

Abstract
The soil nematodes Caenorhabditis briggsae and Caenorhabditis elegans diverged from a common ancestor roughly 100 million years ago and yet are almost indistinguishable by eye. They have the same chromosome number and genome sizes, and they occupy the same ecological niche. To explore the basis for this striking conservation of structure and function, we have sequenced the C. briggsae genome to a high-quality draft stage and compared it to the finished C. elegans sequence. We predict approximately 19,500 protein-coding genes in the C. briggsae genome, roughly the same as in C. elegans. Of these, 12,200 have clear C. elegans orthologs, a further 6,500 have one or more clearly detectable C. elegans homologs, and approximately 800 C. briggsae genes have no detectable matches in C. elegans. Almost all of the noncoding RNAs (ncRNAs) known are shared between the two species. The two genomes exhibit extensive colinearity, and the rate of divergence appears to be higher in the chromosomal arms than in the centers. Operons, a distinctive feature of C. elegans, are highly conserved in C. briggsae, with the arrangement of genes being preserved in 96% of cases. The difference in size between the C. briggsae (estimated at approximately 104 Mbp) and C. elegans (100.3 Mbp) genomes is almost entirely due to repetitive sequence, which accounts for 22.4% of the C. briggsae genome in contrast to 16.5% of the C. elegans genome. Few, if any, repeat families are shared, suggesting that most were acquired after the two species diverged or are undergoing rapid evolution. Coclustering the C. elegans and C. briggsae proteins reveals 2,169 protein families of two or more members. Most of these are shared between the two species, but some appear to be expanding or contracting, and there seem to be as many as several hundred novel C. briggsae gene families. The C. briggsae draft sequence will greatly improve the annotation of the C. elegans genome. Based on similarity to C. briggsae, we found strong evidence for 1,300 new C. elegans genes. In addition, comparisons of the two genomes will help to understand the evolutionary forces that mold nematode genomes.
With the Caenorhabditis briggsae genome now in hand, C. elegans biologists have a powerful new research tool to refine their knowledge of gene function in C. elegans and to study the path of genome evolution
Introduction
Comparative sequence analysis is a global approach toward recognizing much of the functional sequence in a genome. Comparisons of genomes of appropriate evolutionary distance can aid in defining protein-coding genes, in recognizing noncoding genes, and in finding regulatory sequences and other functional elements of a genome.
The soil-dwelling nematode Caenorhabditis elegans has been intensively studied over the past several decades to establish the molecular genetic basis of its development and behavior. The completion of its genome sequence (C. elegans Sequencing Consortium 1998) provides a complete description of the genetic information, but decoding the program embedded in the sequence remains a challenge.
Caenorhabditis briggsae is another soil-dwelling nematode that diverged from C. elegans approximately 100 million years ago (MYA) (Coghlan and Wolfe 2002) and, along with Caenorhabditis remanei, is one of C. elegans' closest known relatives (Jovelin et al. 2003). The two organisms are almost indistinguishable morphologically (Nigon and Dougherty 1949). They follow very similar developmental programs, for example, in sex determination and vulval development (Kirouac and Sternberg 2003; Stothard and Pilgrim 2003). There are, however, subtle differences between the two species. One interesting difference is the inability of C. briggsae to take up and distribute interfering RNAs (M. Montgomery, personal communication). There are also differences in the nature and timing of key events in the development of the vulva (Kirouac and Sternberg 2003), the excretory pore (Wang and Chamberlin 2002), and the male tail (Fitch and Emmons 1995; Fitch 1997).
Genomic sequencing of C. briggsae actually began in the mid-1990s, when the Genome Sequencing Center at Washington University in St. Louis began sequencing large-insert clones from throughout the genome, eventually producing 12 Mbp of finished sequence. Even this relatively small amount of sequence, together with genes cloned by individual labs, has been extremely valuable to researchers, who have used it to identify putative transcription factor binding sites (Gilleard et al. 1997; Kirouac and Sternberg 2003), to infer patterns of evolutionary constraints (Civetta and Singh 1998; Castillo-Davis and Hartl 2002), to probe the dynamics of chromosome evolution (Coghlan and Wolfe 2002), and to study the expansion and contraction of large gene families (Sluder et al. 1999; Robertson 2001).
We have now extended this work to the whole genome level, assembling a “draft”-quality C. briggsae sequence that covers an estimated 98% of the genome. In this report we discuss the characteristics of this draft and give our preliminary analysis of the C. briggsae genome at the nucleotide and protein levels.
Results
We begin by describing the sequencing procedure, followed by the results of our analyses at the nucleotide and protein levels. Data files containing the results of the analyses reported here are found as Supporting Information (the full set is Dataset S1). The C. briggsae sequence and analytic results are also available in interactive format from WormBase (http://www.wormbase.org).
Genomic Sequencing
To sequence the C. briggsae genome, we adopted a hybrid strategy combining whole-genome shotgun sequencing (WGS) with a high-resolution, sequence-ready physical map. The previously finished clone-based sequence was then integrated with the whole-genome assembly to produce the draft sequence.
Fingerprint Map Construction
Physical mapping and sequencing was performed on C. briggsae strain AF16 (Fodor et al. 1983). We constructed a physical map for C. briggsae using a high-throughput fingerprinting scheme (Marra et al. 1997). Our substrates were two large-insert C. briggsae libraries: a 6.5-fold coverage fosmid library (M. A. Marra, unpublished data) and a 10-fold coverage bacterial artificial chromosome (BAC) library (M. A. Marra and P. deJong, unpublished data).
After several rounds of automated assembly and manual review, we developed a physical map consisting of 188 fingerprinted contigs (FPCs) with a mean length of approximately 450 kbp containing 17,885 BAC clones and 16,414 fosmid clones. The longest FPC contig spans more than 4 Mbp of genomic sequence. Because there is little genetic mapping information for C. briggsae, these FPC contigs cannot currently be localized to chromosomal locations.
Shotgun Sequencing, Assembly, and Physical Map Integration
We performed WGS of small-insert plasmid libraries using the paired-end sequencing strategy introduced by Edwards et al. (1990). This was supplemented with end sequencing of clones from the BAC library in order to facilitate integration of the assembled sequence with the physical map. In all, 2.068 million shotgun reads were assembled, giving more than 10-fold coverage of the C. briggsae genome. A total of 82% of the shotgun reads were paired.
The WGS was then assembled using the Phusion assembler (Mullikin and Ning 2003) into a set of 5,341 contigs; these contigs were linked together using the read pair information into a total of 899 gapped “supercontigs” containing 105.6 Mbp of DNA sequence and another 1.9 Mbp of inferred gaps. The N50 (the length x such that 50% of the genome lies in blocks of x or longer) of the contigs was 41 kbp. The supercontigs were substantially larger, as reflected in an N50 of 474 kbp. For reasons discussed below, the actual C. briggsae genome may be slightly smaller than 105.6 Mbp.
To integrate the sequence assembly with the physical map, we performed a conceptual restriction digest of the 5,341 sequence contigs and incorporated them into the fingerprint map using automated assembly followed by manual review. During this process, misassemblies in both the physical map and the sequence assembly were detected and corrected.
Lastly, we integrated the sequence from 272 finished fosmid and BAC clones from many different regions of the C. briggsae genome. We used this finished sequence to fill 264 gaps in the assembly, adding 269 kbp of sequence from 155 finished clones.
In the final analysis, 463 sequence supercontigs from the WGS assembly were placed in 142 FPC contigs. These 142 supercontigs had an N50 length of 1,450 kbp and a total sequence length covering 102,431,873 bp, equivalent to 94% of the summed length of all supercontigs. A total of 436 sequence supercontigs could not be placed onto the FPC map. These unplaced supercontigs were relatively small (largest, 103 kbp; mean, 13.8 kbp; SD, 16.6 kbp; N50, 34 kbp) and covered a total of 6.0 Mbp.
This version of the draft assembly is referred to as cb25.agp8. Data for this assembly are available at ftp://ftp.sanger.ac.uk/pub/wormbase/cbriggsae/cb25.agp8/
Completeness and Accuracy of the Draft
To assess the completeness of the C. briggsae draft sequence, we compared the WGS contigs and supercontigs to the 12 Mbp of previously finished sequence. We did this before incorporating the finished sequence into the assembly. The SSAHA algorithm (Ning et al. 2001), a fast search method for large DNA databases, was used to match previously finished contigs to the corresponding regions of the WGS draft. From this, we estimate that the contigs cover 98% of the previously finished clone-based sequences and that the scaffolds span 99.3% of the clone-based sequence. Thus, we estimate that the draft covers 98% of the C. briggsae genome.
Some 65,000 of the unassembled reads matched three or more other reads in the set. These represent 2.5% of the total reads, suggesting that 2.6 Mbp of additional sequence is represented in these unplaced reads. Among these read clusters are the 5S, ribosomal DNA (rDNA), and mitochondrial sequences. Assembly of these reads yields a 14,420 base mitochondrial sequence, a 7,429–7,431 base rDNA repeat unit, and a 697 and a 938–940 base 5S repeat unit. Based on the total mitochondrial reads, we estimate on average approximately ten copies of the mitochondrial genome per haploid nuclear genome. Most other clusters appear to contain tandem copies of low complexity sequence of the type found scattered throughout the C. elegans genome. Read pair and assembly information allow both the rDNA and 5S clusters to be placed in the current assembly. The rDNA sequences lie adjacent to telomeric repeats as they do in C. elegans. The two arrays of the 5S gene lie adjacent to each other.
To assess overall quality of the assembly and to evaluate the gap size estimate, we again used the comparison of the assembly to the finished clone sequence. We detected no global misassemblies. The 264 gaps that were filled with finished sequence contained a total estimated gap size of 179 kbp before filling. After gap filling, however, the total length of the assembly decreased by 323 kbp. Because of the change in the gap size estimates, we estimate that 3% of the bases in the assembly consist of undetected overlaps. Combined with the estimated coverage of 98%, this yields an estimate for the C. briggsae genome size of 104 Mbp. However, this figure contains substantial uncertainty, in part because the finished sequence is not a random selection of the genome, and this number is certain to be revised as additional finishing is performed.
We assessed sequence accuracy in two ways. First, we aligned the finished sequence to the final WGS assembly and counted discrepancies. Using this method, the accuracy is 99.98%. Second, we examined the consensus quality scores (Ewing and Green 1998; P. Green, unpublished data) across the assembly. These data again suggest a sequencing accuracy of approximately 99.98%.
Content of the C. briggsae Genome
To characterize the C. briggsae genome, we began with the following analyses of the content of the C. briggsae genome: (1) identification of candidate protein-coding genes and development of a canonical gene set; (2) characterization of the predicted protein-coding gene set by domain analysis; (3) comparative analysis of the C. briggsae and C. elegans proteomes using the predicted protein-coding gene set; (4) identification and characterization of candidate noncoding genes; and (5) characterization of the repeat family content of the genome. In later sections we examine the C. briggsae and C. elegans genomes at the long-range, structural level and illustrate the utility of comparative analysis in aiding genome interpretation.
For all C. briggsae characterizations described in the remainder of this section, we used the cb25.agp8 assembly described earlier. With minor exceptions noted below, all comparisons that involved C. elegans used the C. elegans genome and annotations contained in WormBase release version WS77 (WS77), which was current as of April 2002.
Protein-Coding Genes
Different protein-coding gene prediction algorithms generally have high concordance rates for predicting exons, but they tend to disagree on the grouping of exons into genes (Reese et al. 2000). Hence, four different gene prediction programs may give four strikingly different answers across the same region of the genome. An increasingly common procedure for overcoming this problem is to predict gene structures using several ab initio gene prediction programs, to compare their output in order to find a representative prediction, and then to partially or fully confirm the structures with experimental data from expressed sequence tags (ESTs), sequenced cDNAs, or protein similarity matches (Goff et al. 2002; Rogic et al. 2002). To find protein-coding genes in C. briggsae, we developed an enhancement of this procedure that uses the concordance of predictions between C. elegans and C. briggsae to predict the most likely gene model.
We predicted genes in the C. briggsae genome using the programs Genefinder (version 980506; P. Green, unpublished data), FGENESH (Salamov and Solovyev 2000), TWINSCAN (Korf et al. 2001), and the Ensembl annotation pipeline (Clamp et al. 2003). These programs combine a variety of gene prediction methodologies, including ab initio predictions (Genefinder, FGENESH), EST- and protein-based comparisons (Ensembl), and sequence conservation metrics (TWINSCAN). We called genes in the C. elegans genome by combining hand-curated data models from WS77 (Stein et al. 2001) with the ab initio predictions from Genefinder and FGENESH. For technical reasons, we were unable to run TWINSCAN on the C. elegans genome, while the Ensembl methodology of predicting genes based on matches to previously predicted proteins meant that an Ensembl C. elegans gene set would essentially be a duplicate of the hand-curated WormBase set.
As expected, the output of the four gene prediction programs was largely concordant with respect to the position of C. briggsae exons (80% of exons predicted identically by two or more programs, 26% predicted identically by all four programs), but discordant with respect to gene predictions (38% of genes called identically by two or more programs, just 4% called identically by all four programs). A similar pattern was seen in the genes called in C. elegans.
To select among overlapping predictions produced by different programs, we reasoned that the most likely gene model is the one that maximizes the similarity between the gene sets in two related species (Figure 1). For each C. briggsae region that had multiple overlapping but inconsistent predictions, our selection procedure chose the prediction that had the most extensive similarity to the matching C. elegans prediction. The extent of similarity was measured by the fraction of the C. briggsae prediction that aligned to the matching C. elegans prediction at the protein level. Likewise, from all the predictions for a C. elegans gene, we chose the prediction having the most extensive similarity to its C. briggsae match. This selection step produced two gene sets, one each for C. briggsae and C. elegans. The gene sets were then filtered to remove transposons and putative pseudogenes.
Figure 1. Joint Refinement of C. elegans and C. briggsae Gene Models: acy-4.

