

Abstract
Cytoplasmic polyadenylylation is an essential process that controls the translation of maternal mRNAs during early development and depends on two cis elements in the 3′ untranslated region: the polyadenylylation hexanucleotide AAUAAA and a U-rich cytoplasmic polyadenylylation element (CPE). In searching for factors that could mediate cytoplasmic polyadenylylation of mouse c-mos mRNA, which encodes a serine/threonine kinase necessary for oocyte maturation, we have isolated the mouse homolog of CPEB, a protein that binds to the CPEs of a number of mRNAs in Xenopus oocytes and is required for their polyadenylylation. Mouse CPEB (mCPEB) is a 62-kDa protein that binds to the CPEs of c-mos mRNA. mCPEB mRNA is present in the ovary, testis, and kidney; within the ovary, this RNA is restricted to oocytes. mCPEB shows 80% overall identity with its Xenopus counterpart, with a higher homology in the carboxyl-terminal portion, which contains two RNA recognition motifs and a cysteine/histidine repeat. Proteins from arthropods and nematodes are also similar to this region, suggesting an ancient and widely used mechanism to control polyadenylylation and translation.
Early development in many animals depends, to a large extent, on the differential expression of mRNAs that have been stored during oogenesis. One mechanism used to regulate maternal mRNA translation among probably most metazoans is cytoplasmic polyadenylylation (1, 2). The essential nature of this process for development was demonstrated in experiments in which cytoplasmic polyadenylylation of key messages was impaired. In Drosophila embryos, for example, the expression of bicoid mRNA is controlled by cytoplasmic polyadenylylation. Injected bicoid mRNA is able to rescue the bicoid phenotype, an abnormal anterioposterior axis, when it contains the sequences necessary for cytoplasmic polyadenylylation, or a long poly(A) tail (3). Similarly, in Xenopus embryos, the overexpression phenotype of injected activin receptor mRNA, a reduction in mesodermal structures, is dependent upon cytoplasmic polyadenylylation elements within the message (4). Finally, in oocytes of both Xenopus and the mouse, ablation of c-mos mRNA prevents normal meiotic progression, a phenotype that can be corrected by injection of a c-mos mRNA only when it contains the sequences for cytoplasmic polyadenylylation (5, 6).
The cis elements necessary for cytoplasmic poly(A) elongation, at least in vertebrates, are located in the 3′-untranslated regions (UTRs) of mRNAs and include the hexanucleotide AAUAAA, also required for nuclear pre-mRNA cleavage and polyadenylylation, and an upstream cytoplasmic polyadenylylation element (CPE) with the general sequence UUUUUAAU, which is used during oocyte maturation, or U(12–18), which is used in early embryogenesis (5, 7–14). Recent investigations have focused on the isolation and characterization of the factors that recognize these elements. Bilger et al. (15) showed that the nuclear polyadenylylation factors poly(A) polymerase (PAP) and cleavage and polyadenylylation specificity factor (CPSF), a multisubunit complex that recognizes AAUAAA, are able to catalyze CPE-dependent polyadenylylation in vitro, provided these factors are diluted in the reaction. This result lead those investigators to propose that the CPSF is solely responsible for the sequence specificity of both nuclear and cytoplasmic polyadenylylation. Although this experiment demonstrates the interchangeability of at least some nuclear and cytoplasmic polyadenylylation factors in vitro, it does not show that the factors catalyzing both reactions are the same in vivo. In this respect, several PAPs have been cloned from Xenopus oocytes (16, 17), one of which is primarily cytoplasmic (17). Furthermore, a factor identified initially by its ability to bind to the CPE (18) has been isolated and cloned (19). This cytoplasmic protein, CPEB, binds to the CPEs of several Xenopus mRNAs and is required for their polyadenylylation both in vitro and in vivo (18–20).
We are interested in cytoplasmic polyadenylylation in mammalian oocytes. In a previous report, we showed that cytoplasmic polyadenylylation of c-mos mRNA is required for mouse oocyte maturation and that this process is dependent on the presence of two CPEs in the 3′-UTR of this message (5). In Xenopus, polyadenylylation of c-mos mRNA occurs before germinal vesicle breakdown, one of the initial events of maturation (6), while in the mouse it occurs after germinal vesicle breakdown (5). The different timing of polyadenylylation of this message in oocytes of both species suggests that distinct factors may be involved. However, the CPEs of mouse c-mos mRNA are similar to those found in several Xenopus mRNAs that are bound by CPEB (20), raising the possibility that a similar protein exists in the mouse that controls c-mos mRNA polyadenylylation. In this report we show that mouse oocytes do indeed contain a homolog of CPEB, which we have termed mCPEB (for mouse CPEB). mCPEB has an 80% overall identity with its Xenopus counterpart (XCPEB) and is 91% identical in the region of the protein containing the putative RNA recognition domains. Moreover, this region is similar to the putative RNA binding domains in the Drosophila protein orb (21, 22) and in two Caenorhabditis elegans proteins of unknown function (23). mCPEB mRNA is primarily restricted to ovary, testis, and kidney. Within the ovary, it is exclusively present in oocytes. As with Xenopus, mCPEB is a 62-kDa protein that binds to the CPEs of mouse c-mos mRNA. The conservation of CPEB-like sequences from arthropods to mammals suggests a conserved mechanism to control polyadenylylation and translation.
MATERIALS AND METHODS
Amplification of Mouse CPEB cDNA Fragments.
