

Abstract
cagA, a gene that codes for an immunodominant antigen, is present only in Helicobacter pylori strains that are associated with severe forms of gastroduodenal disease (type I strains). We found that the genetic locus that contains cagA (cag) is part of a 40-kb DNA insertion that likely was acquired horizontally and integrated into the chromosomal glutamate racemase gene. This pathogenicity island is flanked by direct repeats of 31 bp. In some strains, cag is split into a right segment (cagI) and a left segment (cagII) by a novel insertion sequence (IS605). In a minority of H. pylori strains, cagI and cagII are separated by an intervening chromosomal sequence. Nucleotide sequencing of the 23,508 base pairs that form the cagI region and the extreme 3′ end of the cagII region reveals the presence of 19 ORFs that code for proteins predicted to be mostly membrane associated with one gene (cagE), which is similar to the toxin-secretion gene of Bordetella pertussis, ptlC, and the transport systems required for plasmid transfer, including the virB4 gene of Agrobacterium tumefaciens. Transposon inactivation of several of the cagI genes abolishes induction of IL-8 expression in gastric epithelial cell lines. Thus, we believe the cag region may encode a novel H. pylori secretion system for the export of virulence determinants.
Keywords: secretion, insertion sequence, inflammation, evolution
Helicobacter pylori is a microaerophilic spiral-shaped lophotrichous Gram-negative bacterium that colonizes the gastric lumen of primates, including humans (1). H. pylori was identified as the cause of chronic active gastritis and peptic ulcer disease in humans and is considered to be a risk factor for the development of gastric adenocarcinoma and MALT lymphoma (2, 3). Strains of H. pylori are grouped into two broad families tentatively named type I and type II, which are based on whether they express or not the vacuolating cytotoxin (VacA) and the CagA antigen (cytotoxin-associated gene A) (4). An increasing body of evidence has shown that patients with duodenitis, duodenal ulcers, and gastric tumors are most often infected by type I strains, which suggests that CagA and the coexpressed cytotoxin play a role in its pathogenicity (4–6). These epidemiological observations are supported by studies in the mouse animal model. Sonic extracts of type I strains, but not those from type II strains, induce gastric damage with histological lesions (epithelial vacuolation, mucosal erosion, necrosis, and ulceration) similar to what is found in biopsies from patients infected by H. pylori (7). Furthermore, while both type I and type II isolates of H. pylori can colonize mice, only the type I strains promote gastric injuries similar to the ones observed in humans (8). In addition, only type I strains were able to induce epithelial cells to secrete Interleukin 8 (IL-8), a mediator of neutrophil migration, in vitro (9, 10).
Genetic analysis shows that the cagA gene is present only in type I strains, while the vacA gene is present in both types (4). An active toxin is produced only by type I strains; however, the linkage between CagA and VacA expression is not yet clear since the cagA and vacA genes are located more than 300 kb apart on the chromosome of the H. pylori NCTC 11638 (11). Moreover, it has been shown that inactivation of the cagA gene has no consequence on expression of VacA or on the ability to induce IL-8 (12–14). These findings suggest that CagA is a marker for the increased virulence observed in type I strains.
To understand better the relationships between type I and type II strains and to dissect some of the virulence traits of H. pylori, we have analyzed the region flanking the cagA gene. We found that type I strains contain an insertion of approximately 40 kb of foreign DNA (the cag region) that has the typical features of a pathogenicity island (PAI) (15–23). This PAI encodes for virulence factors unique to H. pylori strains with enhanced virulence, which suggests that the acquisition of this region is an important event in the evolution of H. pylori and marks the differentiation of a more virulent type of bacterium within this genus.
MATERIALS AND METHODS
Bacterial Strains and Growth Media.
The H. pylori type strain CCUG 17874 [identical to NCTC 11638; Akopyants et al. (24)] was obtained from the Culture Collection of the University of Gotheborg (Sweden). The H. pylori collection was established from clinical samples isolated at the Hospitals of Grosseto and Siena, Italy (35 samples) and from bacterial strains from France (2 samples), England (4 samples), and United States (3 samples) and has been described (4). Escherichia coli DH10B (BRL) and TG1 were used to prepare genetic libraries of H. pylori. pBluescript SK+ (Stratagene) was used as the cloning vector. H. pylori strains were recovered from frozen stocks on Columbia agar plates containing 5% horse blood, 0.2% cyclodextrin, and Dent’s or Skirrow’s antibiotic supplement under microaerophilic conditions (Oxoid, Basingstoke, U.K.) at 37°C for 3 days. After passages onto fresh plates, the bacteria were subcultured in a 5% CO2/95% air atmosphere at 37°C. E. coli strains were cultured in Luria–Bertani medium.
Genomic Libraries.
DNA preparations were performed as described (4), except that minipreparations of DNA were further purified by an ethidium bromide–high salt extraction procedure (25). Chromosomal DNA from H. pylori CCUG 17874 (type I) or G50 and G21 (type II) strains were digested to completion with either HindIII or EcoRI restriction enzymes. Digested DNA were cloned in pBluescript SK+ (Stratagene). DNA ligations, electroporations of DH10B, screenings, and amplifications were performed as described (26–28).