When annotating the C. briggsae and C. elegans acy-4 orthologs, we chose the Genefinder ce-acy-4 prediction and the Genefinder cb-acy-4 prediction because, out of the 12 possible combinations of a C. briggsae and a C. elegans prediction, this pair shows the most similarity to each other. Coding sequence (CDS) conservation between cb-acy-4 and ce-acy-4 provides evidence for as many as 12 additional N-terminal exons in the Genefinder ce-acy-4 prediction, as compared to T01C2.1, the WS77 ce-acy-4 prediction. Subsequently, four of the additional N-terminal exons that were predicted by FGENESH and Genefinder were confirmed by new EST data (marked with asterisks).
We call the gene sets produced by our procedure “hybrid” gene sets because the final gene sets are a mixture of gene predictions from multiple programs. The procedure selected predictions for both species simultaneously, yielding a C. briggsae hybrid gene set and a C. elegans hybrid gene set.
To assess the accuracy of the gene prediction programs in C. elegans, we made a “gold standard” set of C. elegans gene predictions, consisting of 2,257 genes from WS77 for which every base and intron–exon junction had been confirmed by cDNA or EST data. Genefinder made 2,309 predictions that overlapped a gold-standard gene, of which 1,280 (53%) contained all confirmed bases and introns. FGENESH made 2,742 predictions that overlapped a gold-standard gene, of which 1,230 (45%) contained all the confirmed data. We also used the gold standard to assess our selection procedure. For C. elegans genes in the gold-standard set, the selection procedure chose the correct gene model for 92% of gold-standard genes, choosing an alternative (incorrect FGENESH or Genefinder) model 8% of the time. We could not assess the accuracy of the gene prediction programs or selection procedure directly in C. briggsae because we lacked an independent dataset to create a gold standard.
The final C. briggsae gene set contains 19,507 genes, and the hybrid C. elegans gene set contains 20,621 genes. Some of the genes taken from WS77 have alternative splices, so the 20,621 C. elegans genes have 21,578 different splice variants. In the absence of substantial EST data, we are currently unable to call or comment on patterns of alternative splicing in C. briggsae.
In order to compare the C. briggsae and C. elegans hybrid gene sets to the C. elegans WS77 gene set, we also applied our transposon and pseudogene filtering step to the C. elegans WS77 gene set. This removed 619 genes to create a “pruned” WS77 set of 18,808 genes and 19,791 splices. This pruned set is henceforth called WS77*. An important caveat is that some of the predictions discarded by our filtering step may include real exons: 29 (9%) of the 316 putative pseudogenes in C. elegans WS77 that were discarded have been partially or fully confirmed by EST or cDNA data.
Files containing the C. briggsae, C. elegans hybrid, and C. elegans WS77* gene predictions, their genomic positions, and their conceptual translations are available as Dataset S2.
Comparing the C. briggsae and C. elegans Gene Sets
The C. briggsae gene set (19,507 genes), the C. elegans WS77* gene set (18,808 genes), and the C. elegans hybrid gene set (20,621 genes) all contain approximately the same number of genes. The recent WormBase release WS103 (June 2003; approximately 19,600 curated genes) also has a similar number. We next set about examining these sets in more detail.
The unspliced lengths of genes are roughly the same in the two species (C. briggsae median, 1.9 kbp; C. elegans WS77*, 1.9 kbp; Table 1), and the total length of the C. briggsae genome occupied by the 19,507 genes, including their introns, is 56 Mbp (54% of the 102 Mbp assembly)—approximately the same fraction of the C. elegans genome occupied by the WS77* gene set. Thus, the larger size of the C. briggsae genome is not due to an increase in the number or size of protein-coding genes.
Table 1. Comparison of the C. briggsae and C. elegans Protein-Coding Gene Sets.

The C. elegans gene sets have slightly more introns than the C. briggsae hybrid set. Some of this difference might be due to the hand-curation of the WS77 gene set, since curation may add exons that were missed by gene prediction software. However, as shown in the C. briggsae/C. elegans Orthologs section below, a portion of the intron differences can be confirmed as exhibiting true evolutionary changes.
We examined codon usage for both the predicted gene sets as a whole and for C. briggsae/C. elegans ortholog pairs (described below) and found no substantial differences between the protein-coding gene codon usage in the two species. We then used the EMBOSS tool codcmp to test for a significant difference in codon usage. The GC content for the two species differed only slightly for the genome as a whole (35.4% for C. elegans versus 37.4% for C. briggsae) and for protein coding exons (42.7% versus 44.1%).
Domain and Gene Ontology Analysis
We used Pfam analysis to search the C. briggsae gene set for functional domains and other known sequence motifs and to assign InterPro annotation to each such feature found (Zdobnov and Apweiler 2001). These InterPro annotations were translated into Gene Ontology (GO) functional descriptions, and the descriptions were grouped into broader categories according to molecular function (13 categories) and biological process (9 categories) (“GOslim”; http://www.ebi.ac.uk/proteome; Gene Ontology Consortium 2001). We did not classify proteins by intracellular compartment due to the small number of genes in these categories for the C. elegans dataset.
Of 19,507 predicted proteins in the C. briggsae dataset, 10,606 (54.4%) had one or more Pfam annotations, 9,829 were associated with one or more InterPro terms, and 6,526 of these could be assigned one or more GO terms. This annotation process touched 1,443 InterPro terms and 706 GO terms. The results of these classifications are available as Dataset S2.
For comparison, we performed an identical analysis on the C. elegans proteins in the C. elegans hybrid gene set. Of the 20,621 proteins in this set (counting only the longest protein encoded by each alternatively spliced gene), 11,116 (53.9%) had Pfam annotations, 10,460 had InterPro annotations, and 6,696 of these could be assigned GO terms, touching 1,436 InterPro terms and 699 GO terms.
As expected, the distribution of classifications is roughly the same in both species (Table 2), with 75%–76% of classifiable proteins associated with metabolic processes, 15%–16% associated with transport, and 14%–15% associated with cell communication. The molecular function classification showed the majority of proteins classified as having ligand-binding or carrier functions (63%–66%), followed by enzymatic activity (48%) and nucleic acid binding (28%–30%). There is a 4% difference in the proportion of proteins classified as involved in signal transduction in C. elegans relative to C. briggsae. As will be discussed in a later section, this difference is largely due to the difference in the number of predicted chemosensory receptor proteins in the two genomes. The significance of other small differences, such as the approximately 1% increase in C. elegans in the proportion of proteins involved in cell motility, is not known.
Table 2. Classification of Predicted C. briggsae and C. elegans Proteins into GOslim Categories.

The total number of proteins in each species that could be matched to terms in the GO molecular function and biological process ontologies is shown in parentheses. For each GOslim category, the percentage of proteins placed in that category was normalized by dividing it by the total number of proteins that could be matched to any term in the ontology. The values sum to more than 100% because some proteins were placed into two or more categories
C. briggsae/C. elegans Orthologs
We searched for orthologs between the 19,507 C. briggsae genes and the 18,808 C. elegans WS77* genes. Although it is possible for a gene in one species to have multiple orthologs in another species if the gene has duplicated since the two species diverged, for this analysis we used the simpler definition of a pair of genes that have a common ancestor and are in a one-to-one correspondence between the two species.
We found orthologs by searching for C. briggsae/C. elegans gene pairs that were each other's top BLASTP (Altschul et al. 1997) match in the opposite species. We identified 11,255 such gene pairs. We then used conserved gene order (synteny) between the two species to identify nonreciprocal best matches that were supported by the positions of flanking orthologs. This procedure netted an additional 900 orthologs. The final set of 12,155 orthologs corresponds to 62% of the C. briggsae gene set and 65% of the C. elegans WS77* gene set.
To assess the accuracy of this ortholog definition, we compared the results obtained by this procedure to those obtained by building phylogenetic trees of the chemosensory receptor sra protein subfamily (see the Protein Families section below). Phylogenetic tree building identified eight ortholog pairs in this set, seven of which were called identically by the mutual-best BLASTP match procedure. The mutual-best BLASTP match procedure had one false negative and one false positive involving a recent C. briggsae gene amplification. As will be seen, the chemosensory receptor family represents the worst-case scenario of a family that is undergoing rapid evolutionary change. This provides conservative estimates of false positive and false negative rates for the mutual-best BLASTP match procedure of roughly 15%.
The median percent identity between orthologs at the protein level is 80% (mean, 75%; SD, 18%), which is similar to the level of divergence between mouse/human orthologs (median identity, 78.5%; Waterston et al. 2002). The ortholog pairs are very similar in terms of exon length (median in both species, 0.15 kbp), coding length per gene (median, 1.14 kbp in C. elegans versus 1.11 kbp in C. briggsae), and gene length (median, 2.29 kbp in C. elegans versus 2.19 kbp in C. briggsae). However, orthologs are longer than the overall set of predicted genes (median length, 1.90 kbp in C. elegans), which suggests that the nonorthologous gene set includes a population of truncated or split genes.
To pursue the earlier observation that C. elegans has more introns than C. briggsae, we searched for cases in which a C. elegans gene has an intron absent from its C. briggsae ortholog and vice versa. To do this, we aligned orthologous proteins and searched for cases where a single exon in one species aligned to two adjacent exons in the other species.
We found 6,579 species-specific introns among the 60,775 introns in the ortholog pairs: 4,379 C. elegans-specific introns and 2,200 C. briggsae-specific introns. This approximately 2-fold ratio agrees with that reported by Kent and Zahler (2000) using a smaller dataset. Intron gains or losses have occurred at a rate of at least 0.5 per gene in the 80–110 million years (MY) since C. elegans and C. briggsae diverged (see the Estimating the C. briggsae/C. elegans Divergence Date section below). This is similar to the arthropod rate of approximately one intron gain or loss per gene per 125 MY since Drosophila and Anopheles diverged (Zdobnov et al. 2002). In contrast, in mouse and human there have been fewer than 0.01 losses or gains per gene in 75 MY (Roy et al. 2003). Since the average number of introns per gene is quite different among these species, this means that 9% of Caenorhabditis introns are species-specific, in contrast to 50% of Anopheles/Drosophila introns and only 0.05% of mouse/human introns. Thus, intron–exon structure has apparently evolved more rapidly in nematodes and arthropods than in chordates.
The list of ortholog pairs can be found as Dataset S3.
Rate of Neutral Evolution and Estimates of Selective Pressure
Using the set of C. briggsae/C. elegans ortholog pairs, we calculated the rates of nonsynonymous (KA) and synonymous (KS) amino acid substitutions, using a maximum likelihood (ML) algorithm that corrects for reversion events (Yang 1997) and removing pairs where accurate estimates of KA and KS were impossible.
Orthologous genes identified by mutual-best BLASTP hits had an average KS of 1.78 (SD, 0.62) synonymous substitutions per synonymous site and a KA of 0.11 (SD 0.09) nonsynonymous substitutions per nonsynonymous site, while orthologous gene pairs identified by the combination of mutual-best BLASTP hits and colinearity had average KS and KA of 1.73 (SD, 0.68) and 0.12 (SD, 0.131), respectively. The corrected KS rate is almost three times as high as that reported between mouse and human (KS = 0.6; Waterston et al. 2002), despite the fact that the apparent C. briggsae/C. briggsae divergence date is only 5–45 MY before the Mus musculus/Homo sapiens divergence date (see next section). However, the reported KA/KS ratio for mouse/human (0.115) is similar to the ratio that we see in C. briggsae/C. elegans (approximately 0.06), arguing that the levels of purifying selection are similar.
The KA/KS ratio, a measure of selective pressure, is expected to be 1.0 for genes that are under no selective pressure (for example, pseudogenes), less than 1.0 for genes under purifying selection, and greater than 1.0 for genes under positive selection. As expected, we found that nearly all the genes in ortholog pairs are under purifying selection (Figure 2). However, the extent of this purifying selection is more marked in genes with essential functions. For example, ortholog pairs which exhibit an embryonic lethal phenotype in systematic RNA inhibition (RNAi) screens of the C. elegans ortholog partner (Maeda et al. 2001; Piano and Gunsalus 2002; Kamath et al. 2003) show a markedly lower KA/KS ratio than do pairs for which a wild-type phenotype was observed (KA/KS, 0.0445 ± 0.0340 versus 0.0627 ± 0.0494; p < 1 × 10−16 by the Welch two-sample t-test). We also confirmed the findings of Castillo-Davis and Hartl (2002), who showed that genes involved early in development tend to be less prone to duplications and have a lower KS value than late-development genes (data not shown).
Figure 2. Distribution of KA/KS Ratio among Ortholog Pairs.