Xenopus CPEB (XCPEB)-specific oligonucleotides 2 and 3 (see oligonucleotide sequences below), complementary to a conserved region between XCPEB and Drosophila orb, were used to amplify mCPEB sequences by RT/PCR from 0.5 μg of total mouse ovary RNA. An mCPEB cDNA fragment of 96 nt was obtained and sequenced. Using this sequence, mCPEB-specific oligonucleotide 6 was synthesized and used to screen a mouse ovary UniZap (λ Zap) cDNA library (provided by J. Dean, National Institutes of Health, Bethesda). One positive clone was obtained after screening 3 × 105 plaque-forming units. This clone contained 1712 nt, including the sequence encoding the carboxyl-terminal half of mCPEB and a long 3′-UTR. Upstream sequences were obtained from the same library by PCR, using mCPEB-specific oligonucleotides and an oligonucleotide corresponding to the T3 sequence in the vector. In a first PCR, this T3 oligonucleotide and oligonucleotide 10 were used to amplify a fragment of 507 nt. A second fragment was subsequently obtained by nested-PCR using oligonucleotides 22 and 23 and T3. A fragment containing the initiation codon was obtained by 5′ rapid amplification of cDNA ends (RACE) by following the instructions of the GIBCO/BRL 5′ RACE system kit and using 1 μg of ovary poly(A)+ RNA as the template. During this 5′ RACE reaction, oligonucleotide 22 was used for reverse transcription, followed by an oligo(dC) tailing reaction, a PCR amplification with oligonucleotides JLCK-AP and 23, and a subsequent PCR reamplification with oligonucleotides UAP and 24. The overlapping mCPEB fragments encompased a cDNA of 2610 nt.
In addition, the full-length mCPEB open reading frame was amplified from poly(A)+ ovary RNA by PCR using oligonucleotides Startmcpeb and Stopmcpeb (containing the initiation and termination codons, respectively) and cloned into the pT7Blue vector (Novagen). The sequence of this clone was identical to that of the individual cDNA fragments obtained previously.
The sequences of the oligonucleotides mentioned above are as follows: oligonucleotide 2, TATGCTGGGATTGACACAGATAA; oligonucleotide 3, TGAACCTTCTTGGTAAACTT; oligonucleotide 6, CCACAAAAGCAGCGGTGACT; oligonucleotide 10, AAGGTGTTAACCAATCCAGC; oligonucleotide 21, AGAGGGGCTGCTAGAACGGGA; oligonucleotide 22, CCGGGTGTCCAGGCGAGA; oligonucleotide 23, CTTCAGAGTCCTGGAAGTCT; oligonucleotide 24, CGATTTATAGGTGTTGTACAGA; T3, ATTAACCCTCACTAAAGGGA; JLCK-AP, GGCCACGCGTCGACTAGTACGGGIIGGGIIGGGIIG; UAP, AGGCCACGCGTCGACTAGTACG; Startmcpeb, CCGCTCGAGATGGCTTTCTCTCTGGAAGAAG; Stopmcpeb, CCGCTCGAGTCAGTTCTTCTGGTTCCTCATTA.
The cDNA fragments were sequenced by the dideoxynucleotide method (24). In some cases, the Cyclist Exo− Pfu DNA sequencing kit (Stratagene) was used. The sequence was analyzed with the GCG software (gcg package, version 7, Genetics Computer Group, Madison, WI).
Expression of mCPEB in Cos Cells.
The full-length mCPEB open reading frame was cloned into the KpnI/EcoRV sites of the pCMV56 vector (kindly provided by P. Okamoto, Worcester Foundation for Biomedical Research) and was transfected into Cos cells by the LipofectAmine method (GIBCO/BRL). Protein extracts were prepared from transfected and mock-transfected (without DNA) cells by homogenization in Xlink buffer [100 mM KCl/0.1 mM CaCl2/2 mM MgCl2/10 mM Hepes·KOH, pH 7.7/10 mM DTT/heparin (5 mg/ml)/1 mM ATP/7.5 mM creatine phosphate/tRNA (50 μg/ml/BSA (100 μg/ml)/0.5% Nonidet P-40/1 mM phenylmethylsulfonyl fluoride/pepstatin (1 μg/ml)/leupeptin (2 μg/ml). Fifty microliters of Xlink buffer was used per 100-mm diameter dish. The homogenate was centrifuged at 3000 × g for 10 min at 4°C, and the supernatant was adjusted to 10% glycerol, frozen in liquid nitrogen, and stored at −70°C until used.
Expression of XCPEB in Escherichia coli.
His-tagged XCPEB was expressed in E. coli BL21 cells and purified as described (20). XCPEB was dialyzed overnight against 100 mM ammonium bicarbonate (pH 8.0), lyophilized, and stored at −70°C until used.
RNAs.
The c-mos 3′-UTR clones, containing (wt) or lacking (cpe−) CPEs, were as described (5). RNA was synthesized from these clones in vitro using T3 polymerase and [α-32P]UTP as the radiolabeled nucleotide. To increase the specific activity of the radiolabeled RNA, no unlabeled UTP was included in the reaction. The size of wt and cpe− RNAs was 140 nt and 132 nt, respectively. The RNA used for gel mobility-shift assays was gel-purified.
RNA was extracted from mouse tissues by using the RNA STAT-60 reagent (Tel-Test, Friendswood, TX) by following the recommendations of the vendor. Poly(A)+ RNA was selected on oligo(dT) columns as described (25).
Gel Mobility-Shift Assays.
His-tagged XCPEB was mixed with radiolabeled denatured c-mos 3′-UTR (104 cpm, ≈1 fmol) in a final volume of 20 μl containing 100 mM KCl, 0.1 mM CaCl2, 1 mM MgCl2, 10 mM Hepes·KOH (pH 7.7), 5% glycerol, 10 mM DTT, BSA (0.1 mg/ml), tRNA (50 ng/μl), RNasin (0.4 units/μl), and 1 M urea. After 20 min of incubation at room temperature, 2 μl of heparin (50 mg/ml) was added, and the reaction was further incubated for 10 min. The mix was subsequently loaded onto a 4% polyacrylamide/1 M urea gel. After electrophoresis, the results were visualized on a PhosphorImager.