Representational Difference Analysis (RDA) and PCR.
The RDA procedure was performed as described (29), using HindIII restriction endonuclease (New England Biolabs) and DNA isolated from the CCUG 17874 (type I) and from the G50 and the G21 (type II) strains. The iterative hybridization–extension–amplification step was repeated three times. The resulting material was digested with the same restriction endonuclease, was ligated to a dephosphorylated pBluescript SK+ vector, and was transformed into E. coli DH10B competent cells. PCR was performed on genomic DNA using a Perkin–Elmer model 9600 thermo cycler (Perkin–Elmer) and the Expand Long Template PCR System (Boehringer). RDA and PCR products were used for sequencing reactions and Southern blot hybridization with DNA isolated from a collection of H. pylori strains and digested with the HindIII restriction enzyme. Southern blot hybridization was performed as described (26–28).
Molecular Cloning and Nucleotide Sequencing.
General molecular biology techniques were as described in Sambrook et al. (26). Ordered genomic clones that made up the cag region were used for generating unidirectional exonuclease III deletions (Exo-Size deletion kit, New England Biolabs). Nucleotide sequencing (six times, two-strand coverage) was performed by the dideoxynucleotide chain termination method (30) using a T7 sequencing kit (Pharmacia) and by primer walking using an automatic sequencing machine (model 373A stretch, Applied Biosystems). Nucleotide and derived amino acid sequences were assembled and initially analyzed with the Genetics Computer Group (GCG) package (version 8) from the University of Wisconsin (31). Further analyses were carried out using the genemark (32) program and the World-Wide Web at National Center for Biotechnology Information, European Bioinformatics Institute (EBI), and EMBL servers for blasta, blitz, and fasta programs. Data were collected and edited on a Power Macintosh 8500/120.
Transposon Mutagenesis.
The miniTn3-Km-cat delivery system (kindly provided by A. Labigne, Institut Pasteur) was used for random insertional mutagenesis of the cagI region as described (33). Isogenic mutants were generated through allelic replacement in the H. pylori G27 and G39.
IL-8 Assay.
Experiments were performed with KATO-III gastric epithelial cells as described (9, 10). Cell lines routinely were maintained in RPMI 1640 medium, supplemented with 10% fetal calf serum. Suspensions of approximately 5 × 105 cells per ml were cultured for 24 h with 2.5 × 107 bacterial cells per ml in quadruplicate. The culture supernatants were assayed in duplicate for IL-8 by enzyme-linked immunosorbent assay (ELISA). The ELISA was carried as described (34) using a mouse monoclonal antibody to IL-8 and a phosphatase-conjugated goat anti-IL-8 polyclonal antibody (Sandoz Pharmaceutical).
RESULTS
Molecular Tracking of cag.
To define the chromosomal fragment present in type I and missing in type II strains, the region around cagA in the strain CCUG 17874 was cloned and analyzed using chromosome walking on HindIII and EcoRI binary libraries and with RDA. A set of overlapping fragments was ordered and tested by Southern blot analysis on DNA isolated from both types of strains. We found homology to DNA from type II strains downstream from the cagA gene (6), starting with an ORF with strong similarity to bacterial glutamate racemase (glr in Fig. 1). In marked contrast, we found a long region of DNA unique to type I strains upstream from cagA. This region is approximately 40 kb long and is interrupted by an intervening chromosomal sequence that is also present in the genome of type II strains. We defined cagI the region up to the intervening sequence and we defined cagII the region after the intervening sequence (6). The names cagII and cagI were originally proposed by D. E. Berg, who independently characterized the cagII region. We have named the cag ORFs in a sequential way using a one-letter code. Nucleotide sequencing of the ordered clones of the cagI portion revealed the organization shown in Fig. 1. Upstream from cagA, we found nine open-reading frames (B–L) with a transcriptional polarity opposed to that of cagA, followed by two ORFs (M and N) with the same transcriptional polarity of cagA. We found four small ORFs upstream from M, followed by two longer ORFs with opposite polarity (tnpA and tnpB) adjacent to an intervening chromosomal sequence. In the cagII region (Fig. 1), sequences of ORFs S and T and a portion of the 5′ end revealed that the cagII region (Fig. 1) was identical to a region previously sequenced by N. S. Akopyants (personal communication).
Figure 1.