The trend towards higher levels of purifying selection in essential genes has been seen in organisms as distantly related as prokaryotes. For example, Jordan et al. (2002) found similar differences when comparing rates of evolution of essential and nonessential genes in Escherichia coli, Helicobacter pylori, and Neisseria meningitidis.
We also looked for genes with evidence of positive selection. Civetta and Singh (1998) reported generally higher KA/KS ratios for sex determination genes tra-1 and tra-2 than for other sampled genes and interpreted this as positive selection on genes that are involved in speciation. Under the strict definition of positive selection, where KA/KS > 1.0, we do not find evidence for positive selection in these genes, but given the high value of substitution between the two species, genes with a ratio greater than 1.0 may no longer be recognizable as orthologs. Perhaps genes under positive selection might be found among members of gene families or in genes that are no longer recognizably similar at the primary sequence level. Sequence analysis of species of intermediate evolutionary distance, if available, would be revealing. Also, since our tests for positive selection are very conservative, we would not detect any sites under positive selection if they are small in proportion to those of the gene under purifying selection (Yang et al. 2000).
Estimating the C. briggsae/C. elegans Divergence Date
Using the divergence of the nematodes from the arthropods 800–1,000 MYA (Blaxter 1998) to calibrate the molecular clock, we estimated the C. briggsae/C. elegans divergence date from 338 sets of orthologs. Each set comprised a C. elegans gene and its one-to-one orthologs from C. briggsae, A. gambiae, and H. sapiens. When the nematode/arthropod divergence is taken to be 800 MYA, a 95% confidence interval for the median C. briggsae/C. elegans speciation date is 78–90 MYA. If the nematode/arthropod divergence is taken to be 1,000 MYA, the interval becomes 97–113 MYA.
Our best estimate of the C. briggsae/C. elegans speciation date is therefore approximately 80–110 MYA. This confidence interval is tighter than a previous estimate of 50–120 MYA made using 92 sets of orthologs from the then available C. briggsae genome (Coghlan and Wolfe 2002). The current estimate is probably more accurate due to both a larger sample size and improved C. briggsae gene predictions and ortholog assignments. Interestingly, recent studies date the mouse/human divergence to 65–75 MYA (Waterston et al. 2002), so the date of the C. briggsae/C. elegans divergence was between 5 and 45 MY before the rodent/primate divergence.
C. briggsae/C. elegans Paralogs and Orphans
In this section, we look in more detail at those C. briggsae proteins that could not be assigned to C. elegans orthologs. Roughly, a third of the C. elegans and C. briggsae proteins fall into this category. Of these, 4,545 (23%) C. elegans (WS77*) genes and 5,211 (28%) C. briggsae proteins have multiple BLASTP matches in the opposite species. These correspond to paralogous relationships within gene families and are examined in greater detail in the next section (Gene Families).
The remaining 2,108 (11%) C. elegans and 2,141 (11%) C. briggsae genes do not have any BLASTP hit of E-value <10−10 in the opposite genome and therefore represent candidate species-specific genes, or “orphans.” However, many of these are simply genes that have evolved rapidly. Lowering the BLASTP threshold to E-value <10−5 finds 785 C. briggsae proteins that have a weak C. elegans match. An additional 11 proteins have a strong TBLASTN match to the C. elegans genomic sequence, signifying either a C. elegans gene that is missing from the predicted gene set or a pseudogene. These are being examined individually. Another 538 C. briggsae genes were found by TRIBE to belong to shared C. briggsae/C. elegans gene families (see the Gene Families section below) and so are members of rapidly evolving families common to both species.
This leaves 807 C. briggsae proteins that have no BLASTP match (E-value, <10−5) in the opposite species and that do not belong to a shared C. briggsae/C. elegans gene family. A similar analysis yields 1,061 C. elegans orphans. Of these, 695 C. briggsae genes and 963 C. elegans genes have at least two exons and so are less likely than single-exon predictions to be pseudogenes or mispredictions. Of the C. elegans orphans, 208 (22%) have partial or full empirical confirmation of their gene structures in the form of ESTs or cDNA data.
Some of these orphans may be novel genes that have been generated in one of the two genomes since the species diverged (Long 2001). However, we emphasize that some of the candidate orphans may not be real orphans at all, but are either pseudogenes that have not yet been deleted or are very rapidly evolving genes that have diverged so quickly that the BLAST and Smith–Waterman algorithms (used in the Gene Families section) cannot recognize their cross-species matches. In either case, it will be fruitful to look at the orphans in more detail because they are likely to reveal sites of rapid evolution.
A list of the candidate orphans is available as Dataset S3.
Protein Families
In order to identify protein family structure in C. briggsae while simultaneously identifying conserved families in both C. elegans and C. briggsae, we performed all-against-all Smith–Waterman (Smith and Waterman 1981) alignments of the combined C. briggsae and C. elegans (WS77*) predicted protein sets. The pairwise protein similarity data were then used to cluster proteins with the TRIBE-MCL software (Enright et al. 2002). To characterize the clusters, we correlated clusters with the protein family domain analysis of C. briggsae and C. elegans proteins described earlier and kept the domain descriptions that were held in common by the majority of cluster members.
The TRIBE-MCL analysis produced 7,778 clusters, 2,169 of which contained more than two genes. The largest cluster contained 775 genes with a eukaryotic protein kinase protein domain. The next largest clusters were associated with Zn finger, C4-type steroid receptor and ligand-binding domain of nuclear hormone receptor, 7TM receptors, and EGF-like domain. We found 852 clusters of more than two genes (comprising 4,567 genes in total) contained no identifiable Pfam domains. The top ten largest gene families with their percentage of C. elegans and C. briggsae proteins are shown in Table 3.
Table 3. Top Ten Protein Clusters by Size in C. elegans and C. briggsae.

Numbers in parentheses are percentage of proteins relative to whole predicted protein set in that species
While the great majority of TRIBE-MCL clusters had a balanced number of C. elegans and C. briggsae members, we found several interesting exceptions. For clusters of more than two proteins, a total of 328 clusters were made up of only C. briggsae genes; 283 of these did not have identifiable Pfam domains. Similarly there were 111 clusters that contained C. elegans proteins exclusively, 98 of which had no identifiable Pfam domains. Although some of these putative families are undoubtedly artifacts from the gene-calling procedure, some may be true species-specific families, at least at the limit of the sequence similarity criteria used here (see also the C. briggsae/C. elegans Paralogs and Orphans section above).
In other cases, a well-known gene family was found to have greater representation in one species than the other. A prominent example is the chemosensory receptor (also known as the olfactory receptor) family. While there are 718 putative C. elegans chemosensory receptor proteins annotated in WS77*, the TRIBE-MCL clustering and Pfam annotation detects only 429 putative C. briggsae chemosensory receptor proteins. Another example is a large family containing a cyclin-like F-box (usually associated with phosphorylation-dependent ubiquination) which is represented by 243 copies in C. elegans and 98 in C. briggsae. For clusters containing five or more members, there were 202 clusters with compositional differences of at least 2-fold between the two species (118 enriched in C. briggsae proteins, 84 clusters enriched in C. elegans proteins) and 12 clusters with compositional differences of at least 10-fold (ten C. briggsae-enriched; two C. elegans-enriched).
The chemosensory receptor family is subdivided by Pfam 9.0 (Bateman et al. 2002) into six subfamilies: 7TM subfam4, 7TM subfam5, sra, srb, sre, and srg. To determine whether the difference in size of the chemosensory receptor families in the two species affects all subfamilies equally, we undertook a detailed analysis of the clusters using neighbor-joining trees and manual inspection. The results, summarized in Table 4, show that the size differential in the chemosensory receptor family between the two species is not distributed equally but is concentrated in two subfamilies: the 7TM subfam5 subfamily, with 311 and 151 members in C. elegans and C. briggsae respectively, and the sra subfamily, with 36 members in C. elegans and 18 members in C. briggsae.
Table 4. Differential Sizes of Chemosensory Receptor Subfamilies in C. elegans and C. briggsae.
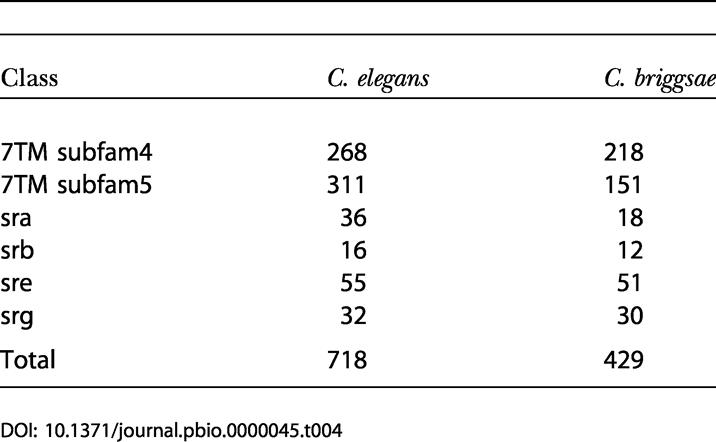
Figure 3 shows a phylogenetic tree for the sra subfamily. Many of the tree's terminal branches have exactly two members, one from the C. elegans and the other from C. briggsae. These cases correspond to putative orthologs. Several branches (arrows in Figure 3) show expansions in C. elegans that presumably represent cases in which a common ancestor in the two species underwent expansion in C. elegans but not in C. briggsae. A more modest expansion of a cluster of C. briggsae genes is also seen.
Figure 3. Tree View of All Chemosensory Receptor Genes in the Sra Subfamily.