UV-Crosslinking and Immunoprecipitation.
Radiolabeled c-mos 3′-UTR was mixed with protein extracts in a final volume of 20 μl of Xlink buffer (see above) containing 0.25% Nonidet P-40. UV-crosslinking was subsequently performed as described by Paris et al. (18).
Immunoprecipitations were performed using the NET-gel buffer procedure described by Sambrook et al. (26). When extracts from transfected and mock-transfected cells were used, the mixture was preincubated for 1 h with beads without the antibody. Samples were analyzed by SDS/polyacrylamide gel electrophoresis.
Northern Blot Analysis and Library Screening.
Fifty micrograms of total RNA from various mouse tissues was loaded in a formaldehyde/0.8% agarose gel and, after electrophoresis, were transferred to nitrocellulose. A random-primed DNA complementary to nt 898-2610 of mCPEB cDNA was used to probe the blot, and the results were visualized using a PhosphorImager. For library screening, oligonucleotide 6 was end-labeled and used to probe phage plaques lysed on filters. In both procedures, nitrocellulose filters were hybridized by following the procedure of Church and Gilbert (27).
In Situ Hybridization.
In situ hybridization to paraffin-embedded ovary sections was performed essentially as described by Sassoon and Rosenthal (28) with the following modifications. The hybridization solution consisted of 50% formamide, 5× standard saline citrate (SSC), 50 mM NaH2PO4 (pH 7), 5 mM EDTA, 2.5× Denhardt’s solution, 10% dextran sulfate, 0.1% SDS, salmon sperm DNA (0.25 mg/ml), tRNA (0.5 mg/ml), and 100 mM 2-mercaptoethanol. After overnight hybridization at 55°C, the slides were washed with 4× SSPE (0.18 M NaCl/10 mM sodium phosphate, pH 7.4/1 mM EDTA) for 30 min at room temperature, 4× SSPE for 30 min at 55°C, and 2× SSPE for 20 min at 55°C. One hundred millimolar 2-mercaptoethanol was included in all washes. The slides were subsequently treated with RNase A (20 μg/ml) in 2× SSPE for 30 min at 37°C, dehydrated, and exposed to liquid emulsion for 1 week. After developing, the tissue was stained with eosin Y/ematoxylin.
Western Blot Analysis and Immunodepletions.
Analysis of proteins by Western blot was performed as described by Harlow and Lane (29) with anti-XCPEB antibody that was affinity-purified as described by Olmsted (30). Goat anti-rabbit horsereadish peroxidase was used as the secondary antibody, and the protein bands were visualized by enhanced chemiluminescence (Renaissence, DuPont/New England Nuclear).
Xenopus egg extracts were immunodepleted of CPEB using antibody coupled to protein A-Sepharose beads, as described by Hake and Richter (19).
Preparation of Extracts and Polyadenylylation Assays.
Xenopus oocyte extracts were prepared as described by Hake and Richter (19). Polyadenylylation competent egg extracts were made as described in McGrew and Richter (9). Polyadenylylation assays were performed as described by Paris et al. (18).
Mouse oocytes, devoid of follicle cells, were collected in a small volume of DPBS supplemented with 150 μM isobutylmethylxanthine to prevent spontaneous meiotic maturation during their isolation. Protein extracts from these oocytes were prepared by repeated freezing/thawing of the sample and brief centrifugation at 4°C to pellet the nuclei. The supernatant was mixed with protease inhibitors [pepstatin (0.5 μg/ml)/leupeptin (1 μg/ml)/0.5 mM phenylmethylsulfonyl fluoride] and was used immediately.
RESULTS
Xenopus CPEB Recognizes the Mouse c-mos CPEs.
The CPEs of mouse c-mos mRNA, which are essential for cytoplasmic polyadenylylation, are similar to those of Xenopus mRNAs that are bound by CPEB (20). These CPEs are all uridine-rich with one or two adenosine residues, a pattern that is also evident in several mouse mRNAs in addition to c-mos (10, 31). Because of this similarity, we reasoned that a protein related to XCPEB could exist in mouse oocytes that was required for c-mos mRNA polyadenylylation. In a preliminary experiment, we analyzed the polyadenylylation of mouse c-mos mRNA 3′-UTR, containing the sequences required for polyadenylylation, in Xenopus egg extracts. The results (Fig. 1A) show that the mouse RNA was efficiently polyadenylylated (lane 2) and that polyadenylylation required the presence of the CPEs (compare lanes 2 and 6). Furthermore, depletion of the extract with anti-XCPEB antibody, but not with preimmune serum, substantially reduced polyadenylylation (lanes 3 and 4), indicating that XCPEB was necessary for this process.
Figure 1.