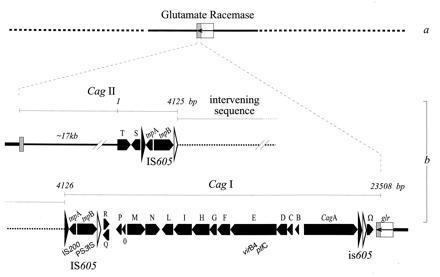
Schematic representation of the cag region as deduced from analysis of strain CCUG 17874. The cag integration site within the glutamate racemase gene (glr) is shaded (a). cag structure: the putative ORFs are represented by arrows (b). Flanking the intervening sequence (dots), the IS605 have a left and right end indicated with large solid and open arrowheads. ORFs with strong similarity are indicated. The cagII region is drawn interrupted but ordered clones and long distance PCR give an approximate indication of the total length, which is indicated by a continuous line. The results of amino acid database searches are included. The name of the genes are in boldface type. LLS, low level of similarity (e−2 > e > e−10); HLS, high level of similarity (>e−50). GenBank, EBI, Swiss-Prot, and Protein Identification Resource releases for December 31, 1995 were used. cagT (LLS), Shigella flexnerii 42-kDa surface antigen IPAC_SHIFL; membrane protein P60 Mycoplasma hominis S42614; Plasmodium falciparum 10b antigen (asparagine rich) J03986 (prokaryotic lipoprotein membrane attachment site). cagS (LLS), Erwinia chrysantemi IIABC component PTS system PTBA_ERWCH; Clostridium perfringens transposase for IS115 TRA1_CLOPE·tnpA (HLS), IS200 from E. coli, S. typhimurium, and Yersinia pestis L25848, U22457, and L25845; S. typhimurium plasmid pSDL2/spvD26 (vsdA–F genes for virulence proteins) X56727. tnpB (HLS), thermophilic bacterium PS3 gene for transposase-like protein D38778; S. typhimurium virulence protein vsdF P24421P24421. cagR (LLS), preprotein translocase secY subunit SECY_STACA; polysialic acid transport protein KPSM_ECOLI; cagQ (LLS) Neisseria gonorrhoeae prepilin peptidase P33566; glutamate/aspartate transporter E. coli GLTJ_ECOLI; 42-kDa membrane antigen precursor Shigella IPAC_SHIFL. cagP (LLS), fimbrial assembly protein (serogroup I) Fmbi_BACNO; type 4 prepilin-like protein-specific leader peptidase LEP3_PSEAE; hydrophobic membrane protein Haemophilus U32720. cagO (LLS), transport system permease P69_Mychr; polysialic acid transporter E. coli KPSN_ECOLI; NADH-ubiquinone oxidoreductase chain 5 (also chain 1, 2) NU1M_ASCSU. cagM (LLS), hook-associated protein type 3 U12817; merozoite surface antigen PFMEZSA1A_1. cagN (LLS), Lmp 1 M. hominis U21963; acidic basic repeat antigen P. falciparum A12521; α-cardiac myosin hc M76598. cagL (LLS), preprotein translocase SECA_ANTSP; proteases secretion protein PRTE_ERWCH. cagI (LLS), CFA/I fimbrial subunit D CFAD_ECOLI; FlaB C. coli A35146;Treponema membrane protein precursor P29721. cagH (LLS), extracellular protease precursor PROA_XANCP; GSP protein d precursor Erwinia chrisantemi GSQD_ERWCH;Treponema membrane protein precursor P29721. cagG (LLS), flagellar motor switch protein FLIM_CAUCR; toxin coregulated pilus biosynthesis protein D (TCP pilus biosynthesis protein TCPD)_VIBCH; GSP protein d precursor Erwinia chrisantemi GSPD_ERWCH. cagF (LLS), ToxB, ToxA_CLODI. cagE (HLS), VirB4 homolog_BPERT (ptlc); VirB4 homolog (plasmid pTiA6)_AGRT9; TraB of IncN plasmid pKM101; TrbE (Tra2 region) of IncP plasmid RP4 (Walker box). cagD (LLS), flagellar hook-associated protein 2 (filament cap) FLID_SALTY; surface presentation SPAO_SHIFL. cagC (LLS), TRAH protein precursor TRH1_ECOLI; SECE_ECOLI protein-export (preprotein translocase); 62-kDa membrane antigen IPAB_SHIDY; sensor protein ENVZ_SALTY. cagB (LLS), sensor protein UHPB_ECOLI; hypothetical 16.6-kDa protein outside the virF region (ORF3)_AGRT9; BACN17G_7 hypothetical protein BACSU; transport system permease protein p69_MYCHR. glr (HLS), glutamate racemase LEPRAE_BREVIS_COLI. Coordinates, headers, and comments are included in the submitted sequence.
Computer analysis of the ORFs sequenced showed that most had weak similarities to proteins included in the data bank; the more significant are reported in Fig. 1. While at first glance, these proteins appear to have little in common, all share motifs present within bacterial inner- and outer-membrane-associated proteins, including translocases, sensors, permeases, and proteins required for pilus or flagellum assembly, which suggests they are part of complex membrane-associated protein structures. The most striking similarity was found between cagE and the virB4 gene of Agrobacterium tumefaciens (35), the ptlC gene of Bordetella pertussis (36, 37), the traB gene of the IncN plasmid pKM101 (38), and the trbE gene of incP plasmid RP4 (39). These homologous molecules are components of a cellular engine responsible for the export of the tDNA (Agrobacterium) and of the pertussis toxin (Bordetella). These genes are thought to originate by an adaptation of a conjugation system present in several self-transmissible plasmids (39). CagE, like VirB4, PtlC, TraB, and TrbE, contains a Walker box (type A nucleotide-binding site) that is required for ATP hydrolysis. tnpA shows high scores when aligned to the transposase gene of IS200 (40) from Escherichia coli, Salmonella enterica serovar Typhimurium, and Yersinia pestis, while tnpB is similar to the transposase of the Thermophylic bacterium PS3 (41). Recently Blaser et al. (42) described two genes, picA and picB, that map within cagI. The picB gene is identical to cagE, but we find two genes, cagC and cagD, rather than a single picA gene. M. Tummuru (personal communication) agrees that as a result of a sequence error, picA is actually composed of two ORFs identical to those described herein.