C. elegans is shown in white background and C. briggsae in light blue background. The arrows indicate regions of C. elegans-specific expansion of the family. The inset shows a schematic of the region of C. elegans chromosome I corresponding to the sra expansion in the upper right of the tree. C. briggsae genes are named using the prefix CBG, while C. elegans genes are numbered consecutively across the cosmid on which they were first identified. The root of this tree is arbitrary.
In the chemosensory receptor family, there is a strong correlation between C. elegans-specific expansions in the similarity tree and regions of tandem duplication in the genome. For example, all the genes found in the large C. elegans-specific expansion in the upper right of Figure 3 are tightly clustered in a tandem array within a single 20 kbp region of C. elegans chromosome I (inset of Figure 3, lower right).
The physical clustering of protein families that was first observed in C. elegans (C. elegans Sequencing Consortium 1998) is also common in C. briggsae. To compare family clustering in the two species, we evaluated sliding windows of 15 protein-coding genes and determined the average number of gene products within the window that belonged to the same TRIBE family. In C. briggsae, the average (mean ± SD) number of family members in a sliding window was 0.37 ± 1.04, while a similar value of 0.55 ± 1.57 was observed in C. elegans, in contrast to values of 0.00008 (C. briggsae) and 0.00058 (C. elegans) that are expected of families that are not clustered. A permutation test indicates that the relationship of families is highly nonrandom (p < 0.001) compared to a reshuffling of the genome on either a chromosome or a contig level.
The presumed mechanism for the observed clustering of family members is one or more cycles of tandem duplication (C. elegans Sequencing Consortium 1998; Gu et al. 2002; Hughes and Friedman 2003).
Noncoding RNAs
Noncoding RNAs (ncRNAs) are a class of gene that produce a functional transcript as a final product, rather than being translated into a protein. We used a combination of ncRNA prediction programs and similarity search algorithms to identify ncRNAs in C. briggsae. For the purposes of comparison, we repeated the analysis with C. elegans.
We found 962 ncRNA genes in C. briggsae and 838 ncRNA genes in C. elegans (Table 5), as well as an additional 191 and 212 fragmentary matches in each species, respectively, that may be pseudogenes. Although the transfer RNA (tRNA) search software we used is able to distinguish pseudogenes from true genes with high sensitivity, this is not true of the other ncRNA assignments, which should therefore be treated with caution in the absence of experimental data.
Table 5. ncRNA Gene Predictions Present in C. elegans and C. briggsae Genome Assemblies.
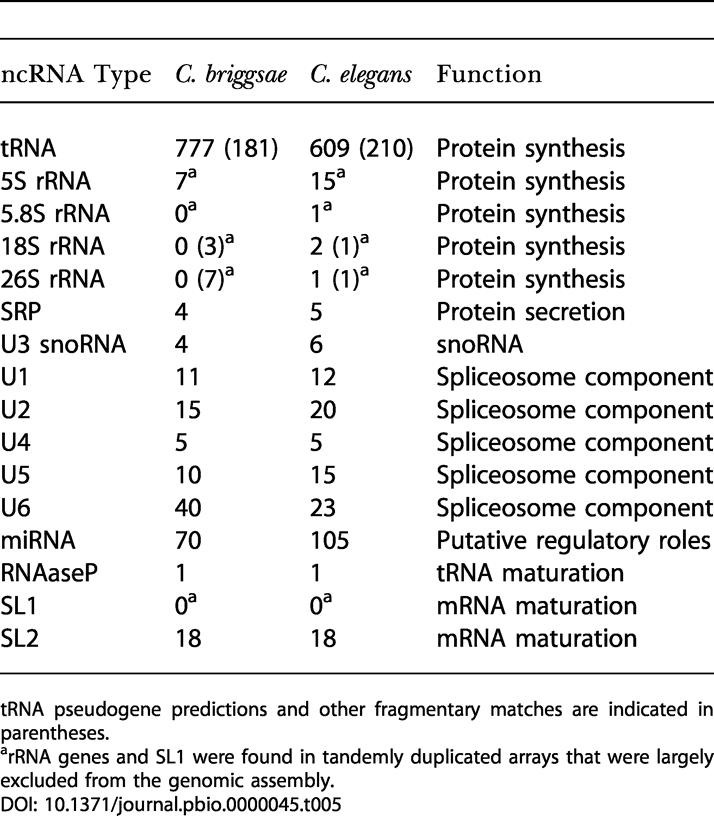
tRNA pseudogene predictions and other fragmentary matches are indicated in parentheses
arRNA genes and SL1 were found in tandemly duplicated arrays that were largely excluded from the genomic assembly
tRNA Genes
We found 777 tRNA genes and 181 tRNA-derived pseudogenes in the C. briggsae genome. In C. elegans we found 609 tRNA genes and 210 tRNA pseudogenes, in good agreement with previous analyses (C. elegans Sequencing Consortium 1998).
The C. briggsae tRNA set included two putative selenocysteine tRNAs. Furthermore we also identified a selenocysteine insertion sequence (SECIS) in the 3′ untranslated region (UTR) of a putative C. briggsae thioredoxin reductase gene. This C. briggsae gene, CBG05747, is orthologous to C. elegans gene C06G3.7, and was previously reported to contain a conserved SECIS in the 3′ UTR (Buettner et al. 1999).
The approximately 170 extra tRNA genes in C. briggsae are distributed across the amino acyl-tRNA families. We constructed neighbor-joining trees of the amino acyl-tRNA families and identified several C. briggsae/C. elegans orthologs (data not shown). The codon wobble patterns predicted by Guthrie and Abelson (1982) and described for the human genome (International Human Genome Sequencing Consortium 2001) are conserved in both nematodes.
Ribosomal RNA Genes
The genes for three of the four ribosomal RNA (rRNA) components, 18S, 5.8S, and 26S, are known to occur in large, tandem-repeat structures in C. elegans and other higher eukaryotes (Ellis et al. 1986). There are thought to be 100–150 copies of this repeat on chromosome I in C. elegans. The 5S rRNA genes are strikingly conserved between C. elegans and C. briggsae, with the genes in the two species identical in sequence (Butler et al. 1981; Nelson and Honda 1989). An estimated 100 copies of 5S rRNA lie in a tandem array on chromosome V in C. elegans.
The C. briggsae rDNA repeat measured 7,429–7,431 bases with variations in length of a poly(A) tract and single nucleotides. The unit repeat contained one copy each of 5.8S, 28S, and 18S rDNA genes. In agreement with previous reports (Nelson and Honda 1989), two distinct 5S unit repeats were found of 697 bases and 938–940 bases. Each version contains a single copy of the 5S gene. Several minor variants of each version were apparent. Based on the number of reads present for each repeat, the rRNA array extends 410 kbp (55 copies), and the 5S arrays extend 20 kbp (30 copies) for the shorter unit and 70 kbp (70 copies) for the longer. The latter two estimates are in reasonable agreement with previous estimates (Nelson and Honda 1989).
Spliced Leader RNA Genes
A characteristic of many C. elegans genes is the presence of a trans-spliced leader, a short RNA that is spliced onto the 5′ end of the primary transcript prior to further processing. Two types of spliced leader, SL1 and SL2, have been described.
The SL1 RNA gene that donates the SL1 spliced leader is present on the 938 bp 5S rRNA repeat units (Nelson and Honda 1989), as it is in C. elegans. There are 18 known SL2 genes in C. elegans. They vary somewhat in sequence and are scattered throughout the genome (Evans et al. 1997; T. Blumenthal, unpublished data). We searched the C. briggsae genome for the SL2 RNA genes and found 18 matches. These matches encode four of the eleven SL2 sequence variants known in C. elegans as well as two variants not found in C. elegans. In both species, roughly half of the SL2 RNA genes are found in a few small clusters. A phylogenetic tree of the SL2 RNA genes in the two species indicates that four SL2 RNA genes in the last common ancestor expanded after separation of the species to create the 18-member gene families that exist today.
MicroRNA Genes
The microRNA Registry (version 1.4; http://www.sanger.ac.uk/Software/Rfam/mirna/) contains 105 C. elegans microRNA (miRNA) sequences reported by several groups (Lau et al. 2001; Lee and Ambros 2001; Ambros et al. 2003; Grad et al. 2003; Lim et al. 2003). We find close homologs of 70 of these genes in the C. briggsae genome, with greater than 90% sequence identity of the mature miRNA sequence and predicted ability of the flanking regions to form a precursor hairpin of around 70 nucleotides using ViennaRNA (Wuchty et al. 1999). Lim et al. (2003) report that 46 of 48 C. elegans miRNA families extend to C. briggsae using more relaxed sequence identity criteria.
Several miRNAs are clustered in the C. elegans genome (for example, mir-42, mir-43, and mir-44) and may be processed from the same primary transcript. Of nine clusters of two or more miRNA genes within regions of around 1 kb in C. elegans, we find seven with conserved gene order and orientation in the C. briggsae genome. An additional cluster contains six C. elegans paralogs to mir-35 and mir-41, versus eight in C. briggsae, as previously reported (Lau et al. 2001).
Other ncRNA Genes
Our analysis identified 178 putative non-tRNA, non-rRNA genes in C. briggsae. The majority of classes, including U1, U2, U5, and SRP RNA, showed slightly higher copy numbers in C. elegans.
The Rfam search failed to find C. elegans or C. briggsae RNAase P and U3 small nucleolar RNA (snoRNA) sequences. However, RNAase P from C. elegans has recently been identified by others (R. Klein, personal communication), and with aid of this sequence, the matching C. briggsae sequence was then easily found using BLASTN. BLASTN searches with six C. elegans U3 snoRNAs (T. Jones, personal communication) identified four matching genes in the C. briggsae genome.
It is interesting to note that there are roughly twice as many putative U6 small nuclear RNAs (snRNAs) in C. briggsae as in C. elegans. Upon further examination, we found that the U6 Rfam family appears to contain large numbers of pseudogenes, and thus the observed expansion may reflect recent pseudogene duplication. The putative U6 snRNA sequences are remarkably conserved, with 22 clusters in C. briggsae and 16 in C. elegans having identical sequences. Two regions of around 10 kbp on the same FPC contig in C. briggsae (cb25.fpc2888) contain six and three U6 snRNA genes and correspond to two similar clusters located on C. elegans chromosome IV. Additional highly similar U6 hits can be found flanking these regions and in additional clusters. However, a neighbor-joining tree of all 63 putative U6 snRNAs did not show any obvious C. briggsae-specific subfamily expansion (data not shown).
Missing ncRNA Genes
We found no homologs of the U12 spliceosome components U4atac, U6atac, U11, and U12 in the C. briggsae sequence. This spliceosome has not been previously identified in C. elegans or other nematodes. We found no obvious homologs of telomerase RNA or of several snoRNAs in the C. briggsae or C. elegans genomes, suggesting significant divergence from previously identified examples.
Repetitive Elements
To characterize the repeat content of C. briggsae, we applied RECON, an ab initio repetitive element identification algorithm (Bao and Eddy 2002) to identify repetitive sequences with more than ten copies in the genome. Putative repetitive elements were then screened to remove RNA and protein families not associated with transposable elements. For the purposes of comparison, we applied the same algorithm to C. elegans.
The RECON-constructed library for C. briggsae contains 473 consensus repeat sequences, and the library for C. elegans contains 377. Previously reported transposons with more than ten copies in the C. briggsae genome, Tcb1 (Harris et al. 1990) and Tcb2 (Prasad et al. 1991) for C. briggsae, as well as C. elegans transposons Tc1, Tc2, Tc3, Tc6, and Tc7 (Plasterk and von Luenen 1997), were all recovered in the corresponding library.
When the RECON libraries were used as the substrate to RepeatMasker, 22.4% of the C. briggsae and 16.5% of the C. elegans genomes were masked. Extrapolation to the expected 104 Mbp of the complete C. briggsae genome indicates that 23.3 Mbp of the C. briggsae genome is repetitive, as opposed to 16.5 Mbp of C. elegans, which has a contiguous genome size of 100.3 Mbp. Hence, the differential repeat content accounts almost entirely for the different observed sizes of the species' genomes.
Comparison of the repetitive portion of the two genomes confirms the early observation from c0t curves that closely related species contain similar repetitive sequences (Britten and Kohne 1968): the C. briggsae library masks 4.6% of the C. elegans genome, while the C. elegans library masks 6.6% of the C. briggsae genome. In contrast, the RECON-constructed library for Arabidopsis thaliana (Z. Bao and S. R. Eddy, unpublished data) can only mask 0.2% of the C. elegans genome and 0.1% of the C. briggsae genome.
The top ten repeat families in the two genomes are shown in Tables 6 and 7. Interestingly, a single C. briggsae repeat family, Cb000047, is present more than 20,000 times and constitutes 4.7 Mbp of the C. briggsae genome. In both genomes, the majority of repeat families are either short, nonautonomous DNA transposons or tandem arrays. The presence of Cb000047 is an interesting difference between C. briggsae and C. elegans, the largest of whose repeat families is present in roughly 3,000 copies.
Table 6. Top Ten Most Abundant Repeat Element Families in C. briggsae.

Key to types: DNA, DNA transposon; tandem, tandemly duplicated element
Table 7. Top Ten Most Abundant Repeat Element Families in C. elegans.

Key to types: DNA, DNA transposon; tandem, tandemly duplicated element
Despite their general similarities, we were not able to systematically identify ortholog pairs among the C. briggsae and C. elegans repeats. When we used RepeatMasker to compare the sequences of one library against the other, we found no simple one-to-one mapping between them. Furthermore, similarity between repeat element pairs was generally restricted to subparts of the consensus sequences, presumably reflecting domains important for the propagation of the elements. It is not yet clear whether the overall similarity in repeat composition between the two genomes is due to orthology or because related genomes tend to be permissive for similar types of repeats (in particular, transposable elements). The relatively small amount of similarity between the repeat libraries in the two species suggests that most observed dispersed repeat elements postdate the divergence of the two species.
Genome Organization of C. briggsae and C. elegans
To begin to compare the organization of the C. briggsae and C. elegans genomes, we created whole-genome alignments of the two genomes at the nucleotide level using the WABA algorithm (Kent and Zahler 2000). WABA produced a total of 1,340,518 blocks of alignment. After adjusting for overlaps, the WABA alignments were found to cover 52.3% of the C. elegans genome (52.4/100.2 Mbp) and 50.1% of the C. briggsae assembly (52.9/105.6 Mbp).
To characterize the alignments, we compared them with the positions of annotations on the C. elegans genome, using the WS77* annotations for the positions of genes, coding regions, and introns, and the WS87 annotations (October 2002) for the positions of 5′ and 3′ UTRs (the annotations for which were not available in WS77). In addition, we also scored aligned regions that fell within the upstream and downstream regions of genes, which were defined arbitrarily as the region 1,000 bp 5′ to the translational initiation codon for upstream regions and the region 1,000 bp 3′ of the translational stop codon for downstream regions.
Table 8 summarizes the relationship among the 1.3 million raw WABA alignments and annotated C. elegans genes. The WABA algorithm produces pairwise alignments that contain smaller aligning blocks of three types: “coding,” “strong,” and “weak.” Coding blocks have the characteristic match-2/skip-1 pattern of diverged coding regions, while strong and weak blocks have high and low levels of similarity, respectively. Remarkably, only a third of aligned bases overlap known coding exons or their 5′ or 3′ UTRs. Another third lie in introns, and the final third lie in intergenic regions. More than half of the latter class lie more than 1,000 bases outside of known protein-coding genes.
Table 8. Bases Covered by Aligned WABA Blocks, Stratified by Relationship to Annotated C. elegans Genes.

Percentages in the “Total” column and “Total” row indicate the number of aligned bases in each category divided by the total number of aligned bases. Percentages in the body of the table indicate the number of aligning bases in the indicated category divided by the number of bases in the compartment. For example, 42.1% of C. elegans coding bases are covered by WABA alignments of the “coding” type
As indicated by Kent and Zahler (2000), although there is a preference of WABA coding blocks for coding regions and strong blocks for intergenic regions, there is not a clear delineation. Figure 4 shows a C. elegans gene (snt-1) and its WABA alignment to part of C. briggsae supercontig cb25.fpc2454. WABA blocks covered the snt-1 CDS and predicted 3′ UTR. Coding WABA blocks (dark blue in Figure 4) correlate well with exons of both of the alternatively spliced transcripts of snt-1. However, there are also small coding blocks between exons 1 and 2 and between exons 2 and 3. These could be missed exons in the C. elegans gene model or conserved functional elements, but there are no experimental data to support either supposition.
Figure 4. A WABA Alignment over a Known C. elegans Gene (snt-1).