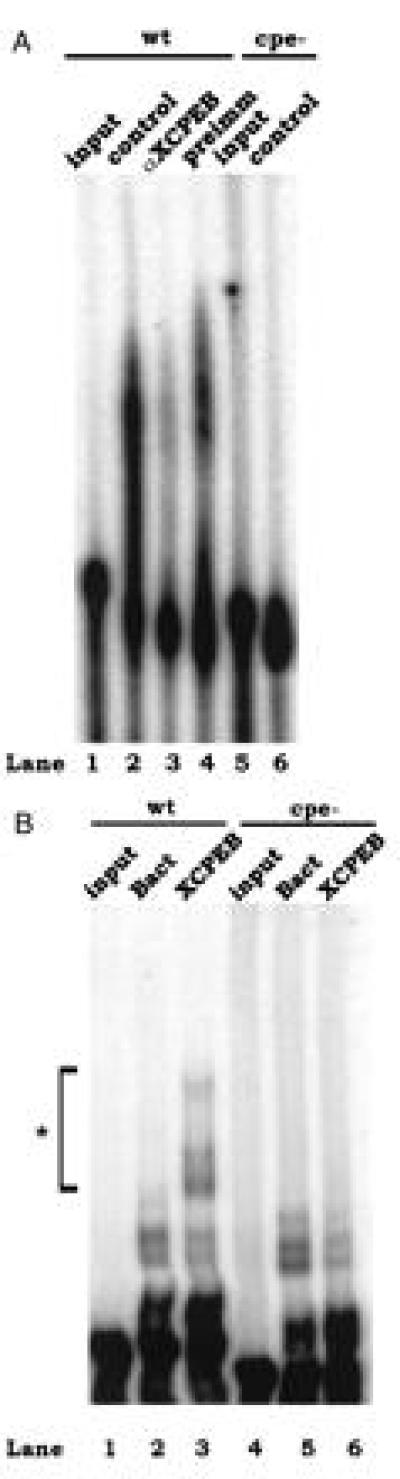
Xenopus CPEB recognizes the mouse c-mos CPEs. (A) Polyadenylylation of c-mos mRNA 3′-UTR in Xenopus egg extracts. Radiolabeled c-mos 3′-UTR was incubated with egg extracts that were untreated (control), mock-depleted (preimm), or immunodepleted of CPEB (anti-XCPEB, αXCPEB). The RNA was then extracted and analyzed for polyadenylylation by denaturing gel electrophoresis and autoradiography. Polyadenylylation assays were performed with wild-type RNA (lanes 1–4) or a mutant RNA that lacked CPEs (lanes 5 and 6). (B) Gel mobility-shift assay with His-tagged XCPEB. Radiolabeled wild-type (lanes 1–3) or cpe− (lanes 4–6) c-mos 3′-UTR was incubated with protein preparations from bacteria expressing (lanes 3 and 6) or lacking (lanes 2 and 5) XCPEB or was incubated without protein (lanes 1 and 4). The RNA was resolved in a 4% polyacrylamide gel containing 1 M urea.
We next determined whether XCPEB directly recognized the CPEs of mouse c-mos mRNA by measuring the binding of bacterially expressed XCPEB to c-mos mRNA 3′-UTR in a gel mobility-shift assay. Fig. 1B shows the presence of unique shifted bands when the c-mos RNA was incubated in the presence of XCPEB (lane 3), but not in the presence of bacterial extracts processed in a similar manner (lane 2). In addition, these bands were not present when a mutant c-mos 3′-UTR lacking the CPEs (cpe−) was incubated with XCPEB (lane 6). These data demonstrate that XCPEB binds to the CPEs of mouse c-mos mRNA and raise the possibility that a similar protein exists in mouse oocytes with an identical function.
Cloning Mouse CPEB cDNA.
The sequence in the carboxyl-terminal half of XCPEB is highly homologous to that of Drosophila orb (19), a protein involved in mRNA localization (21, 22). Using this conserved region, we designed oligonucleotide primers to amplify mCPEB sequences from mouse ovary RNA by reverse transcription-couple PCR. A short fragment was amplified and used to design an oligonucleotide specific for mouse sequences, which was subsequently employed to screen a UniZap mouse ovary cDNA library (kindly provided by J. Dean). Sequencing of one positive clone revealed that it contained an insert encoding the carboxyl-terminal half of mCPEB and a long 3′-UTR. The sequence upstream of this fragment was obtained by PCR from the same library and by 5′ RACE using poly(A)+ ovary RNA as the template. The full-length mCPEB open reading frame was amplified from poly(A)+ ovary RNA by reverse transcription-couple PCR using oligonucleotides containing the initiation and termination codons. The sequence of this clone was identical to that of the individual cDNA fragments obtained previously.
The sequence contained an open reading frame encoding a 561-amino acid protein and was 73% identical to that coding for XCPEB. At the amino acid level, mCPEB shows 80% overall identity with its Xenopus counterpart (Fig. 2). The highest homology is found in the carboxyl-terminal region, which contains two ribonucleoprotein-type RNA-recognition motifs and a cluster of cysteine/histidine residues that could constitute a zinc-finger domain (Fig. 2A). Proteins from other organisms are also homologous to the carboxyl-terminal half of mCPEB, including Drosophila orb and two open reading frames from C. elegans (Fig. 2B).
Figure 2.
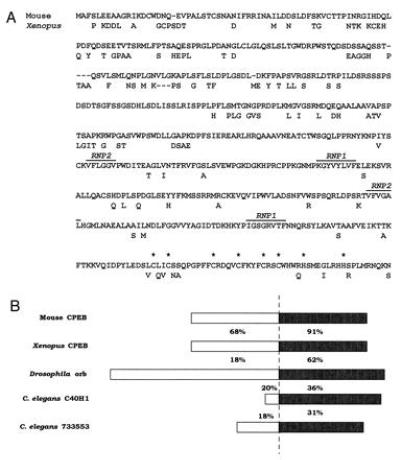
mCPEB amino acid sequence and its comparison with related proteins. (A) Predicted amino acid sequence of mouse CPEB compared with Xenopus CPEB. The conserved ribonucleoprotein-type RNA recognition motifs are denoted. The asterisks indicate the cysteine and histidine residues that could form a zinc finger. (B) Schematic representation of a comparison between mCPEB and related proteins from other organisms. The numbers indicate the percentage of amino acid identity of the various proteins with respect to mCPEB. The highest identity (solid box) is found in the carboxyl-terminal portion.