The IS605, a New IS from H. pylori.
On the basis of amino acid sequence similarities, tnpA and tnpB could encode for transposases. They are flanked by two short nucleotide sequences with a dyadic symmetry and a common core sequence CTTTAG (Fig. 2A). The left (L) and the right (R) ends are 41 and 33 bp long, respectively, and were identified after alignment of several independent insertions. This structure is repeated twice within the cag region (Fig. 1) and at least five more times in the genome of strain CCUG 17874, which suggests that this may represent a novel IS, named IS605 (independently identified by N.S. Akopyants, personal communication). Usually IS are characterized by the presence of a single transposase flanked by inverted repeats. IS605 has an unusual structure that includes two coexisting transposases: one related to those found in the IS200 from Gram-negative bacteria and the other resembling the IS1341 from a Gram-positive thermophilic bacterium. Recently, an IS sequence, IS1253, with a structure close to the IS605, has been described in the ovine pathogen Dichelobacter nodosus (43). Interestingly, in this case the IS also was found to be associated with a virulence region responsible for chromosomal rearrangements, including deletions and transpositions. The two transposases of IS605 may form a heterodimeric complex acting on both of the ends; alternatively, they are not associated and each is highly specific for only one end. IS605 was found in more than 56% of the 44 clinical isolates tested. Some strains had at least seven copies per genome (data not shown). The maximal frequency of insertions was in type I strains, which revealed a close association between cag and IS605. Single copies of the IS605 also were detected in type I strains with an intermediate phenotype and a defective cagI (a partial deletion; see Fig. 4). Type II strains were negative for cag and also negative for the IS605. We have identified a variant of IS605 located at nt 22476–22735 (Fig. 1). This element is composed of two arms of IS605 without any associated ORFs (Fig. 2A). This small IS605 (is605) may represent a remnant of a transposition event or a mobile element that depends on transposases supplied in trans. The cagI deletions seen in certain H. pylori strains might have been mediated by an IS.
Figure 2.
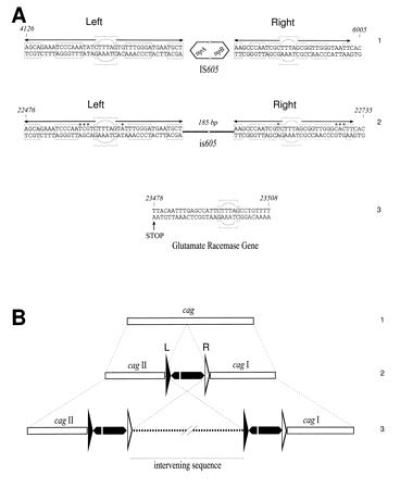
(A) Sequences of the left (L) and right (R) end of IS605 (1) and is605 (2) and the general structure of both elements. The core is formed by a conserved hexanucleotide, inscribed into a circle. The arrows indicate the dyadic structure of the ends. Astericks are nonconserved nucleotides. The sequences included in the boxes are specular. The last 31 bp of glutamate racemase gene, representing the duplicated sequence, are shown (3). The core is marked. Stop is the stop codon of glutamate racemase gene. Coordinates are relative to the cag nucleotide sequence. (B) Summarizes the structure of cag in a collection of strains previously described (4). cag is represented by a horizontal white bar (1). IS605 (2 and 3) and the intervening sequence (3) are indicated as in Fig. 1.
Figure 4.
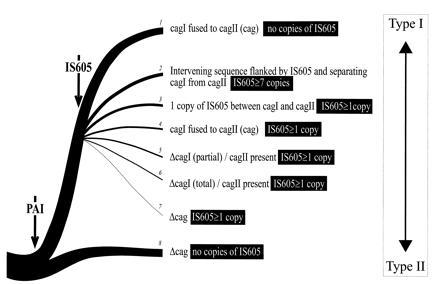
Evolutionary tree that describes the hypothetical emergence of H. pylori type I as a result of an event of gene conversion (PAI acquisition). Presumably, it is only after the integration of an IS605 that subpopulations of intermediate strains with attenuated virulence are differentiated. cag retention is indicated by arrows. Type II strains are distinguishable from type I strains with a complete cag deletion by the absence of IS605. The following strains have the genotypes listed in the tree. Genotypes: 1, G11, G33, G46, G89, G103, G109, 932, Ba99, Ba 137, and Ba158; 2, CCUG 17874, G32, G56, G106, and G204; 3, 60190, G20, G27, G29, G65, Ba179, and Ba 211; 4, G39, D933, Ba167, Ba182, Ba194, and Ba212; 5, G12 and G25; 6, G104 and D925; 7, Tx30a and G50; 8, Ba82, Ba142, G21, G198, 2U+, and 2U-.
cag Represents a PAI of H. pylori.
