synapse number but are still involved in the stabilization of dendritic spine shape. Consistent with this result, we find that EphB2 expression remains elevated as neurons mature (14–28DIV), but at levels lower than that seen during the initial phase of EphB-dependent synapotogenesis (7–14DIV). These data suggest that, when expressed at its highest levels, EphB acts to initiate synaptogenesis and spinogenesis, and when expressed at lower levels, EphB regulates spine shape. These findings imply that, at mature spine synapses, other trans-cellular factors likely play the principle role in maintaining pre- and postsynaptic terminal adhesion. Interestingly, the morphology of dendritic protrusions following late EphB2 knockdown (Figure 4E) is reminiscent of the overabundant elongated filopodia-like processes found on dendrites in patients with fragile X syndrome and a fragile X mouse model (Bagni and Greenough, 2005; Irwin et al., 2001; Pfeiffer and Huber, 2007). Along with results demonstrating that inhibition of PAK rescues abnormalities in fragile X mice (Hayashi et al., 2007), these data suggest that further examination of the link between EphB, PAK, and filopodia motility might yield insights into fragile X syndrome.
Conclusions
Our work addresses two important principles of CNS synapto-genesis. First, the time during which synapses form might be an important determinant of how they are made. Numerous synaptogenic factors have been identified in the mammalian CNS, but the purpose of having so many is unclear. The temporally restricted nature of EphB as a signal for synapse formation raises the possibility that other synaptogenic molecules may share similar specificity, with each organizing distinct populations of inputs during different windows of development. Moreover, contacts organized by one molecule might rely on dendritic filopodia motility and be induced by the postsynaptic neuron, while others are motility independent and induced by the presynaptic axon. Second, EphB provides a link between filopodia motility, cell-cell interactions, and synapse formation. The requirement of the EphB-ephrinB interaction in this process is a clear demonstration that the ability of a neuron to explore a maximal amount of extracellular space for a presynaptic partner must be coupled with an ability to recognize and stabilize a contact with that partner.
EXPERIMENTAL PROCEDURES
Cortical Neuronal Culture
Dissociated cortical neurons were prepared from E17–18 rats and cultured as described previously (Kayser et al., 2006). Dissociated cortical neurons were also made from postnatal day 1–3 EphB1•/•, EphB2•/•, EphB3•/•and EphB1•/•, EphB3•/• mice generated by Dr. Mark Henkemeyer (Henkemeyer et al., 2003) and wild-type CD1 mice, cultured in NB-A (GIBCO) plus supplements and 10 mm FUDR (5-Fluoro-2-deoxyuridine and Uridine [Sigma]). Cortical brain slices were prepared and cultured as described previously (Kayser et al., 2006).
Antibodies
The following antibodies were used: mouse aPSD-95 (Affinity Bioreagents); rabbit aEphB2, a-phospho-EphB2 (Dalva et al., 2000); goat aEphB2 (R&D Systems); guinea pig avGlut1 (Chemicon); chicken aGFP (Chemicon). Cy2, Cy3, and Cy5 secondary antibodies were obtained from Jackson Immuno Research and used at 1:250.
Immunocytochemistry
Dissociated cortical neurons were fixed in 4% PFA/2% sucrose in PBS for 8 min at room temperature. Neurons were then washed three times for 5 min in PBS and blocked for 2 hr at room temperature with either 5% normal goat serum or 1% albumin from chicken egg white (Sigma) + 0.2% gelatin from cold water fish skin (Sigma), permeabilized with 0.1% saponin (Sigma). Neurons were immunostained with primary antibodies overnight at 4°C and secondary antibodies for 45 min at room temperature.