Tissue Distribution of mCPEB mRNA.
Northern blot analysis of total RNA from various mouse tissues was used to determine the distribution of mCPEB mRNA (Fig. 3). This message was most abundant in the ovary, and somewhat less prevalent in the testis and kidney. As a control, we probed ovary RNA that was depleted of poly(A)+ RNA; as expected, no mCPEB mRNA signal was detected [ovary(A−)]. In addition, to ensure that equivalent amounts of RNA were loaded, the blot was probed for 18S ribosomal RNA, which was present in similar amounts in all lanes (Fig. 3 Lower).
Figure 3.
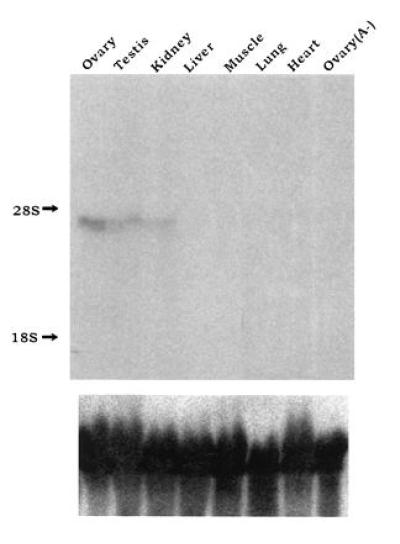
Tissue distribution of mCPEB mRNA. (Upper) Fifty micrograms of total RNA from the indicated mouse tissues was examined by Northern blot analysis using a random-primed probe complementary to nt 898-2610 of mCPEB cDNA. Ovary RNA that was depleted of poly(A)+ RNA [ovary (A−)] was included as a negative control. The positions of the 18S and 28S ribosomal RNAs are indicated as markers at the left. To ensure that equivalent amounts of RNA were loaded in each lane, the filter was stripped and rehybridized with a probe complementary to the 18S ribosomal RNA (Lower).
The mouse ovary is composed of several different cell types including oocytes, follicle cells, and thecal cells, among others. In the mature follicle, for example, there are about 50,000 follicle cells per oocyte. To determine which cell type(s) in the ovary contained mCPEB mRNA, we performed in situ hybridization to mouse ovary sections (Fig. 4). When an antisense mCPEB sequence was used as a probe, a hybridizing signal was clearly evident in oocytes of both preantral and antral follicles but in no other cell type (Fig. 4A). Under higher magnification, it is again obvious that only oocytes contain mCPEB sequences (Fig. 4B). Therefore, mCPEB mRNA is restricted mostly to tissues containing germ cells and, in the ovary, is only present in oocytes.
Figure 4.
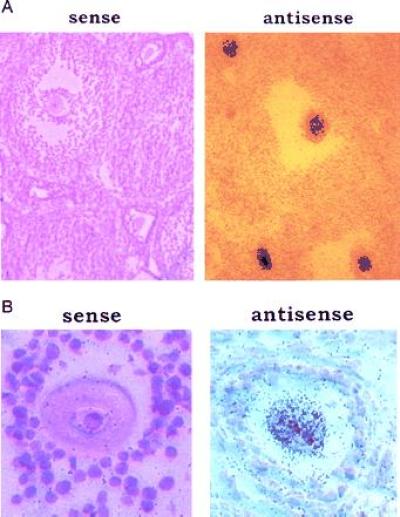
Distribution of mCPEB mRNA in the ovary. Low (A) and high (B) magnifications of an in situ hybridization to ovary sections, using as a probe an 35S-labeled RNA complementary to nt 1424–1520 of mCPEB cDNA (antisense). The sense labeled RNA was used as a negative control.
mCPEB Is Present in Oocytes and Recognizes the CPEs of Mouse c-mos mRNA.
Given the homology between Xenopus and mouse CPEBs, an anti-XCPEB antibody (19) was expected to cross-react with the mouse protein. We therefore used affinity-purified anti-XCPEB antibody to determine whether mCPEB was expressed in mouse oocytes by Western blot analysis. The results show that a major band of 62 kDa was present in oocytes, which correlated well with the predicted size of the product translated from our cloned open reading frame (Fig. 5). In addition, minor bands of high molecular weight (more than 101 kDa) were also detected. None of these bands were present when the secondary antibody alone was used to probe an identical blot (Fig. 5).
Figure 5.
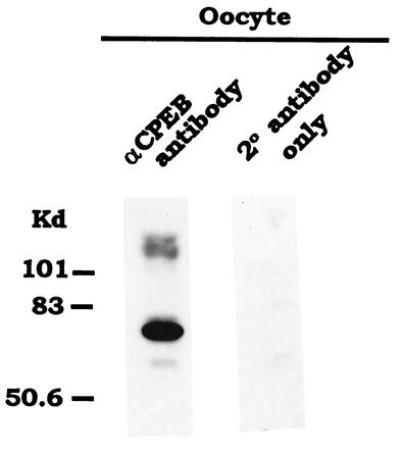
mCPEB is present in oocytes. Immunoblot of a mouse oocyte extract with affinity-purified anti-XCPEB antibody. An extract derived from 385 oocytes was used. As a negative control, a similar extract was only incubated with the secondary antibody. The protein bands were visualized by enhanced chemiluminescence.