The genetic organization of the cag region was studied in a collection of 44 clinical isolates using Southern blot hybridization, long-distance PCR, RDA, and nucleotide sequencing. Clones isolated from DNA libraries of the type II strains G21 and G50 were also sequenced and mapped. In all type I strains analyzed, the cag region was inserted at the 3′ end of the glutamate racemase gene and flanked by a 31-bp direct repeat, presumably derived from the duplication of the 3′ end of the gene itself (Fig. 2A). Interestingly, the duplicated 31 bp had the same CTTTAG core sequence identified in the right and left arms of the IS605. The presence of the direct repeats suggested that the cag region has been acquired by H. pylori through a recombination event. Analysis of the G+C content of the cag region revealed a G+C abundance of approximately 35% compared with the 38–45% calculated from the chromosomal genes of H. pylori deposited into the data banks. A similar G+C content is also present in a 8.1-kb plasmid that we have recently identified in 33% of H. pylori strains (S.C., S. Guidotti, R.R., and A.C., unpublished results). These observations suggest to us that the cag region may be derived from a plasmid or a phage, possibly through horizontal transmission (44–46). The structure of the cag region is not identical in all type I strains, which indicates that after the initial integration event, this region has gone through a series of rearrangements in different strains. In the simplest organization (Fig. 2B1), cag is found as a single uninterrupted unit (strains G11, G33, G46, G89, G103, G109, D932, Ba99, Ba137, and Ba158). We suggest that this structure is likely to be the one initially acquired by H. pylori. In some strains, such as G20 or 60190, the cag region is divided into the cagI and cagII regions by the insertion of one copy of the IS605 (Fig. 2B2). In other strains, including the one from which we determined the map shown in Fig. 1 and the nucleotide sequence, cagI and cagII are separated by a large piece of chromosomal DNA, flanked by two IS605 sequences (Fig. 2B3). Finally, we have six strains where the recombination events have deleted part or all of the cagI, cagII, or both regions (Fig. 4, genotypes 5–7). Insertion of IS elements in functionally silent regions of PAIs has been reported (47). Additionally, models of prokaryotic and eukaryotic transposition mechanisms predict that a single IS inserted into a target may generate a second symmetric IS with an intervening sequence following homologous recombination (48).
The features described for the cag region, the presence only in disease-associated strains, the G+C content different from the mean average of chromosomal sequences, the flanking direct repeats, the presence of IS elements, and the genes packed at high density that encode a secretion system, lead us to conclude that cag is a PAI of H. pylori (15–23).
The cag Region Is Required for H. pylori-Mediated IL-8 Induction in Gastric Epithelial Cells.
The functional characterization of the cagI region was assessed by gene inactivation. Mutants in each of the major identified ORFs were generated by random insertion of a mini-Tn3 transposon and null mutations were transferred into the recipient strains G27 and G39. The set of mutants were tested using KATO-III gastric epithelial cells for their ability to induce IL-8, a proinflammatory chemokine believed to play a major role in the pathogenesis of gastritis and gastroduodenal ulceration. As shown in Fig. 3, mutations affecting cagE, cagG, cagH, cagI, cagL, and cagM all show a marked reduction in the ability of H. pylori to induce IL-8 secretion from gastric epithelial cells, which supports the view that the cag PAI is important for H. pylori virulence. These data confirm and extend the recent observation that inactivation of picB (cagE) knocks out IL-8 induction (42). Mutations in cagN and cagA did not affect IL-8 induction. We cannot rule out the existence of a polar effect in some of our mutants or the possibility that an IL-8-inducing molecule is encoded by one of the inactivated genes. The data are consistent with a model where the different proteins encoded by the cagI PAI form a contact-dependent and pilus-like secretion apparatus responsible either for direct injection of a bacterial molecule that triggers de novo synthesis of IL-8 mRNA or for IL-8 induction mediated by the membrane-anchored transfer complex that interacts with a receptor. The physical nature of this inducer is at present unknown, but bacteria other than H. pylori (Yersinia, Shigella and Salmonella species, for example) secrete factors involved in virulence and bacterial–host cell interactions through specialized and functionally distinct secretion engines commonly referred to as type III secretion systems (49–53). The presence of a set of structurally similar genes in Bordetella, Agrobacterium, and H. pylori suggests conservation of a secretion system associated with virulence analogous to conservation of type III secretion systems in bacterial pathogens of plants and animals (49).
Figure 3.
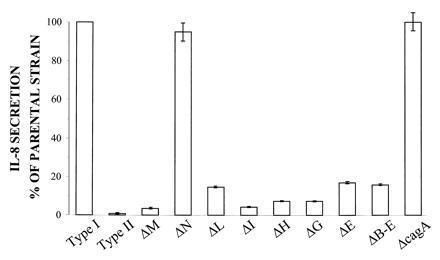
Schematic diagram indicating the IL-8 secretion from KATO-III gastric epithelial cells induced by different mutants as compared with the parenteral strain G27. Mutants are single gene inactivation by mini-Tn3 insertion and are indicated by the name of the affected ORF. A type II strain is included as a negative control. The results represent the mean (SEM) of 7–11 experiments.