cDNA and shRNA Constructs
EphB2 shRNAs and EphB2-YFP were described previously (Kayser et al., 2006). B2-R, B2KI-R, and B2Deb-R were generated by making silent mutations within the region targeted by EphB2 shRNA in either a full-length FLAG-tagged EphB2, the previously described FLAG-tagged kinase-inactive EphB2 (Dalva et al., 2000), or the previously described FLAG-tagged EphB2 globular domain deletion mutant (Kayser et al., 2006). B2DebKI-R was generated based on B2Deb-R by making a kinase-inactivating point mutation in the EphB2 kinase domain (Dalva et al., 2000) and silent mutations to the EphB2 shRNA target region. PFUG vector containing SynRFP was a generous gift from Dr. Mark Lush and Dr. Jonathan Raper (University of Pennsylvania). Constitutively active aCaMKII was a generous gift from Dr. Steve Moss (University of Pennsylvania). Lentivirus was produced and purified by the Gene Therapy Program Penn Vector Core Facility at the University of Pennsylvania. Myc-tagged wild-type and constitutively active PAK were generous gifts from Dr. Jonathan Chernoff (Fox Chase Cancer Center).
Transfection/Infection of Neuronal Cultures
Three DIV rat or mouse cortical neurons were transfected using a modified version of the calcium phosphate precipitation method (Xia et al., 1996) with washes in DMEM acidified with CO2. Cortical neurons were transfected in suspension using Lipofectamine 2000 (Takasu et al., 2002), as were cortical neurons transfected at 10–14DIV. Two DIV cortical neurons were infected with lentivirus for 16–18 hr at a titer of • 2 viral particles per cell. Neurons in cultured brain slices were transfected using the Helios Gene Gun as described previously (Kayser et al., 2006).
Imaging and Analysis
Fixed cortical neurons were imaged using confocal scanning microscopy (Leica). Images are z projections of • 2–4 images taken at 0.3–0.4 mm step intervals. All images were acquired blind to experimental condition or analyzed blind to condition in NIH ImageJ using custom programming. Puncta cocluster analysis was described previously (Kayser et al., 2006), and dendritic arbor length was measured using NeuronJ plugin. Total number of synapses at each time point was calculated by multiplying average synapse density by total dendritic arbor length on a cell-by-cell basis. Rate of synapse addition was calculated as the slope of the best-fit line between specified data points. Spine and filopodia density was determined by manually counting and classifying protrusions as “spine,” “filopodia,” or “unsure” along a minimum of 40 mm of dendrite per cell. Live cell motility experiments in dissociated neurons were imaged with confocal microscopy using a 633 oil-immersion objective, with coverslips inverted on a flow chamber (Warner), bathed in HEPES-buffered ACSF (in mM concentrations: 140 NaCl, 5 KCl, 1 MgCl2, 2 CaCl2, 20 glucose, 10 HEPES, pH 7.2) at 37°C. To reduce photobleaching, laser levels were set as low as possible to still permit detection of cells/puncta, the pinhole opened to 3AU, and scanning minimized by taking 0.5–0.8 mm steps to create z projections. Images were collected every 3 min for 30 min for GFP motility experiments and every 5 min for 2 hr for PSD-95-GFP ± SynRFP experiments. For each, the time between images was chosen to minimize laser exposure of the field while allowing resolution of protrusions or puncta. Motility data in cultured brain slices were acquired in 1 mm steps with a two-photon laser-scanning confocal microscope described previously (Kayser et al., 2006). Brain slices were imaged using a 403 water-immersion objective in an open flow chamber (Warner), bathed in ACSF described above at 37°C. In all motility experiments, frames were aligned in ImageJ and protrusion tips or puncta tracked throughout the series using Manual Tracking plugin. For all analysis, statistical measures were conducted on a per cell or per protrusion basis as indicated and collected from a minimum of three independent experiments.
Supplementary Material
ACKNOWLEDGMENTS
We thank R. Balice-Gordon, G. Bashaw, P. Haydon, J. Raper, and members of the Dalva Lab for helpful discussions and advice; A. McClelland for help with ImageJ programming; J. Chernoff for providing PAK constructs; M. Lush and J. Raper for SynRFP; and S. Moss for aCaMKII. This work was supported by an NIH Ruth L. Kirschstein National Research Service Award NS051894-01 (M.S.K.), the Training Program in Developmental Biology (5T32HD007516) (M.J.N.), and the Whitehall Foundation, Philadelphia Foundation/MRDDRC (HD-026979-0), Edward Mallinckrodt, Jr., Foundation, the CRCNS program and NIMH (MH073357), and National Institute of Drug Addiction (DA022727) (M.B.D.).