To determine whether endogenous mCPEB recognizes the CPEs of c-mos mRNA, we incubated radiolabeled c-mos RNA, containing (wt) or lacking (cpe−) the CPEs, with protein extracts prepared from mouse oocytes. Because our data in Fig. 1 indicated that XCPEB recognized the mouse c-mos CPEs, parallel reactions were performed using protein extracts from Xenopus oocytes. After an incubation of 30 min, the extract was UV-irradiated and treated with RNase A, and CPEB was immunoselected with the anti-XCPEB antibody. An autoradiogram of the crosslinked and immunoprecipitated material shows that a protein of the expected size of CPEB (62 kDa) was crosslinked in both mouse and Xenopus extracts, but only when the RNA contained CPEs (Fig. 6, compare lanes 1 and 3 with lanes 2 and 4, respectively). In addition, other bands of smaller size were also crosslinked and immunoprecipitated, which probably correspond to CPEB degradation products because they frequently appear after freezing/thawing or long storage of the protein extract (data not shown). Thus, these data show that endogenous mCPEB recognizes the CPEs of c-mos mRNA.
Figure 6.
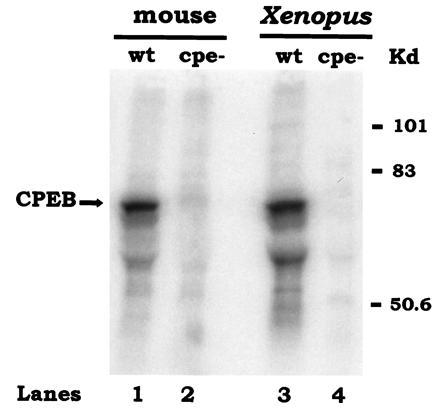
Endogenous mCPEB recognizes the CPEs of c-mos mRNA. Radiolabeled c-mos mRNA 3′-UTR containing (wt) or lacking (cpe−) CPEs was incubated with protein extracts from 265 mouse oocytes (lanes 1 and 2) or one Xenopus oocyte (lanes 3 and 4). After an incubation of 30 min at room temperature, the extract was UV-irradiated, digested with RNase A, and immunoselected with anti-XCPEB antibody. Radiolabeled proteins were resolved by SDS/polyacrylamide gel electrophoresis and visualized using a PhosphorImager.
To ensure that the cDNA we cloned did indeed encode mCPEB, we tested whether recombinant mCPEB could recognize the CPEs of c-mos mRNA. Because mCPEB expressed in bacteria was insoluble, we expressed this protein in Cos cells after transient transfection. Cytoplasmic extracts were prepared from both transfected and untransfected cells and examined by Western blot analysis with affinity-purified anti-XCPEB antibody. Fig. 7A shows that mCPEB was synthesized in transfected Cos cells (compare lanes 1 and 2, transfected, with lanes 3 and 4, untransfected). Although most of the protein copurified with the pellet during the preparation of the extract (lane 1), sufficient protein remained in the supernatant (lane 2) to perform RNA-binding assays. Supernatants from transfected (T) and untransfected (UT) cells were incubated with wild-type (wt) or CPE-lacking (cpe−) c-mos 3′-UTRs and used for UV-crosslinking experiments. After immunoselection with anti-XCPEB antibody, the material was resolved by SDS/polyacrylamide gel electrophoresis and autoradiography. Fig. 7B shows that a crosslinked protein of the size of mCPEB was detected only from transfected cells, and only when the labeled c-mos RNA contained CPEs. Therefore, mCPEB binds to the same regulatory elements, the CPEs, that control c-mos mRNA polyadenylylation and translation during oocyte maturation (5).
Figure 7.
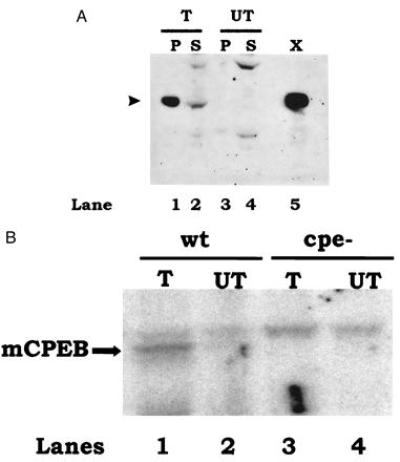
Recombinant mCPEB recognizes the CPEs of mouse c-mos mRNA. (A) Immunoblot of mCPEB expressed by transient transfection of Cos cells. Cells were transfected with mCPEB cDNA (T) or mock-transfected (UT, no DNA). After an incubation of 36 h, the cells were homogenized and the nuclei were pelleted by centrifugation. Equivalent amounts of pellet (P) and supernatant (S) (approximately one-third of a 100-mm diameter dish) were loaded in a 10% polyacrylamide gel and examined by Western blot analysis using affinity-purified anti-XCPEB antibody. A protein extract corresponding to one-fourth of a Xenopus oocyte (X) was used as a positive control. The size of CPEB is indicated by an arrowhead at the left. (B) UV-crosslinking and immunoselection of mCPEB expressed in Cos cells. Protein extracts from Cos cells that were transfected with mCPEB cDNA (T) or mock-transfected (UT) were UV-crosslinked to radiolabeled c-mos 3′-UTR containing (wt) or lacking (cpe−) CPEs. A supernatant corresponding to 50% of a 100-mm dish was used per lane. Crosslinked products were immunoselected with anti-XCPEB antibody and resolved by SDS/polyacrylamide gel electrophoresis. The results were visualized in a PhosphorImager.
DISCUSSION
De novo translation of c-mos mRNA is required for vertebrate oocyte maturation. In Xenopus, ablation of c-mos mRNA with antisense oligonucleotides prevents germinal vesicle breakdown (32), while in the mouse a similar treatment results in a failure of oocytes to progress to metaphase II and in subsequent parthenogenetic activation (33). The translation of c-mos mRNA is regulated by cytoplasmic polyadenylylation (5, 6), a process that depends, at least in Xenopus, on the binding of CPEB to the 3′-UTR of the message (20).