DISCUSSION
We have classified H. pylori strains into those associated with severe disease pathology (type I) and attenuated in virulence (type II) according to the presence/absence of the cagA gene and the vacuolating activity on epithelial cells (4). In this work, we have shown that the difference between type I and type II is not only restricted to the cagA and vacA genes but is due to the presence in type I strains of a 40-kb PAI that contains the cagA gene. Clearly, one cannot reduce the whole pathogenicity of H. pylori to a PAI and VacA. More functions are yet to be discovered. Nevertheless, the cag PAI encodes for proteins with similarities to several prokaryotic secretory pathways, including the type IV (VirB4, PtlC, TraB, TraH, and TrbE) secretion systems involved in the export of virulence factors. Preliminary experiments show that mutants in the cag region may have an increased hemolytic activity, a reduced hemagglutination activity, and a different growth rate (C.L., S.C., S. Guidotti, R.R., and A.C., unpublished results). PAIS have been described recently in other pathogenic bacteria such as Salmonella, Yersinia, and E. coli. In all cases, they encode for a cluster of genes associated with virulence that are believed to be acquired through horizontal transmission and confer a selective advantage. Some of them are flanked by direct repeats (17), encode for a secretion system (23), and harbor IS elements as is the case in H. pylori (15). The presence of IS elements and of the direct repeats may be responsible for rearrangements and lead to partial or total deletions of the H. pylori PAI that we observed.
The acquisition and the subsequent rearrangements of the cag PAI could have played a major role in the evolution of this pathogen. For instance, the evolution of the cytotoxin vacA gene may have been driven by the acquisition of the PAI. The gene is present in type I and also in type II strains, where it is silent or encodes for a nontoxic but immunoreactive molecule (4). The VacA nucleotide sequence is well conserved within each H. pylori type but not between the types. Intratype similarity is close to 87% but intertype similarity is in the range of 50–60% (54). A consequence of the acquisition of a PAI could be a selective pressure for recruiting additional virulence determinants through mutations of ancestral molecules with a gain-of-function mechanism. Similar evolutionary mechanisms are possible for other virulence determinants. We propose that following the initial PAI acquisition, the insertion of the IS605 and the subsequent rearrangements have generated the H. pylori quasispecies that we observe today in clinical isolates. A scheme reflecting this model of H. pylori population dynamics is presented in Fig. 4. As shown in this model, the PAI acquisition generated the more virulent bacterium (type I) that today represents the dominant subpopulation. IS605-driven transpositions and deletions have then generated a number of bacteria with intermediate phenotypes that still retain most of the virulence traits. In rare cases, however, large deletions of cagI, cagII, or both occurred. In these cases, the phenotype has returned almost entirely to that of a type II strain. We believe that the molecular dissection of the PAI cag will be instrumental for the understanding of H. pylori pathogenicity mechanisms.
Acknowledgments
We thank Stanley Falkow, Lucy Tompkins, and members of both laboratories at Stanford University for helpful discussions, encouragement, and for sheltering one of us (A.C.); A. Labigne (Institut Pasteur, Paris) for the mini-Tn3 system; C. Montecucco (University of Padova, Padova, Italy), D. Holden (Hammersmith Hospital, London), and D. Unutmatz (Skirball Center, New York) for helpful suggestions; and D. E. Berg (Washington University, St. Louis) for exchanging data prior to publication. We gratefully acknowledge the National Center for Biotechnology Information, EBI, EMBL, and Georgia Institute of Technology (Atlanta) for granting access to the WWW servers for sequence analysis; I. J. D. Lindley (Sandoz Pharmaceutical, Basel, Switzerland) for IL-8 antibodies; S. Perry for excellent technical assistance; C. Mallia for reading the manuscript; and Dr. N. Figura (University of Siena, Siena, Italy) for H. pylori clinical isolates. We gratefully acknowledge G. Corsi for excellent graphic work and S. Fisher (Stanford University, Stanford) for excellent editorial assistance. We also thank A. Ruspetti for media and buffers. C.L. is supported by an EMBO long-term fellowship.