WABA coding segments are shown as dark blue, strong alignments as medium blue, and weak alignments as grey. Regions that do not align are shown as dotted lines. The alignments of three sequenced C. elegans mRNA sequences are also shown for comparison.
Strong blocks are found in exons and in the region immediately upstream of the gene. Weak blocks are frequently found in introns and in intergenic regions. Intriguingly, the intergenic region 2,000 bp upstream of snt-1 includes regions of alignment that contain strong, weak, and coding blocks. Such regions, which are numerous in the alignment of the two genomes, do not correspond to repeats or other known features and beg investigation to determine whether they are footprints of unknown functional elements.
A file containing the raw WABA alignments is available as Dataset S4.
Colinearity between C. briggsae and C. elegans Genomes
We used the C. briggsae/C. elegans WABA alignment data and a simple set of algorithms to identify regions of long-range colinearity between the two genomes. We first merged alignments whose coordinates overlapped in both the C. elegans and C. briggsae genomes. We then filtered these data to remove regions in which an excessive number (more than five) of segments of C. briggsae sequence were aligning to the same region of C. elegans or vice versa. This was followed by a simple merge, in which adjacent blocks of colinear alignments were merged, and a second round of merging using a dynamic programming algorithm to bridge small transpositions and inversions by finding runs of monotonically increasing alignment blocks. The resulting set of 13,467 candidate synteny blocks was then filtered to remove small blocks of alignment of less than 1.8 kbp.
The final list of candidate synteny blocks contained 4,837 alignments covering 84.6% and 80.8% of the C. elegans and C. briggsae genomes (mean, 37,472 bp; median, 5,557 bp). The largest such synteny block was a 1.68 Mbp segment involving the center of C. elegans chromosome II and C. briggsae supercontig cb25.fpc0058. The next largest blocks are on C. elegans chromosome X, where there are two adjacent synteny blocks of roughly 1.3 Mbp each. However, these blocks cannot be merged without introducing a disproportionately large gap in the corresponding C. briggsae alignment.
To understand the nature of the unaligned areas, we examined the ten longest gaps, which ranged in size from 51 kbp to 85 kbp. In four cases, the gaps were occupied by expansions of one or more C. elegans gene families, none of which had strong matches to C. briggsae. Three cases corresponded to a region of genes that were unrelated to each other, but none had strong matches to C. briggsae. Two cases were gene deserts with no genes or just a single gene without a C. briggsae match. In the last case, we found that half the interval was occupied by a set of unrelated genes without strong C. briggsae matches, while the other half contained a large set of WABA alignments that overlapped a set of orthologous genes. This last case therefore represented an instance in which our procedure did not extend an alignment block as far as possible.
Changes in gene order during genome evolution are typically classified as inversions, in which a region of the genome is flipped without loss of genetic material; reciprocal translocations, in which two regions are swapped without loss of genetic material; and transpositions, in which a nonreciprocal movement of genetic material takes place. We classified the colinear blocks by searching for the signature breakpoints of each of these events and counted 1,384 putative inversions, 244 putative translocations, and 2,735 putative transposition events. The remaining 476 blocks could not be classified because they abutted the ends of C. briggsae supercontigs.
We refer to the junctions between adjacent colinear blocks within a C. briggsae supercontig as “breakpoints,” because they correspond to a putative rearrangement between ancestral chromosomes during the divergence of C. briggsae from C. elegans. Table 9 is a tally of such breakpoints, broken down by the two C. elegans chromosomes matching either end of the breakpoint junction on a C. briggsae supercontig. There is a clear bias towards rearrangements within the same ancestral chromosome, which occur roughly four times more frequently than those between chromosomes (p < 10−4, χ2 test). When the relative sizes of the intra- versus interchromosomal compartments are considered, the overall density is approximately ten times higher for intrachromosomal rearrangements: 29.7 intrachromosomal rearrangements per megabase pair versus 3.6 interchromosomal rearrangements per megabase pair (p < 10−4, χ2 test). Overall, the X chromosome has a lower density of colinearity breakpoints than the autosomes (31.1 versus 52.1 rearrangements per megabase pair in X versus the autosomes). This conclusion applies equally to rearrangements within X and between X and the autosomes.
Table 9. Chromosomal Partners at Colinearity Breakpoints.
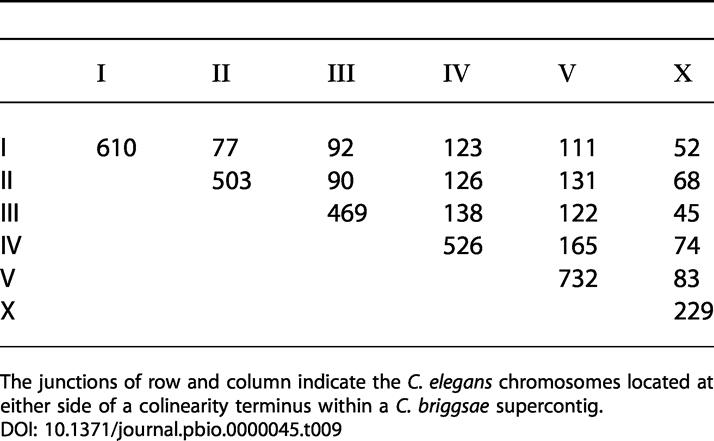
The junctions of row and column indicate the C. elegans chromosomes located at either side of a colinearity terminus within a C. briggsae supercontig
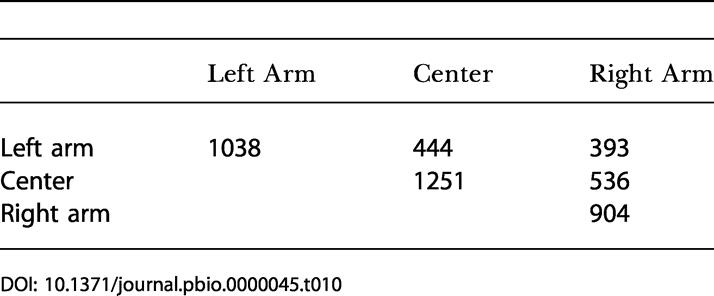
We also examined the frequency of breakpoints within and between C. elegans chromosomal arms. The C. elegans autosomes are regionally divided into a “central cluster,” in which the meiotic recombination rate is low, and two “arms,” in which the meiotic rate is high (Barnes et al. 1995). The arms are located roughly one third of the distance from the two telomeres. Because the chromosomal arms are asymmetrical with respect to the meiotic pairing region (Sanford and Perry 2001), for this analysis we reversed C. elegans chromosomes I and V to place the meiotic pairing region on the left arm of all chromosomes.
We found that breakpoints between regions of colinearity are strongly biased towards junctions that are within the same arm of the same chromosome (Table 10; p < 10−4, χ2 test). We also found a regional difference in the distribution of the lengths of colinear blocks. The mean block length increases gradually from a mean of 25.5 kbp in the first 10% of C. elegans chromosome length to 44 kbp in the center and then decreases to a mean length of 27.5 kbp in the last 10% of the chromosome. This distribution is nonrandom at p < 10−4 (Student's t-test comparing the first 10% to the center or the center to the last 10%). This correlation between synteny block lengths and chromosomal position was first noted by Kent and Zahler (2000). Coghlan and Wolfe (2002) attempted to reproduce this finding but were unable to show statistical significance with the data available at the time.
Table 10. Chromosomal Arms at Colinearity Breakpoints.
The list of candidate synteny blocks is available as Dataset S5.
Ordering of C. briggsae Supercontigs by C. elegans Synteny Block
We used the merged synteny blocks to order the C. briggsae supercontigs by placing each supercontig into the order and orientation dictated by its single largest block of C. elegans colinearity. To maximize the number of supercontigs for which there was ordering information, we used the unfiltered set of 13,467 alignment blocks. With this technique we were able to position all but one of the 142 supercontigs that had been placed on the C. briggsae physical map and 241 of the 436 unplaced contigs. The total amount of sequence that could be placed in this way was 104.5 Mbp of the 105.6 Mbp C. briggsae assembly.
The C. briggsae supercontigs that could not be placed onto C. elegans by synteny block were small (mean, 5,462 bp; median, 3,479 bp). We examined the ten largest of the unplaced supercontigs, including the supercontig that had been placed onto the C. briggsae physical map (cb25.fpc4500; 8 kbp) but not onto the reconstruction. We found the unplaced contigs to be generally gene-poor and largely devoid of nucleotide-level alignments to C. elegans.
Figure 5 shows a graphical representation of this ordering. Of note are the maintenance of large regions of colinearity between C. elegans chromosome X and C. briggsae supercontigs fpc0045, fpc3857, fpc2397, fpc4044, fpc4033, fpc4069, and fpc0929 and a marked preference for intrachromosomal versus interchromosomal rearrangements within the autosomes. Also of note is the high level of concordance between synteny blocks predicted by nucleotide-level synteny and the positions of protein-level orthologs. Small areas of discordance, scattered throughout the genome, may be misassignments among the orthologs or small regions of colinearity that were not detected at the nucleotide level.
Figure 5. Representation of the C. briggsae WGS Assembly on a C. elegans Scaffold Using Colinearity Relationships.

C. briggsae supercontigs are shown on the y-axis, and C. elegans chromosomes from WS77 are shown on the x-axis. Red dots and lines indicate regions of colinearity identified by WABA alignments between the two genomes. Blue dots are the positions of protein orthologs. Green areas show where blue and red intersect, indicating concordance between the positions of ortholog pairs and colinearity blocks.
It is important to emphasize that this ordering of C. briggsae supercontigs is arbitrary and gives only an approximate idea of their chromosomal assignments. Accurate chromosomal assignment of the supercontigs and a comparison of large-scale rearrangements between the chromosomes of the two species must await empirical determination.
The ordering of C. briggsae supercontigs on the C. elegans genome is available as Dataset S5.
Genomic Breakpoint Rate
As noted in the previous section, we identified 1,384 putative inversions, 244 putative translocations, and 2,735 putative transposition events. As described in Sankoff (1999), each inversion or translocation implies two breakpoint events (at either end of the conserved block), while each transposition implies three breakpoint events (two at the source and one at the destination of the conserved block). This gives an estimated 11,461 genomic breakpoint events during the divergence of C. elegans and C. briggsae, or 57 breakpoint events per megabase pair per species.
Using our previous estimate of a divergence time of 80–110 MYA between the two species, we calculate a breakpoint rate of between 0.5 and 0.7 breakpoints per megabase pair per million years. Although our synteny block counts must be viewed as approximate, since modest changes in the parameter choices can affect the number of small blocks counted by as much as a factor of two, our estimate is in good agreement with previous estimates based on the available finished sequence (Coughlan and Wolfe 2002). A minor difference is that we found relatively fewer translocations than were found in the earlier study. It is likely that differences in methodologies are responsible for this discrepancy, as we defined conserved blocks on the basis of merged regions of nucleotide-level similarity, whereas Coghlan and Wolfe (2002) used protein-level similarity of sequenced genes to identify orthologous regions. The C. briggsae/C. elegans breakpoint rate is roughly five times the rate reported by Ranz et al. (2001) for Drosophila species.
Conservation of Operon Structure
C. elegans contains roughly 1,000 trans-spliced operons (Blumenthal and Gleason 2003), most of which have been identified. On the basis of the colinearity blocks described above, we examined the conservation of the 800 operons present in WS77 and found that 768 (96%) are preserved intact in the C. briggsae genome, as judged by the criterion that all the genes in the C. elegans operon could be found in the same order and orientation in the corresponding region of C. briggsae. By comparison with the preservation of order of nonoperonic genes, we would have expected that only 60% of the operons would be preserved if no selection were operating to preserve gene order. Hence, we conclude that the structures of operons are under purifying selection.
Of the 32 C. elegans operons that were disrupted in C. briggsae, seven were broken by large expansions of intercistronic distance, five had transpositions of the first gene in the operon to another location, nine had rearrangement breakpoints within two-gene operons, four were breakpoints between the last gene and the rest of the operon, two were breakpoints between the first gene and the rest of the operon, two were internal breakpoints, two were breakpoints within a gene in the operon, and one was a general scattering of all the genes in the operon.
Position-Specific Variations in Caenorhabditis Chromosomes
When the distribution of synteny blocks, orthologs, low-stringency orphans, and silent site substitution rate is projected onto the C. elegans sequence map, an intriguing pattern appears (Figure 6; see also Poster S1). When normalized for the distribution of genes, there is a marked increase in the frequency of orthologs in the center of the chromosomes versus in the arms (Figure 6C; 74.8 versus 53.2 orthologs per 100 genes, p < 1 × 10−12 by Welch's two sample t-test) and an increase in the frequency of “orphan” genes in the chromosome arms versus the centers (Figure 6D; 6.15 versus 3.36 orphans per 100 genes in the arms versus the central regions, p < 1 × 10−8 by t-test). This pattern is correlated with the ratio of nonsynonymous to synonymous substitutions (the KA/KS ratio) between ortholog pairs, which is higher in the chromosomal arms than in the centers (Figure 6G; mean KA/KS, 0.065 versus 0.059; p < 4.6 × 10−9 by t-test). This is due in part to elevated rates of KA on chromosome arms versus centers (mean KA, 0.138 versus 0.128; p < 7.9 × 10−5 by t-test.). Although there is considerable regional variation in the rate of silent site substitutions (the KS value), there is no significant difference in the mean or variance between chromosome arms and the central regions.
Figure 6. Evolutionary Divergence across C. elegans Chromosome V.