The distinct timing of c-mos mRNA polyadenylylation and translation in mouse versus Xenopus oocytes suggested that different factors might be involved. However, the similarity of the CPEs between Xenopus and mouse RNAs suggested a common binding factor. Based on this observation, it was possible that mouse oocytes contained a CPEB homolog that regulated c-mos mRNA polyadenylylation. Indeed, the 3′-UTR of mouse c-mos mRNA is polyadenylylated efficiently in Xenopus egg extracts (Fig. 1A). This result is in agreement with a previous report in which the 3′-UTR of mouse hypoxanthine phosphoribosyltransferase (HPRT), which contains a CPE-like sequence, was polyadenylylated in maturing Xenopus oocytes (10) as it was in maturing mouse oocytes (34). Importantly, immunodepletion of CPEB from Xenopus egg extracts renders them, for the most part, incapable of polyadenylylating exogenous mouse c-mos mRNA (Fig. 1A). In addition, Xenopus CPEB binds to the mouse c-mos CPEs (Fig. 1B). These results strongly suggested that the mechanism of cytoplasmic polyadenylylation was conserved and that a factor similar to Xenopus CPEB existed in the mouse.
Consistent with these observations, we have isolated a mouse cDNA encoding the homolog of XCPEB. This cDNA contains an open reading frame of 1683 nt that is 73% identical to that encoding the Xenopus protein. There is no homology, however, in the 5′ and 3′-UTRs. Although there is no stop codon upstream of the putative initiation codon, the protein detected in oocytes by Western blot analysis (Fig. 5) has a similar size to that translated from our open reading frame, indicating that the initiation codon is correct.
mCPEB contains 561 amino acids and shares 80% overall identity with its Xenopus counterpart. However, the identity is much higher (91%) in a region that contains two RNA recognition motifs and a conserved spacing of cysteine and histidine residues that could potentially form a zinc finger (19). Indeed, at least with the Xenopus protein, all of these motifs appear to be important for binding to the CPE (L. E. Hake and J.D.R., unpublished results). Interestingly, the homology in this region extends to invertebrates. Drosophila orb, a protein involved in mRNA localization (21, 22) is 62% identical to mCPEB in its carboxyl-terminal portion. In addition, two open reading frames from C. elegans that were identified in the genome sequencing project (23) also show significant homology (36% and 31% identity) to mCPEB in this region. Finally, the surf clam Spisula solidissima contains a protein, potentially involved in masking (35), that is 67% identical (to XCPEB) in the carboxyl-terminal region (N. Standart, personal communication). Thus, because animals as diverse as mammals, amphibians, arthropods, nematodes, and molluscs contain proteins with significant homology to CPEB, we believe it is likely to be present in most metazoans. With respect to the function of these proteins, it is tempting to speculate that they bind similar RNA sequences. At least this is the case with the Xenopus and the mouse proteins (see below).
In addition to CPEB, a protein greater than 100 kDa reacts with the anti-XCPEB antibody (Fig. 5). While the identity of this protein is unknown, we have identified additional upstream open reading frames continuous with mCPEB using 5′ RACE (data not shown). These data suggest that there may be additional CPEB-like proteins in the mouse, which we have so far been unable to detect in Xenopus.
In Xenopus, XCPEB binds to the CPE of c-mos mRNA, leading to polyadenylylation and subsequent translation of this message (20). Similarly, mCPEB binds to the CPEs of mouse c-mos mRNA (Figs. 6 and 7) and most likely regulates polyadenylylation in this species as well. However, given the paucity of mouse oocytes that can be obtained, further biochemical analysis of mCPEB activity is very difficult. As an alternative approach, we mapped the chromosomal location of mCPEB to determine whether it corresponded with a known genetic defect. Unfortunately, this was not the case. Therefore, we are in the process of disrupting the mCPEB gene to assess whether this prevents cytoplasmic polyadenylylation and early development.
Acknowledgments
We thank J. Dean for the generous gift of the mouse ovary library, P. Okamoto for her gift of the pCMV plasmid, N. Standart for communication of unpublished results, and J. Valcárcel for useful comments on the manuscript. This work was supported by grants from the National Institutes of Health and the March of Dimes Birth Defects Foundation (to J.D.R.).
Footnotes
Abbreviations: UTR, untranslated region; CPE, cytoplasmic polyadenylylation element; CPEB, CPE binding protein; m, mouse; X, Xenopus; RACE, rapid amplification of cDNA ends.
Data deposition: The sequence reported in this paper has been deposited in the GenBank data base (accession no. Y08260Y08260).