Footnotes
References
- 1.Solnick J V, Tompkins L S. Infect Ag Dis. 1993;1:294–309. [PubMed] [Google Scholar]
- 2.Parsonnet J, Friedman G D, Vandersteen D P, Chang Y, Vogelman J H, Orentreich N, Sibley R K. N Engl J Med. 1991;325:1127–1231. doi: 10.1056/NEJM199110173251603. [DOI] [PubMed] [Google Scholar]
- 3.Parsonnet J, Hansen S, Rodriguez L, Gelb A B, Warnke R A, Jellum E, Orentreich N, Vogelman J H, Friedman G D. N Engl J Med. 1994;330:1267–1271. doi: 10.1056/NEJM199405053301803. [DOI] [PubMed] [Google Scholar]
- 4.Xiang Z Y, Censini S, Bayeli P F, Telford J L, Figura N, Rappuoli R, Covacci A. Infect Immun. 1995;63:94–98. doi: 10.1128/iai.63.1.94-98.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Weel J F, van der Hulst R W M, Gerrits Y, Roorda P, Feller M, Dankert J, Tygat N J G, van der Ende A. J Infect Dis. 1996;173:1771–1775. doi: 10.1093/infdis/173.5.1171. [DOI] [PubMed] [Google Scholar]
- 6.Covacci A, Censini S, Bugnoli M, Petracca R, Burroni D, Macchia G, Massone A, Papini E, Xiang Z, Figura N, Rappuoli R. Proc Natl Acad Sci USA. 1993;90:5791–5795. doi: 10.1073/pnas.90.12.5791. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Telford J L, Ghiara P, Dell’Orco M, Comanducci M, Burroni D, Bugnoli M, Tecce M F, Censini S, Covacci A, Xiang Z Y, Papini E, Montecucco C, Parente L, Rappuoli R. J Exp Med. 1994;179:1653–1658. doi: 10.1084/jem.179.5.1653. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Marchetti M, Arico B, Burroni D, Figura N, Rappuoli R, Ghiara P. Science. 1995;267:1655–1658. doi: 10.1126/science.7886456. [DOI] [PubMed] [Google Scholar]
- 9.Crabtree J E, Covacci A, Farmery S M, Xiang Z, Tompkins D S, Perry S, Lindley I J D, Rappuoli R. J Clin Pathol. 1995;48:41–45. doi: 10.1136/jcp.48.1.41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Crabtree J E, Farmery S M, Lindley J D, Figura N, Peichl P, Tompkins D S. J Clin Pathol. 1994;47:945–950. doi: 10.1136/jcp.47.10.945. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Bukanov N O, Berg D E. Mol Microbiol. 1994;11:509–523. doi: 10.1111/j.1365-2958.1994.tb00332.x. [DOI] [PubMed] [Google Scholar]
- 12.Tummuru M K R, Cover T L, Blaser M J. Infect Immun. 1994;62:2609–2613. doi: 10.1128/iai.62.6.2609-2613.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Sharma S A, Tummuru M K R, Miller G G, Blaser M. Infect Immun. 1995;63:1681–1687. doi: 10.1128/iai.63.5.1681-1687.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Crabtree J E, Xiang Z, Lindley I J D, Tompkins D S, Rappuoli R, Covacci A. J Clin Pathol. 1995;48:967–969. doi: 10.1136/jcp.48.10.967. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Fetherston J D, Schuetze P, Perry R D. Mol Microbiol. 1992;6:2693–2704. doi: 10.1111/j.1365-2958.1992.tb01446.x. [DOI] [PubMed] [Google Scholar]
- 16.Gouin E, Mengaud J, Cossart P. Infect Immun. 1994;62:3550–3553. doi: 10.1128/iai.62.8.3550-3553.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Hacker J, Bender L, Ott M, Wingender J, Lund B, Marre R, Goebel W. Microb Pathog. 1990;8:213–225. doi: 10.1016/0882-4010(90)90048-u. [DOI] [PubMed] [Google Scholar]
- 18.Blum G, Ott M, Lischewski A, Ritter A, Imrich H, Tschape H, Hacker J. Infect Immun. 1994;62:606–614. doi: 10.1128/iai.62.2.606-614.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.McDaniel T K, Jarvis K G, Donnenberg M S, Kaper J B. Proc Natl Acad Sci USA. 1995;92:1664–1668. doi: 10.1073/pnas.92.5.1664. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Knapp S, Hacker J, Jarchau T, Goebel W. J Bacteriol. 1986;168:22–30. doi: 10.1128/jb.168.1.22-30.1986. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Morschhauser J, Vetter V, Emody L, Hacker J. Mol Microbiol. 1994;11:555–566. doi: 10.1111/j.1365-2958.1994.tb00336.x. [DOI] [PubMed] [Google Scholar]
- 22.Bajaj V, Hwang C, Lee C A. Mol Microbiol. 1995;18:715–727. doi: 10.1111/j.1365-2958.1995.mmi_18040715.x. [DOI] [PubMed] [Google Scholar]
- 23.Shea J E, Hensel M, Gleeson C, Holden D. Proc Natl Acad Sci USA. 1996;93:2593–2597. doi: 10.1073/pnas.93.6.2593. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Akopyants, N. S., Jiang, Q., Taylor, D. E. & Berg, D. E. (1996) Helicobacter, in press. [DOI] [PubMed]
- 25.Stemmer W P C. BioTechniques. 1991;10:726. [PubMed] [Google Scholar]