Each panel corresponds to a C. elegans chromosome, and the individual tracks show different measurements of evolutionary divergence.
(A) Regions of synteny (colinearity) between C. elegans and C. briggsae. White areas correspond to areas where the two genomes could not be aligned owing to divergence and are more abundant in the chromosome arms.
(B) C. elegans gene density and genetic map position. Gene density is plotted as a histogram, showing a relatively uniform distribution of genes across each chromosome. The relationship of the position of genes on the genetic map to their position on the sequence is superimposed on the y-axis. Steeper slopes in this plot indicate higher rates of meiotic recombination. Inflection points in the genetic map plot reflect the division of the chromosomes into recombinationally active “arms” and recombinationally slow “centers.”
(C) C. briggsae/C. elegans orthologs normalized for gene density in 100 kbp sliding windows. Prominent regions of low ortholog density are seen on chromosome arms.
(D) C. elegans “orphans,” genes with no significant protein similarity in C. briggsae or the non-C. elegans portion of SwissProt. This histogram has been normalized for gene density in 100 kbp sliding windows. Spikes in orphan density seem to correlate with regions of low ortholog density.
(E) C. elegans genes that mutate to lethality or are lethal in RNAi screens, in 100 kbp sliding windows normalized to overall gene density. This track shows the distribution of essential genes and demonstrates their tendency to cluster in the chromosome centers.
(F) Repetitive elements, binned in 100 kbp sliding windows. Repeat-rich regions correlate with both the absence of significant syntenic coverage and ortholog-poor regions.
(G) The KA/KS ratio in ortholog pairs. Lower values indicate greater levels of purifying selection.
(H) The rate of KS within ortholog pairs, in 100 kbp sliding windows.
The difference between arms and centers is also reflected in the distribution of blocks of nucleotide-level similarity between C. elegans and C. briggsae (Figure 6A). As noted in the previous section, synteny blocks are longer in the centers than in the arms. These patterns are pronounced in all the autosomes, but present to a lesser degree in the X chromosome as well.
The arm/center dichotomy seen in the comparison between the two species is reflected in a number of intrinsic features of the C. elegans chromosomes, many of which were reported at the time of whole-genome sequencing (C. elegans Sequencing Consortium 1998). The centers are gene-rich and have a lower meiotic recombination rate than the arms (Figure 6B). Transposons and other repeat elements are greatly enriched in the arms and largely excluded from the centers (Figure 6F). Lastly, the frequency of essential genes, as judged by mutant phenotype or lethal outcomes in genome-wide RNAi screens (Fraser et al. 2000; Gonczy et al. 2000; Kamath et al. 2003), is higher in the chromosomal centers than in the arms even after normalization for gene number (Figure 6E).
Together, these data suggest that the arms of the C. elegans chromosomes are evolving more rapidly than the centers. Consistent with previous work on C. elegans (C. elegans Sequencing Consortium 1998; Cutter and Payseur 2003; Kamath et al. 2003) and in contrast with the genomic organization of Saccharomyces (Mewes et al. 1997), Arabidopsis (Tabata et al. 2000), Drosophila (Myers et al. 2000), and vertebrates (International Human Genome Sequencing Consortium 2001; Waterston et al. 2002), nematode chromosomes appear to be organized in such a way that essential, highly conserved genes are preferentially confined to the centers, whereas the arms are where much of the evolutionary experimentation occurs. This is supported by the increased rate of KA amongst ortholog pairs on chromosome arms, indicating increased tolerance of mutation among genes on the arms.
Mechanistically, the increased rate of species-specific genes in the chromosomal arms could be explained by the preponderance of transposable element insertions in these locations. Transposable elements have been identified as an engine driving exon shuffling (Ejima and Yang 2003) and gene evolution (Lev-Maor et al. 2003). Transposable elements and other repeats are also associated with an increased rate of illegitimate chromosomal rearrangement (Gray 2000; Stakiewicz and Lupski 2002), which may be responsible for the trend toward shorter synteny blocks in the arms.
The observation of large positional variances in neutral substitution rate (KS) has been seen in recent mouse/human comparisons (Waterston et al. 2002), and it appears that the same phenomenon is seen in Caenorhabditis. However, while there is a significant difference in KA/KS in chromosome centers versus arms, the variation in KS alone is more localized in nature.
Using C. briggsae Sequence to Improve C. elegans Annotation
The C. elegans genome now totals 100,273,501 bases (WS103 release; June 2003) and consists of six contiguous segments of DNA corresponding to the C. elegans chromosomes. The last gap in the sequence was closed in November 2002. Since the publication of the C. elegans genome (C. elegans Sequencing Consortium 1998), the gene set has been extensively hand curated. Between the WormBase WS17 release in April 1999 and the WS77 release in April 2002 (the release used in this paper), WormBase curators made manual changes to approximately 6,300 genes (D. Lawson, personal communication).
To investigate the potential of the C. briggsae/C. elegans comparison for improving the C. elegans gene annotations, we compared the C. elegans hybrid gene set of 20,621 genes derived from our comparison of the two species to the set of 18,808 protein-coding genes in WS77* derived from WormBase. The majority (14,011) of the hybrid gene set predictions overlapped perfectly with WS77* gene predictions. Many of these, of course, are derived indirectly through Ensembl from WS77. From the remainder we derived 1,275 well-supported suggestions for new C. elegans genes, 1,763 new exons in 1,100 existing genes, 2,093 exon deletions in 1,583 existing genes, 1,675 exon truncations in 1,502 existing genes, and 1,115 exon extensions in 1,008 existing genes (Table 11).
Table 11. Updating the C. elegans Gene Set Using C. briggsae Similarity.
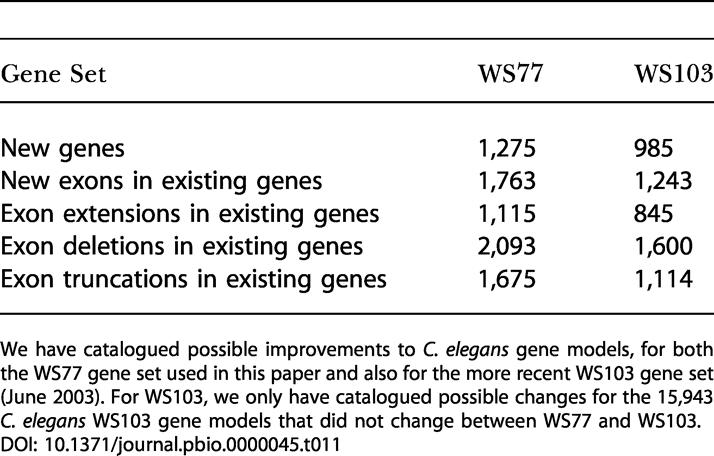
We have catalogued possible improvements to C. elegans gene models, for both the WS77 gene set used in this paper and also for the more recent WS103 gene set (June 2003). For WS103, we only have catalogued possible changes for the 15,943 C. elegans WS103 gene models that did not change between WS77 and WS103
Most of the corrections suggested for the WS77 gene set using C. briggsae similarity are still applicable to WS103, even after the manual correction of approximately 3,800 C. elegans genes between the WS77 and WS103 WormBase releases, prompted in part by the open reading frame (ORF) sequence tag (OST) dataset of Reboul et al. (2003). Only 290 of the 1,275 proposed new hybrid set genes overlap new WormBase gene predictions made since WS77, and 4,802 of the 6,646 proposed exon changes are in gene structures that have not been edited between WS77 and WS103.
The automated analysis presented above indicates that the C. briggsae sequence will suggest many changes to the C. elegans set of gene predictions. To clarify the nature of these changes, we subjected several areas of colinearity to careful hand curation. In one carefully inspected area involving three C. elegans cosmids (ZK632, R10E11, and T05G5) and 33 C. elegans predicted genes, the syntenic C. briggsae region has 38 predicted genes (Figure 7). Inversions have broken the syntenic region into three conserved segments, within which gene order and orientation are largely conserved, except for one single-gene inversion (ZK632.9). There are 30 one-to-one orthologs in the syntenic block and two one-C. elegans-to-two-C. briggsae orthologs (T05G5.6 versus CBG10003 and CBG09979, and T05G5.8 versus CBG10002 and CBG09978), where the two C. briggsae orthologs seem to have been duplicated as a block since speciation. The remaining C. elegans gene (CEG09285) belongs to a C. elegans-specific gene family; its closest C. elegans paralog is F40F12.3, a gene of unknown function that is located nearby on chromosome III.
Figure 7. A Region on C. elegans Chromosome III Containing 33 Genes, and the Syntenic C. briggsae Region, Which Has 38 Genes.

Inversions have broken the syntenic region into three conserved segments. Genes that do not have an ortholog in this syntenic region are in grey; orthologs are joined by lines. In C. elegans, genes that differ substantially in structure between the WS77 and hybrid gene sets are marked with an asterisk.
The remaining four C. briggsae genes include two members of a C. briggsae-specific gene family (CBG10004 and CBG09973), one that has a C. elegans ortholog on chromosome X (CBG09992) and one that has no clear C. elegans ortholog but has a match on chromosome X (CBG09973). Compared to the C. elegans WS77 gene set, the C. elegans hybrid set has two extra gene predictions: the C. elegans-specific genes CEG09285 and CEG09299, which are the orthologs of C. briggsae gene CBG09988. The other 31 C. elegans genes in this region are in both the C. elegans WS77 and hybrid gene sets. However, for seven of these genes, there are substantial differences between the WS77 and C. elegans hybrid gene structure that are supported by C. briggsae similarity. These include extra exons (in T05G5.10 and T05G5.4), deletions of WS77 exons (in T05G5.1, T05G5.9, and ZK632.7), truncations of WS77 exons (in R10E11.5), and extensions of WS77 exons (in T05G5.1, T05G5.4, and R10E11.7). In summary, the analysis of 33 C. elegans genes suggested corrections to seven gene models, proposed two missed genes, and confirmed the other 24 gene models.
We also evaluated a set of C. elegans WS77 gene predictions where the new C. elegans hybrid gene set suggests a strong likelihood of a change to the WS77 prediction. We found examples of hybrid gene set predictions that are pseudogenes and possible splitting and fusing of existing WS77 genes, as well as the addition of new exons.
Extrapolation of these results to the whole C. elegans genome suggests that the C. elegans gene set will be increased by at least approximately 1,300 gene predictions and that approximately 2,800 exons will be extended or truncated in existing WS77 predictions, based on C. briggsae similarity. Thus, despite five years of extensive manual curation, the comparative genomic approaches made possible by the C. briggsae sequence can provide significant improvements to the C. elegans gene models. Manual inspection of these discrepancies will be necessary, but especially for less highly expressed genes, where EST and messenger RNA (mRNA) data are not available, and for initial and terminal exons where signals can be difficult to detect, sequence conservation with C. briggsae will now provide a primary pointer for C. elegans gene structure refinement.
Discussion
We have completed a draft of the C. briggsae genome that is now 98% complete in sequence contigs and over 99% complete in map contigs. We have performed an initial characterization of the genome, including an analysis of the gene content and a comparison of the C. briggsae genome with its cousin C. elegans.
Comparing Comparisons
It is interesting to contrast the C. briggsae/C. elegans comparison to the recent whole-genome comparison of mouse and human (Waterston et al. 2003). Both pairs of species diverged at roughly comparable times (80–110 MYA for C. briggsae/C. elegans, 75 MYA for mouse/human) and show similar levels of amino acid identity between orthologs (80% for C. briggsae/C. elegans, 78.5% for mouse/human). Both genome pairs show large amounts of new repetitive elements, balanced presumably by deletions so that genome size between the pairs remains similar. A consequence of this for both pairs is that they share only approximately 50% of the nonfunctional sequence from their last common ancestors.
However, as evidenced by multiple measures, C. briggsae/C. elegans are evolving more rapidly than mouse/human. In mouse/human, 80% of predicted proteins could be assigned to a 1: 1 ortholog pair, whereas fewer than 65% of C. briggsae genes could be assigned an ortholog in C. elegans. The flip side of this relationship is that protein families are more dynamic in the two nematodes; as many as several hundred families are either novel or have diverged so far that their common origin cannot be recognized and another 200 have expanded or contracted by more than 2-fold. This contrasts with the mouse/human comparison, in which the great majority of the genes are in identical order and orientation in the two species and in which just 25 instances of gene expansion due to local duplication were found. By a similar token, the number of genes lacking a sequence match in its opposite species (orphans) is 4% in C. briggsae/C. elegans, but less than 1% in mouse/human.
C. briggsae/C. elegans are also evolving more rapidly at the nucleotide level, with a rate of synonymous substitution of 1.78 substitutions per synonymous site versus 0.6 substitutions per synonymous site in mouse/human. This is mirrored in a dramatic difference in chromosomal rearrangement rate, which is roughly an order of magnitude higher in the nematodes (4,837 conserved syntenic blocks of mean size 37 kbp) than in mouse/human (342 syntenic blocks of mean size 6.9 Mbp).
None of this should be too surprising, as evolutionary rate is better measured in generations than in years, and the generation time of the two nematodes is presently an order of magnitude or more faster than the two mammals. What is surprising is that despite their abundant differences at the genomic level, C. briggsae and C. elegans remain morphologically almost indistinguishable, whereas mouse and human are more similar genetically, but show dramatic anatomic and behavioral differences.
The C. briggsae Draft as a Research Tool
The 12 Mbp of finished C. briggsae genome sequence released in recent years has been exploited extensively by the worm community. The C. briggsae genome has been used to find C. briggsae orthologs of C. elegans genes, to identify candidate conserved cis-regulatory elements in genes, and to test for differences in expression pattern and function. For example, sequence from the genome project, along with sequence for genes cloned by individual labs, has been used to compare C. briggsae/C. elegans orthologs for gut-specific esterases (Marshall and McGhee 2001), vulval-expressed genes (Cui and Han 2003; Kirouac and Sternberg 2003), acetylcholinesterase genes (Culetto et al. 1999), cuticle collagen genes (Gilleard et al. 1997), inositol 1,4,5-trisphosphate receptor genes (Gower et al. 2001), and myogenic regulatory factor genes (Krause et al. 1994). At the 14th International C. elegans Conference held in the summer of 2003, multiple researchers presented work that utilized C. briggsae, including its use to dissect the mechanisms of RNAi (M. Montgomery, personal communication), to identify novel miRNAs (N. Lau, personal communication; V. Ambros, personal communication), to identify the potential targets of miRNAs (S.-Y. Lin, personal communication), to find putative cis-regulatory regions that control pharyngeal development (J. Gaudet, personal communication), and to identify candidate transcription factor-binding motifs in genes involved in vulval development (E. Schwarz, personal communication).
Outside the nematode research community, the C. briggsae genome sequence has also generated much interest among molecular evolutionists. For example, the C. briggsae genome has been used to study evolution of large protein families (Hope et al. 2003) and ncRNA gene families (Lim et al. 2003), evolution of introns (Kent and Zahler 2000), molecular evolution through ontogeny (Castillo-Davis and Hartl 2002), nucleotide substitution patterns along chromosomes and genes (Shabalina and Kondrashov 1999; Cutter and Payseur 2003), and patterns of chromosomal rearrangement (Kent and Zahler 2000; Coghlan and Wolfe 2002).
One intriguing prospect for further research is raised by the observation that although the C. briggsae and C. elegans genomes have diverged considerably, their developmental programs have not. This raises the possibility of searching the two species' genomes to identify compensatory and coevolutionary changes in genes involved in the same regulatory pathway.
The C. briggsae sequence will also be heavily used to refine C. elegans gene models by correcting gene models, adding and removing genes, and verifying gene predictions. The comparative results reported in this paper are currently being used by WormBase curators to improve the gene set, and it is anticipated that, by the end of this process, the C. elegans and C. briggsae predicted gene sets will have attained a level of confidence unprecedented among animal genomes.
We have also produced a clone-based physical map of C. briggsae. The cosmid-based physical map of C. elegans has been very useful to the research community as a substrate for sequence-based experiments (for example, mutant rescues), and we expect that the C. briggsae physical map will also become an important resource by facilitating molecular analysis of genes that are discovered by studying mutants and for use in transformation and rescue experiments in C. elegans as well as C. briggsae.
Looking Forward
At present, we have a high-quality draft sequence of C. briggsae. Would finishing the C. briggsae genomic sequence add significantly to its utility?
With the high-quality data generated by today's sequencing machines and chemistries and the deep coverage of the current draft (more than 10-fold), the quality of the existing C. briggsae sequence is sufficient for addressing most of the research questions described in the preceding paragraphs. However, for genomic evolutionary studies, gene family studies, and other analyses that are dependent on correct long-range ordering of C. briggsae genes, some automated finishing with gap closure will be of significant value. In such a process, directed reads are selected automatically to close gaps and to improve regions of low quality. Some gaps and complex regions will be refractory, but many of the remaining problems will be solved at relatively low cost. Aligning the current contigs to the C. briggsae chromosomes would also add long-range continuity and, if carried out through the use of single nucleotide polymorphisms and sequence-tagged sites, would lead to a detailed genetic map.
Although we are confident that researchers will be successful in exploiting the conservation of noncoding sequence between the two species to identify candidate cis-regulatory elements, this use is complicated by the presence of other sources of sequence similarity between the two species. Many of the approximately 900,000 conserved noncoding alignments will ultimately prove to be fragmented pseudogene matches, conserved transposons, low complexity sequence matches, or just the tail-end of neutral evolution. Adding the genome sequence of one or two additional nematodes to the alignment, however, will greatly reduce the noise, because pseudogenes, repeats, and other artifacts in one species will tend to cancel out in another. This strategy of identifying conserved regulatory elements by the alignment of sequence from multiple related species has been used effectively in fungi (Cliften et al. 2003; Kellis et al. 2003) and shows promising early results in cloned regions surrounding egl-30, lin-11, and mab-5 from the nematodes C. elegans, C. briggsae, CB5161, and PS1010 (E. Schwarz, personal communication).
Another argument for sequencing additional nematodes is the species-specific differences among the Caenorhabditis species. The known species (Figure 8) vary considerably in key behavioral and developmental processes (Fitch 1997), and it would be of great interest to relate genome-specific differences to these phenotypic differences. For example, C. remanei is roughly equidistant from C. elegans and C. briggsae (D. H. A. Fitch, unpublished data), but unlike either of the latter two species, C. remanei is strictly sexually dimorphic. To explore these differences, Haag et al. (2002) recently cloned the C. remanei sex-determining genes fem-3 and tra-2 and were able to relate species-specific changes in sex determination to genetic changes in the coding and cis-regulatory regions of these genes. Because of its distance from C. elegans and C. briggsae and its amenability to laboratory propagation and manipulation, C. remanei would make an ideal candidate for genome sequencing.
Figure 8. Phylogeny of Caenorhabditis.