REFERENCES
- Ahmari SE, Buchanan J, Smith SJ. Assembly of presynaptic active zones from cytoplasmic transport packets. Nat. Neurosci. 2000;3:445–451. doi: 10.1038/74814. [DOI] [PubMed] [Google Scholar]
- Bagni C, Greenough WT. From mRNP trafficking to spine dysmorphogenesis: the roots of fragile X syndrome. Nat. Rev. Neurosci. 2005;6:376–387. doi: 10.1038/nrn1667. [DOI] [PubMed] [Google Scholar]
- Blue ME, Parnavelas JG. The formation and maturation of synapses in the visual cortex of the rat. II. Quantitative analysis. J. Neurocytol. 1983;12:697–712. doi: 10.1007/BF01181531. [DOI] [PubMed] [Google Scholar]
- Dailey ME, Smith SJ. The dynamics of dendritic structure in developing hippocampal slices. J. Neurosci. 1996;16:2983–2994. doi: 10.1523/JNEUROSCI.16-09-02983.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dalva MB, Katz LC. Rearrangements of synaptic connections in visual cortex revealed by laser photostimulation. Science. 1994;265:255–258. doi: 10.1126/science.7912852. [DOI] [PubMed] [Google Scholar]
- Dalva MB, Takasu MA, Lin MZ, Shamah SM, Hu L, Gale NW, Greenberg ME. EphB receptors interact with NMDA receptors and regulate excitatory synapse formation. Cell. 2000;103:945–956. doi: 10.1016/s0092-8674(00)00197-5. [DOI] [PubMed] [Google Scholar]
- Dalva MB, McClelland AC, Kayser MS. Cell adhesion molecules: signalling functions at the synapse. Nat. Rev. Neurosci. 2007;8:206–220. doi: 10.1038/nrn2075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ding M, Chao D, Wang G, Shen K. Spatial regulation of an E3 ubiquitin ligase directs selective synapse elimination. Science. 2007;317:947–951. doi: 10.1126/science.1145727. [DOI] [PubMed] [Google Scholar]
- Ethell IM, Pasquale EB. Molecular mechanisms of dendritic spine development and remodeling. Prog. Neurobiol. 2005;75:161–205. doi: 10.1016/j.pneurobio.2005.02.003. [DOI] [PubMed] [Google Scholar]
- Ethell IM, Irie F, Kalo MS, Couchman JR, Pasquale EB, Yamaguchi Y. EphB/syndecan-2 signaling in dendritic spine morphogenesis. Neuron. 2001;31:1001–1013. doi: 10.1016/s0896-6273(01)00440-8. [DOI] [PubMed] [Google Scholar]
- Fiala JC, Feinberg M, Popov V, Harris KM. Synaptogenesis via dendritic filopodia in developing hippocampal area CA1. J. Neurosci. 1998;18:8900–8911. doi: 10.1523/JNEUROSCI.18-21-08900.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Flanagan JG, Vanderhaeghen P. The ephrins and Eph receptors in neural development. Annu. Rev. Neurosci. 1998;21:309–345. doi: 10.1146/annurev.neuro.21.1.309. [DOI] [PubMed] [Google Scholar]
- Friedman HV, Bresler T, Garner CC, Ziv NE. Assembly of new individual excitatory synapses: time course and temporal order of synaptic molecule recruitment. Neuron. 2000;27:57–69. doi: 10.1016/s0896-6273(00)00009-x. [DOI] [PubMed] [Google Scholar]
- Gerrow K, Romorini S, Nabi SM, Colicos MA, Sala C, El-Husseini A. A preformed complex of postsynaptic proteins is involved in excitatory synapse development. Neuron. 2006;49:547–562. doi: 10.1016/j.neuron.2006.01.015. [DOI] [PubMed] [Google Scholar]
- Goda Y, Davis GW. Mechanisms of synapse assembly and disassembly. Neuron. 2003;40:243–264. doi: 10.1016/s0896-6273(03)00608-1. [DOI] [PubMed] [Google Scholar]