References
- 1.Richter J. In: Translational Control. Hershey J W B, Mathews M B, Sonenberg N, editors. Plainview, NY: Cold Spring Harbor Lab. Press; 1996. pp. 481–503. [Google Scholar]
- 2.Vassalli J, Stutz A. Curr Biol. 1995;5:476–479. doi: 10.1016/s0960-9822(95)00095-9. [DOI] [PubMed] [Google Scholar]
- 3.Sallés F, Lieberfarb M, Wreden C, Gergen J, Strickland S. Science. 1994;266:1996–1998. doi: 10.1126/science.7801127. [DOI] [PubMed] [Google Scholar]
- 4.Simon R, Wu L, Richter J D. Dev Biol. 1996;179:239–250. doi: 10.1006/dbio.1996.0254. [DOI] [PubMed] [Google Scholar]
- 5.Gebauer F, Xu W, Cooper G, Richter J. EMBO J. 1994;13:5712–5720. doi: 10.1002/j.1460-2075.1994.tb06909.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Sheets M, Wu M, Wickens M. Nature (London) 1995;374:511–516. doi: 10.1038/374511a0. [DOI] [PubMed] [Google Scholar]
- 7.McGrew L, Dworkin-Rastl E, Dworkin M, Richter J. Genes Dev. 1989;3:803–815. doi: 10.1101/gad.3.6.803. [DOI] [PubMed] [Google Scholar]
- 8.Fox C, Sheets M, Wickens M. Genes Dev. 1989;3:2151–2162. doi: 10.1101/gad.3.12b.2151. [DOI] [PubMed] [Google Scholar]
- 9.McGrew L, Richter J. EMBO J. 1990;9:3743–3751. doi: 10.1002/j.1460-2075.1990.tb07587.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Paris J, Richter J. Mol Cell Biol. 1990;10:5634–5645. doi: 10.1128/mcb.10.11.5634. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Simon R, Tassan J-P, Richter J. Genes Dev. 1992;6:2580–2591. doi: 10.1101/gad.6.12b.2580. [DOI] [PubMed] [Google Scholar]
- 12.Sallés F, Darrow A, O’Connell M, Strickland S. Genes Dev. 1992;6:1202–1212. doi: 10.1101/gad.6.7.1202. [DOI] [PubMed] [Google Scholar]
- 13.Simon R, Richter J. Mol Cell Biol. 1994;14:7867–7875. doi: 10.1128/mcb.14.12.7867. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Stebbins-Boaz B, Richter J. Mol Cell Biol. 1994;14:5870–5880. doi: 10.1128/mcb.14.9.5870. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Bilger A, Fox C A, Wahle E, Wickens M. Genes Dev. 1994;8:1106–1116. doi: 10.1101/gad.8.9.1106. [DOI] [PubMed] [Google Scholar]
- 16.Ballantyne S, Bilger A, Astrom J, Virtanen A, Wickens M. RNA. 1995;1:64–78. [PMC free article] [PubMed] [Google Scholar]
- 17.Gebauer F, Richter J. Mol Cell Biol. 1995;15:1422–1430. doi: 10.1128/mcb.15.3.1422. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Paris J, Swenson K, Piwnica-Worms H, Richter J. Genes Dev. 1991;5:1697–1708. doi: 10.1101/gad.5.9.1697. [DOI] [PubMed] [Google Scholar]
- 19.Hake L, Richter J. Cell. 1994;79:617–627. doi: 10.1016/0092-8674(94)90547-9. [DOI] [PubMed] [Google Scholar]
- 20.Stebbins-Boaz B, Hake L, Richter J. EMBO J. 1996;15:2582–2592. [PMC free article] [PubMed] [Google Scholar]
- 21.Lantz V, Chang J, Horabin J, Bopp D, Schedl P. Genes Dev. 1994;8:598–613. doi: 10.1101/gad.8.5.598. [DOI] [PubMed] [Google Scholar]
- 22.Christersen L, McKearin D. Genes Dev. 1994;8:614–628. doi: 10.1101/gad.8.5.614. [DOI] [PubMed] [Google Scholar]
- 23.Wilson R, Ainscough R, Anderson K, Baynes C, Berks M, et al. Nature (London) 1994;368:32–38. doi: 10.1038/368032a0. [DOI] [PubMed] [Google Scholar]
- 24.Sanger F, Nicklen S, Coulson A. Proc Natl Acad Sci USA. 1977;74:5463–5467. doi: 10.1073/pnas.74.12.5463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Aviv H, Leder P. Proc Natl Acad Sci USA. 1972;69:1408–1412. doi: 10.1073/pnas.69.6.1408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Maniatis T, Fritsch E, Sambrook J. Molecular Cloning: A Laboratory Manual. 2nd Ed. Plainview, NY: Cold Spring Harbor Lab. Press; 1989. [Google Scholar]
- 27.Church G, Gilbert W. Proc Natl Acad Sci USA. 1984;81:1991–1995. doi: 10.1073/pnas.81.7.1991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Sassoon D, Rosenthal N. Methods Enzymol. 1993;225:384–404. doi: 10.1016/0076-6879(93)25027-y. [DOI] [PubMed] [Google Scholar]
- 29.Harlow E, Lane D. Antibodies: A Laboratory Manual. Plainview, NY: Cold Spring Harbor Lab. Press; 1988. [Google Scholar]
- 30.Olmsted J B. Methods Enzymol. 1986;134:467–472. doi: 10.1016/0076-6879(86)34112-0. [DOI] [PubMed] [Google Scholar]
- 31.Sallés F J, Darrow A L, O’Connell M L, Strickland S. Genes Dev. 1992;6:1202–1212. doi: 10.1101/gad.6.7.1202. [DOI] [PubMed] [Google Scholar]
- 32.Sagata N, Daar I, Oskarsson M, Showalter S, Vande Woude G. Science. 1989;245:643–645. doi: 10.1126/science.2474853. [DOI] [PubMed] [Google Scholar]
- 33.O’Keefe S, Wolfes H, Kiessling A, Cooper G. Proc Natl Acad Sci USA. 1989;86:7038–7042. doi: 10.1073/pnas.86.18.7038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Paynton B, Bachvarova R. Mol Reprod Dev. 1994;37:172–180. doi: 10.1002/mrd.1080370208. [DOI] [PubMed] [Google Scholar]
- 35.Walker J, Dale M, Standart N. Dev Biol. 1996;173:292–305. doi: 10.1006/dbio.1996.0024. [DOI] [PubMed] [Google Scholar]