- 26.Maniatis T, Fritsch E F, Sambrook J. Molecular Cloning: A Laboratory Manual. 2nd Ed. Plainview, NY: Cold Spring Harbor Lab. Press; 1989. [Google Scholar]
- 27.Davis R H, Botstein D, Roth J R. Advanced Bacterial Genetics. Plainview, NY: Cold Spring Harbor Lab. Press; 1980. [Google Scholar]
- 28.Ausubel F M, Brent R, Kingston R E, Moore D D, Seidman J G, Smith J A, Struhl K. Current Protocols in Molecular Biology. New York: Wiley; 1987. Vols. 1–3. [Google Scholar]
- 29.Lisitsyn N, Wigler M. Methods Enzymol. 1995;254:291–304. doi: 10.1016/0076-6879(95)54021-0. [DOI] [PubMed] [Google Scholar]
- 30.Sanger F, Nicklen S, Coulson A R. Proc Natl Acad Sci USA. 1977;74:5463–5467. doi: 10.1073/pnas.74.12.5463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Devereux J, Haeberli P, Smithies O. Nucleic Acids Res. 1984;12:387–399. doi: 10.1093/nar/12.1part1.387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Borodovsky M, Rudd K E, Koonin E V. Nucleic Acids Res. 1994;22:4756–4767. doi: 10.1093/nar/22.22.4756. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Suerbaum S, Josenhans C, Labigne A. J Bacteriol. 1993;175:3278–3288. doi: 10.1128/jb.175.11.3278-3288.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Crabtree J E, Peichl P, Wyatt J I, Stachl U, Lindley I J. Scand J Immunol. 1993;36:65–70. doi: 10.1111/j.1365-3083.1993.tb01666.x. [DOI] [PubMed] [Google Scholar]
- 35.Ward J E, Akiyoshi D E, Regier D, Datta A, Gordon M P, Nester E W. J Biol Chem. 1988;263:5804–5814. [PubMed] [Google Scholar]
- 36.Covacci A, Rappuoli R. Mol Microbiol. 1993;8:429–434. doi: 10.1111/j.1365-2958.1993.tb01587.x. [DOI] [PubMed] [Google Scholar]
- 37.Weiss A A, Johnson F D, Burns D. Proc Natl Acad Sci USA. 1993;90:2970–2974. doi: 10.1073/pnas.90.7.2970. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Winans S C, Burns D L, Christie P J. Trends Microbiol. 1996;4:64–68. doi: 10.1016/0966-842X(96)81513-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Lessl M, Lanka E. Cell. 1994;77:321–324. doi: 10.1016/0092-8674(94)90146-5. [DOI] [PubMed] [Google Scholar]
- 40.Haack K R, Roth J R. Genetics. 1995;141:1245–1252. doi: 10.1093/genetics/141.4.1245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Murai N, Kamata H, Nagashima Y, Yagisawa H, Hirata H. Gene. 1995;163:103–107. doi: 10.1016/0378-1119(95)00384-i. [DOI] [PubMed] [Google Scholar]
- 42.Tummuru M K R, Sharma S A, Blaser M J. Mol Microbiol. 1995;18:867–876. doi: 10.1111/j.1365-2958.1995.18050867.x. [DOI] [PubMed] [Google Scholar]
- 43.Billington S J, Sinistaj M, Cheetham B F, Ayres A, Moses E K, Katz M E, Rodd J. Gene. 1996;172:111–116. doi: 10.1016/0378-1119(96)00032-7. [DOI] [PubMed] [Google Scholar]
- 44.Groisman E A, Sturmoski M A, Solomon F R, Lin R, Ochman H. Proc Natl Acad Sci USA. 1993;90:1033–1037. doi: 10.1073/pnas.90.3.1033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Aitmeyer R M, McNern J K, Bossio J C, Rosenshine I, Finlay B B, Galan J E. Mol Microbial. 1993;7:89–98. doi: 10.1111/j.1365-2958.1993.tb01100.x. [DOI] [PubMed] [Google Scholar]
- 46.Li J, Ochman H, Groisman E A, Boyd E F, Soloman F, Nelson K, Selander R K. Proc Natl Acad Sci USA. 1995;92:7252–7256. doi: 10.1073/pnas.92.16.7252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Portnoy D A, Falkow S. J Bacteriol. 1981;148:877–883. doi: 10.1128/jb.148.3.877-883.1981. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Kleckner N. In: Mobile DNA. Berg D E, Howe M H, editors. Washington, DC: Am. Soc. Microbiol.; 1989. pp. 227–268. [Google Scholar]
- 49.Salmond G P C, Reeves P J. Trends Biochem Sci. 1993;18:7–12. doi: 10.1016/0968-0004(93)90080-7. [DOI] [PubMed] [Google Scholar]
- 50.Van Gijsegem F, Genin S, Boucher C. Trends Microbiol. 1993;1:175–180. doi: 10.1016/0966-842x(93)90087-8. [DOI] [PubMed] [Google Scholar]
- 51.Galan J E, Curtiss R. Proc Natl Acad Sci USA. 1989;86:6383–6387. doi: 10.1073/pnas.86.16.6383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Groisman E A, Ochman H. EMBO J. 1993;12:3779–3787. doi: 10.1002/j.1460-2075.1993.tb06056.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Ginocchio C C, Olmsted S B, Wells C L, Galan J E. Cell. 1994;76:717–724. doi: 10.1016/0092-8674(94)90510-x. [DOI] [PubMed] [Google Scholar]
- 54.Atherton J C, Cao P, Peek R M, Tummuru M K R, Blaser M J, Cover T. J Biol Chem. 1995;270:17771–17777. doi: 10.1074/jbc.270.30.17771. [DOI] [PubMed] [Google Scholar]