Courtesy of Karin Kiontke and David H. A. Fitch (unpublished data). This phylogeny is based on weighted-parsimony analysis of DNA sequences from three genes, concatenated: 18S and 28S rRNA genes, and the RNA polymerase II gene. The root of this tree is arbitrary.
With genome sequences from several equidistant species, we cannot infer the directionality of evolutionary changes that we observe. That is, when we observe species differences such as amino acid substitutions, nucleotide substitutions within promoters, or chromosomal rearrangements, we cannot distinguish in which species the change has occurred, and therefore we cannot correlate the change with species-specific behavior or developmental patterns. More distantly related species act as outgroups to provide directionality. The genome of the non-Caenorhabditis nematode Brugia malayi is currently being sequenced (http://www.tigr.org/tdb/e2k1/bma1) and may be useful in this role. From within the Caenorhabditis clade, candidate outgroup species include C. japonica and C. drosophilae.
Thus, while the two completely sequenced Caenorhabditis genomes are an achievement that will be of great use in the coming years, they represent only a beginning. Sequencing additional nematode genomes will reap even further rewards.
Materials and Methods
Fingerprint map construction
Details of the DNA preparation, restriction enzyme digestion, agarose gel electrophoresis and data acquisition, and computer analysis and contig construction can be found in Marra et al. (1997). A brief description is provided here.
A C. briggsae fosmid library (strain AF16 [Fodor et al. 1983]; mean size, 39 kbp; SD, 8 kbp; median, 40 kbp; 6.5-fold coverage) was courteously provided by M. A. Marra and the BAC library by M. A. Marra and P. deJong (http://bacpac.chori.org/cbriggsea94.htm; strain AF16; mean size, 66 kbp; SD, 30 kbp; median, 73 kbp; 10-fold coverage).
C. briggsae fosmid clones were propagated in XL1-Blue MR cells (Stratagene, La Jolla, California, United States). BAC clones were propagated in DH10B cells (Life Technologies, Carlsbad, California, United States). For DNA preparation, culture volumes of 2× YT (Sambrook et al. 1989) containing chloramphenicol were inoculated with a single colony from a freshly streaked plate. After culture growth in 96-well blocks, glycerol stocks were prepared and sealed. Bacterial cell cultures were pelleted by centrifugation, excess culture media was removed, and the cells were placed immediately on ice. We prepared DNA using a modified alkaline lysis procedure (Sambrook et al. 1989). The cell pellet was thoroughly resuspended by addition of GET/RNAase, the cells were lysed, and cell debris was pelleted by centrifugation. After centrifugation, supernatant-containing DNA was separated from the cell debris and transferred to a filter. The DNA was precipitated and the DNA pellet washed, dried, resuspended, and collected in the bottom of the well.
Individual restriction digests for BAC and fosmid DNA contained ddH20, HindIII, PstI, and the DNA. Components of the digestion cocktail were assembled in plates and digested by incubation in a 96-well thermocycler. After digestion, the DNA was collected in the bottom of the wells by brief centrifugation, and loading dye was added to each well. We prepared and cooled 1% agarose gels and then poured them into UV-transparent trays. The comb was inserted until the gel solidified. Gels were placed into electrophoresis units. The restriction enzyme digestion/loading dye mixture was loaded into each well, with standard marker DNA added in the first well and in every fifth well thereafter. Samples were electrophoresed, and buffer was recirculated under constant temperature. The total electrophoresis time was 8 h. After electrophoresis, gels were removed to plastic trays containing SYBR (FMC BioProducts, Rockland, Maryland, United States) or Vistra (Molecular Probes, Eugene, Oregon, United States) Green and imaged using a Molecular Dynamics FluorImager (Amersham Biosciences, Uppsalla, Sweden. The FluorImager was also used to measure DNA yield.
The restriction fragment bands were identified interactively using IMAGE (D. Platt, F. Wobus, and R. Durbin, Sanger Institute; http://www.sanger.ac.uk/Software/Image/). Band cell data were collected and loaded into the fingerprint-mapping software FPC (Soderlund et al. 1997, 2000). Experimentally determined parameters for initial automated “binning” of related clones within FPC were: tolerance = 7, cutoff score = 10−8 (for fosmids) and 10−13 (for BACs), Diff = 0.3, minbands = −3, Diffbury = 0.10, MinEnd = 8. The “tolerance” is a window size; for example, if the tolerance is set to 7, then two restriction fragments occurring in different fingerprints must have relative mobilities within 0.7 mm to be considered equivalent fragments. The “cutoff score” is a threshold representing the maximum allowable probability of a chance match between any two clones (the “Sulston score“; see Sulston et al. 1988). The lower the cutoff score, the lower the probability that a match has arisen by chance and the more extensive the overlap between any two clones. Practical experience with our human fingerprint data has led us to apply a cutoff score of 10−8.
After initial “binning” of related clones into contigs, manual ordering of clones was performed within FPC. Ordering proceeded as follows: (1) a complex clone was chosen to be the initial clone to seed the contig (clone A); (2) the clone exhibiting the best match to the seed clone (by Sulston score) was selected (clone B); (3) if clone B had no unique restriction fragments, it was hidden under clone A; (4) if clone B had unique fragments, we repeated the search against the clones within the contig to find the best match (clone C); (5) the fingerprint of clone C was compared manually against the fingerprints of clones A and B; (6) to incorporate clone C into the contig, we required that the restriction fragments unique to clone B were present in clone C.
In this repeating, stepwise manner, we proceeded both to the “left” and “right” through the contig from the initial seed clone. After ordering within all contigs, contigs were searched against one another at reduced stringency to find potential joins between contigs. Contigs were only joined after it had been manually verified that they overlap.
Genomic library preparation
Genomic libraries used at the Genome Sequencing Center (GSC) (Washington University School of Medicine, St. Louis, Missouri, United States of America) were prepared by randomly shearing C. briggsae strain AF16 genomic DNA to a fragment size between 4.0 and 4.4 kbp. The fragments were size-selected using gel electrophoresis and ligated into a pOT4 vector. Transformation, DNA isolation, sequencing, and data collection were all performed using the current GSC protocols. These protocols are described in detail at the following web site: http://genome.wustl.edu/tools/protocols/.
Genomic libraries used at the Wellcome Trust Sanger Institute (Hinxton, United Kingdom) were prepared by random shearing of whole-genome C. briggsae DNA using sonication (for 2–3 kbp libraries) or by needle shearing (for 11 kbp libraries). Sheared DNA was end-repaired and phosphorylated using T4 DNA polymerase, Klenow, and T4 polynucleotide kinase and then size-fractionated by double gel purification. The size-selected blunt-ended DNA was ligated into the SmaI site of either pUC18 for the 2–3 kbp libraries or the low-copy number vector pTrueBlue-rop for the 11 kbp libraries. The ligations were purified by phenol extraction and transformed into E. coli DH10B by electroporation. Random clones were sequenced using the appropriate primers for each vector system.
BAC end sequencing
Details of the sequencing process can be found at http://genome.wustl.edu/. In brief, the process began with purification of DNA from selected clones from the physical map. The DNA was sheared mechanically, and after size selection, the resulting fragments were subcloned into M13 or plasmid vectors. Random subclones were shotgun sequenced, and data were generated with fluorescent dye-labeled primers or terminators.
BAC and fosmid clone sequencing
Raw sequence traces were processed through our read-processing tool GASP (Wendl et al. 1998), which uses PHRED (Ewing and Green 1998; Ewing et al. 1998) for processing and integrates the data into the central production database. All traces were then assembled using PHRAP (P. Green, unpublished data). We manually edited the data and selected primers (for improving data quality or for gap closure) using CONSED (Gordon et al. 1998). Gaps were closed and sequence ambiguities were resolved by sequencing longer reads, by directed sequencing reactions using custom oligonucleotide primers on chosen templates, or by additional chemistries. All data were made available on the Internet within 24 h of assembly.
We submitted each finished sequence to a series of quality control tests, including verification that the assembly is consistent with restriction digest information. In addition, the raw traces were completely reassembled using a different assembly algorithm, and any discrepancies in assembly or sequence were manually reviewed. New data were collected if necessary.
Phusion assembly
Of the 2.068 million shotgun reads, the initial Phusion read-grouping step clustered 1.93 million reads into 16,300 groups. Each group was assembled independently using the RPphrap step in the Phusion assembler. The RPphrap contigs were then joined together if reads and read pairs indicated that the contigs overlapped and overlapping sequence confirmed the joins. After merging contigs, we had 105.6 Mbp of sequence in 5,341 contigs, with an N50 contig size of 41 kbp (indicating that half the assembly is represented in contigs larger than 41 kbp). These 5,341 contigs were scaffolded using read-pair information, resulting in 107.5 Mbp of scaffold in 899 supercontigs with an N50 size of 474 kbp.
Integration of the WGS assembly with the physical map
All WGS supercontigs were “cut” into simulated overlapping BACs (66 kb) and fosmids (39 kb), each overlapping the previous clone by 40%, and digested in silico with HindIII and PstI. These simulated clones were compared to the clones in the physical map. We calculated the Sulston score and used it as a measure of possible overlap, thereby positioning the simulated BACs onto the physical map. Additionally, BAC end reads were placed onto the physical map by virtue of their name. This process allowed integration of map information, which could then be sorted by supercontigs across FPC contigs or by FPC contigs across supercontigs. We then evaluated the consistency of this mapping information. A lack of consistency suggested possible breaks in either the sequence assembly or the physical map. Similarly, consistency of data at contig ends suggested possible joins in the sequence assembly or the physical map.
Protein-coding gene prediction
We predicted protein-coding genes in the C. briggsae genome using Genefinder (version 980506; P. Green, unpublished data), FGENESH (Salamov and Solovyev 2000), TWINSCAN (Korf et al. 2001), and the Ensembl annotation system (Clamp et al. 2003). We also ran Genefinder and FGENESH on the C. elegans genome.
The four gene prediction programs yielded a combined total of 430,575 exon predictions and 73,997 gene predictions in the C. briggsae assembly. Many of the predictions from different programs overlapped, so the actual number of exons and genes is far fewer. The C. elegans data consisting of WS77 gene models and FGENESH and Genefinder predictions totaled 393,529 exon predictions and 61,525 gene predictions.
To select among overlapping predictions produced by different programs, we developed a selection procedure that worked as follows.
First, many of the exons predicted by different programs overlapped. We took only the longer of any two exons that overlapped by greater than 75% of their lengths and were in the same ORF.
Second, we clustered the exons within each species. Two exons were put in the same exon cluster if more than one gene prediction program placed them together in a gene prediction. Each exon cluster consisted of more than one overlapping gene prediction.