- Harris KM. Structure, development, and plasticity of dendritic spines. Curr. Opin. Neurobiol. 1999;9:343–348. doi: 10.1016/s0959-4388(99)80050-6. [DOI] [PubMed] [Google Scholar]
- Hayashi ML, Rao BS, Seo JS, Choi HS, Dolan BM, Choi SY, Chat-tarji S, Tonegawa S. Inhibition of p21-activated kinase rescues symptoms of fragile X syndrome in mice. Proc. Natl. Acad. Sci. USA. 2007;104:11489–11494. doi: 10.1073/pnas.0705003104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Henderson JT, Georgiou J, Jia Z, Robertson J, Elowe S, Roder JC, Pawson T. The receptor tyrosine kinase EphB2 regulates NMDA-dependent synaptic function. Neuron. 2001;32:1041–1056. doi: 10.1016/s0896-6273(01)00553-0. [DOI] [PubMed] [Google Scholar]
- Henkemeyer M, Itkis OS, Ngo M, Hickmott PW, Ethell IM. Multiple EphB receptor tyrosine kinases shape dendritic spines in the hippo-campus. J. Cell Biol. 2003;163:1313–1326. doi: 10.1083/jcb.200306033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Irie F, Yamaguchi Y. EphB receptors regulate dendritic spine development via intersectin, Cdc42 and N-WASP. Nat. Neurosci. 2002;5:1117–1118. doi: 10.1038/nn964. [DOI] [PubMed] [Google Scholar]
- Irwin SA, Patel B, Idupulapati M, Harris JB, Crisostomo RA, Larsen BP, Kooy F, Willems PJ, Cras P, Kozlowski PB, et al. Abnormal dendritic spine characteristics in the temporal and visual cortices of patients with fragile-X syndrome: a quantitative examination. Am. J. Med. Genet. 2001;98:161–167. doi: 10.1002/1096-8628(20010115)98:2<161::aid-ajmg1025>3.0.co;2-b. [DOI] [PubMed] [Google Scholar]
- Kayser MS, McClelland AC, Hughes EG, Dalva MB. Intra-cellular and trans-synaptic regulation of glutamatergic synaptogenesis by EphB receptors. J. Neurosci. 2006;26:12152–12164. doi: 10.1523/JNEUROSCI.3072-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ko J, Kim S, Chung HS, Kim K, Han K, Kim H, Jun H, Kaang BK, Kim E. SALM synaptic cell adhesion-like molecules regulate the differentiation of excitatory synapses. Neuron. 2006;50:233–245. doi: 10.1016/j.neuron.2006.04.005. [DOI] [PubMed] [Google Scholar]
- Lendvai B, Stern EA, Chen B, Svoboda K. Experience-dependent plasticity of dendritic spines in the developing rat barrel cortex in vivo. Nature. 2000;404:876–881. doi: 10.1038/35009107. [DOI] [PubMed] [Google Scholar]
- Miller M, Peters A. Maturation of rat visual cortex. II. A combined Golgi-electron microscope study of pyramidal neurons. J. Comp. Neurol. 1981;203:555–573. doi: 10.1002/cne.902030402. [DOI] [PubMed] [Google Scholar]
- Papa M, Bundman MC, Greenberger V, Segal M. Morphological analysis of dendritic spine development in primary cultures of hippocampal neurons. J. Neurosci. 1995;15:1–11. doi: 10.1523/JNEUROSCI.15-01-00001.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Penzes P, Beeser A, Chernoff J, Schiller MR, Eipper BA, Mains RE, Huganir RL. Rapid induction of dendritic spine morphogenesis by trans-synaptic ephrinB-EphB receptor activation of the Rho-GEF kalirin. Neuron. 2003;37:263–274. doi: 10.1016/s0896-6273(02)01168-6. [DOI] [PubMed] [Google Scholar]
- Pfeiffer BE, Huber KM. Fragile X mental retardation protein induces synapse loss through acute postsynaptic translational regulation. J. Neurosci. 2007;27:3120–3130. doi: 10.1523/JNEUROSCI.0054-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rao A, Kim E, Sheng M, Craig AM. Heterogeneity in the molecular composition of excitatory postsynaptic sites during development of hippocampal neurons in culture. J. Neurosci. 1998;18:1217–1229. doi: 10.1523/JNEUROSCI.18-04-01217.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scheiffele P. Cell-cell signaling during synapse formation in the CNS. Annu. Rev. Neurosci. 2003;26:485–508. doi: 10.1146/annurev.neuro.26.043002.094940. [DOI] [PubMed] [Google Scholar]
- Small JV, Stradal T, Vignal E, Rottner K. The lamellipodium: where motility begins. Trends Cell Biol. 2002;12:112–120. doi: 10.1016/s0962-8924(01)02237-1. [DOI] [PubMed] [Google Scholar]
- Tada T, Sheng M. Molecular mechanisms of dendritic spine morphogenesis. Curr. Opin. Neurobiol. 2006;16:95–101. doi: 10.1016/j.conb.2005.12.001. [DOI] [PubMed] [Google Scholar]
- Takasu MA, Dalva MB, Zigmond RE, Greenberg ME. Modulation of NMDA receptor-dependent calcium influx and gene expression through EphB receptors. Science. 2002;295:491–495. doi: 10.1126/science.1065983. [DOI] [PubMed] [Google Scholar]
- Tashiro A, Yuste R. Regulation of dendritic spine motility and stability by Rac1 and Rho kinase: evidence for two forms of spine motility. Mol. Cell. Neurosci. 2004;26:429–440. doi: 10.1016/j.mcn.2004.04.001. [DOI] [PubMed] [Google Scholar]
- Tolias KF, Bikoff JB, Kane CG, Tolias CS, Hu L, Greenberg ME. The Rac1 guanine nucleotide exchange factor Tiam1 mediates EphB receptor-dependent dendritic spine development. Proc. Natl. Acad. Sci. USA. 2007;104:7265–7270. doi: 10.1073/pnas.0702044104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Varoqueaux F, Aramuni G, Rawson RL, Mohrmann R, Missler M, Gott-mann K, Zhang W, Sudhof TC, Brose N. Neuroligins determine synapse maturation and function. Neuron. 2006;51:741–754. doi: 10.1016/j.neuron.2006.09.003. [DOI] [PubMed] [Google Scholar]
- Waites CL, Craig AM, Garner CC. Mechanisms of vertebrate synaptogenesis. Annu. Rev. Neurosci. 2005;28:251–274. doi: 10.1146/annurev.neuro.27.070203.144336. [DOI] [PubMed] [Google Scholar]
- Xia Z, Dudek H, Miranti CK, Greenberg ME. Calcium influx via the NMDA receptor induces immediate early gene transcription by a MAP kinase/ERK-dependent mechanism. J. Neurosci. 1996;16:5425–5436. doi: 10.1523/JNEUROSCI.16-17-05425.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang H, Webb DJ, Asmussen H, Niu S, Horwitz AF. A GIT1/ PIX/Rac/PAK signaling module regulates spine morphogenesis and synapse formation through MLC. J. Neurosci. 2005;25:3379–3388. doi: 10.1523/JNEUROSCI.3553-04.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zito K, Knott G, Shepherd GM, Shenolikar S, Svoboda K. Induction of spine growth and synapse formation by regulation of the spine actin cytoskeleton. Neuron. 2004;44:321–334. doi: 10.1016/j.neuron.2004.09.022. [DOI] [PubMed] [Google Scholar]
- Ziv NE, Smith SJ. Evidence for a role of dendritic filopodia in synaptogenesis and spine formation. Neuron. 1996;17:91–102. doi: 10.1016/s0896-6273(00)80283-4. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.