Third, for each exon cluster X, we found the most homologous exon cluster Y in the other species. Cluster Y was the exon cluster with the top BLASTP (Altschul et al. 1997) hit from any of the exons in X. For example, for the C. elegans exon cluster containing the ce-acy-4 gene, its top homolog was the C. briggsae exon cluster containing the cb-acy-4 gene (see Figure 1).
Fourth, each exon cluster X consisted of n overlapping gene predictions x1, x2, x3 … xn, where n ≥ 1. We chose one best prediction, x*, for X in this way: (1) We aligned proteins x1, x2, x3 … xn to each of the m predicted proteins y1, y2, y3 … ym in the homologous exon cluster Y, using T-COFFEE (Notredame et al. 2000); (2) from each pairwise alignment, we calculated a similarity score Sxy = 0.5(a/Lx + a/Ly), where a was the number of aligned (not necessarily conserved) amino acids and Lx and Ly the lengths of proteins x and y; (3) the best prediction x* for X was that having the highest S score when aligned to any of y1, y2, y3 … ym; (4) if X was a C. elegans exon cluster, the best prediction x* had to agree with experimentally confirmed coding bases or intron–exon junctions in WS77. This step produced gene sets for C. briggsae and for C. elegans, which we called the G1 gene sets.
Fifth, some exon clusters did not have a sequence match in the other species. We chose one best prediction for each such exon cluster by ranking the gene prediction programs by the fraction of predictions from each program that was selected for gene set G1. The ranking for C. briggsae was Ensembl, Genefinder, FGENESH, TWINSCAN. The ranking for C. elegans was the WS77 prediction set, FGENESH, Genefinder.
Sixth, the predictions chosen were added to the G1 gene sets, to produce the G2 gene sets for C. elegans and C. briggsae.
It is worth noting that there is an unavoidable bias in the way in which our selection procedure produced the G1 gene sets, which will have affected the ranking of gene predictions programs. predicted genes in C. briggsae by using similarity to C. elegans WS77 genes; therefore C. briggsae Ensembl and C. elegans WS77 predictions will tend to have similar structures. Likewise, the C. briggsae and C. elegans FGENESH predictions will tend to be similar, because FGENESH used the same parameters (for example, intron size distribution) to predict both gene sets. Thus, the selection procedure will have selected some C. briggsae and C. elegans FGENESH predictions for the G1 gene sets not because they are more accurate than a C. briggsae TWINSCAN and C. elegans Genefinder prediction for that C. briggsae/C. elegans ortholog pair, but rather because both were predicted by FGENESH. Therefore, while we used the ranking within our selection procedure, it cannot be used as a comparison of the four gene prediction programs' performance.
The G2 gene sets were filtered to remove transposons and putative pseudogenes. First, as described in the Repeat Families section below, we removed transposable element genes. Second, a prediction was taken to be a pseudogene if it was very short or lacked any sequence match: (1) if it could only be aligned using T-COFFEE (Notredame et al. 2000) to less than 25% of the lengths of its top two matches in Caenorhabditis or in SwissProt 40.38 (Boeckmann et al. 2003); or (2) if it did not have any BLASTP hit in Caenorhabditis or SwissProt of E-value <10−10 with the SEG filter on (Wootton and Federhen 1996) or E-value <10−20 with SEG off; or (3) if it had a within-species sequence match, but no cross-species sequence match and was less than 50 amino acids long.
This yielded the final G3 gene sets for C. elegans and C. briggsae.
Protein domain analysis
For the analysis of orthologs, gene families, and functional domains, a unique gene set was identified for each species in which every alternatively spliced gene was represented only once by the form predicted to give the longest ORF.
Each predicted protein in the C. briggsae and C. elegans unique gene sets was analyzed with the Pfam 9.0 database (Bateman et al. 2002) using the hmmPfam program (version 2.2g; S. Eddy, unpublished data; http://hmmer.wustl.edu/) to identify functional domains and other known sequence motifs. An InterPro annotation was assigned to each such feature (Zdobnov and Apweiler 2001). These were translated into GO functional descriptions (ftp.ebi.ac.uk/pub/databases/interpro/interpro2go), and the descriptions were grouped into broader “GOslim” categories for molecular function and biological process (http://www.ebi.ac.uk/proteome; Gene Ontology Consortium 2001).
C. briggsae/C. elegans orthologs
We ran NCBI BLASTP (Altschul et al. 1997) with the C. briggsae protein set as the query database and the C. elegans WS77* protein set as the target database and vice versa. For C. elegans WS77* genes that have alternative transcripts, we only took the longest splice variant.
We found orthologs in the following way. First, we found C. briggsae/C. elegans gene pairs that were each other's top BLASTP hits. We required the BLASTP hits to have an E-value of <10−10 with the SEG filter (Wootton and Federhen 1996) on or <10−20 with SEG off. Furthermore, to avoid assigning paralogs to ortholog pairs, the top hit had to have an E-value 105 times lower (more significant) than the next best hit.
Second, we found additional orthologs by analyzing conserved gene order. We found syntenic blocks by looking for orthologs A (found in step 1) that were near orthologs B (also found in step 1) in both species. We identified C. briggsae/C. elegans gene pairs within the A–B syntenic block that were each other's top BLASTP hits. To avoid assigning paralogs to ortholog pairs, the top hit had to have an E-value 105 times lower (more significant) than the next best hit in the A–B syntenic block.
Third, we identified C. briggsae/C. elegans gene pairs that were each other's top BLASTP hits and that were within 100 kbp of orthologs C (found in step 1) in both species.
Intron gain and loss
We used T-COFFEE (Notredame et al. 2000) to align all C. briggsae/C. elegans ortholog pairs. We then searched the alignments for cases in which exon i in species A aligned well to two adjacent exons j and k in species B. To ensure that orthologous exons were matched properly, we required that exons i and j as well as exons i and k had to consist of identical or conserved amino acids across at least 20% of the shorter exon.
Codon usage
We used the EMBOSS (Rice et al. 2000) program cusp to calculate codon usage in the predicted CDS from both species. We then used the EMBOSS tool codcmp to test for a significant difference in codon usage.
Synonymous and nonsynonymous substitution rates
Mutual-best hit and syntenically assigned pairs of orthologous protein sequences were aligned using the “needle” program from EMBOSS (Rice et al. 2000). Alignments of the corresponding CDSs were then produced using software written in Perl and available in the Bioperl toolkit (Stajich et al. 2002). An ML calculation of KA and KS for each orthologous pair of CDSs was calculated using PAML (Yang 1997). Only those pairs where the Nei-Gojobori (1986) method could estimate a KA and KS value and the PAML ML values fell within KA <4 and KS <9 were processed further to reduce the amount of noise due to spurious alignments.
A dataset of genes classified through RNAi knockouts as lethal, phenotype, no phenotype, and untested were obtained from WormBase based on the work of Maeda et al. (2001), Piano and Gunsalus (2002), and Kamath et al. (2003). The R statistical package (Ihaka and Gentleman 1996) was used to calculate t-tests and plots.
Estimating the C. briggsae/C. elegans divergence date
We downloaded human and Anopheles gambiae proteins from http://www.ensembl.org/ in December 2002 (human release 9.30 and mosquito release 9.1; Hubbard et al. 2002). We took the longest alternative splice for each of the 22,980 human genes and 15,088 Anopheles genes. To identify C. elegans/human orthologs, we compared the C. elegans WS77* protein set to the human proteins using BLASTP (Altschul et al. 1997) with the SEG filter (Wootton and Federhen 1996). A C. elegans gene and a human gene were considered one-to-one orthologs if they were each other's top BLASTP hits and hit each other with E-values of <10−20, where the second-best hit in each species had to have an E-value a factor of >1020 greater (less significant) than the best hit. In this way we identified 1,914 C. elegans/human orthologs and 2,498 C. elegans/A. gambiae orthologs, while 11,255 C. briggsae/C. elegans one-to-one orthologs were found by identifying mutual-best BLASTP hits as described above. For 1,397 C. elegans genes, we had a C. briggsae, a human, and a mosquito ortholog. For each of the 1,397 quartets, we aligned the four proteins using ClustalW (Thompson et al. 1994) and made a guide-tree using protdist and neighbor from the PHYLIP package (Felsenstein 1989). For each ortholog set, the alignment and guide-tree were used as input for Gu and Zhang's (1997) program GAMMA, which estimated an α parameter for the à distribution used to correct for rate variation among amino acid sites. For 148 trees, GAMMA could not estimate the α parameter. For the remaining 1,249 trees, we used the two-cluster test (Takezaki et al. 1995) to check for unequal rates between lineages, taking human to be the outgroup to Anopheles and Caenorhabditis (Aguinaldo et al. 1997); 338 trees passed the test at the 5% significance level. For each of these 338 trees, the branch lengths were re-estimated under the assumption of rate constancy, using Takezaki and Nei's (1995) program with the à correction for multiple hits. We calibrated the linearized trees by taking the nematode/arthropod divergence to be 800–1,000 MYA (Blaxter 1998).
Gene family clustering
Gene families were identified utilizing the TRIBE-MCL method (Van Dongen 2000; Enright et al. 2002). Briefly, TRIBE-MCL identifies gene families using a Markov Clustering (MCL) procedure operating on a matrix of expectation values computed from a similarity search of all versus all of C. briggsae and C. elegans protein sequences. We used the Smith–Waterman algorithm as implemented in SSEARCH (Smith and Waterman 1981; Pearson 1991) to achieve a greater measure of sensitivity than BLASTP. The MCL clustering was executed with an inflation value of 1.6, chosen to minimize the number of orphaned or mispaired putative orthologs without greatly expanding the total number of clusters. Where more than one splice variant existed for a gene, only the longest transcript was chosen as a representative for the gene. TRIBE cluster annotation was created by merging the information from the InterPro (Zdobnov and Apweiler 2001) and Pfam 9.0 (Bateman et al. 2002) domains that occurred in at least two of the genes in a family. The full output from the TRIBE-MCL clustering and the Pfam domains for each cluster are available as Dataset S6.
To develop a phylogenetic tree of the sra chemosensory receptor protein family, we combined chemosensory receptor genes belonging to the same Pfam subfamily from both species into a joint file that was then aligned using ClustalW (Thompson et al. 1994). The output file was fed into the PHYLIP package (programs used include: seqboot, protdist, neighbor, and consense; Felsenstein 1989) to generate a tree file. TreeView, part of the PHYLIP package, was used to visualize the family relationships.
To identify phylogenetic-tree-based ortholog pairs of the sra protein family, we chose C. briggsae/C. elegans protein pairs that were each other's closest neighbors in greater than 900 of 1,000 bootstrap repetitions.
Physical clustering of genes in a family along the chromosomes was tested by a permutation test counting the number of genes in a sliding window of 15 genes that are members of the same TRIBE family cluster. The average per window in each species was compared to the averages for 1,000 simulated genomes of randomly ordered genes. The maximum observed value for the shuffled genomes was always found to be smaller than the observed genome state, indicating that physical clustering of gene families is significantly nonrandom.
Prediction of ncRNA genes
We predicted tRNA genes using tRNAscan-SE version 123 (Lowe and Eddy 1997) in eukaryotic mode with default parameters and a threshold of 20 bits. To predict rRNA and miRNA genes, we extracted sequences from the sequence databases and searched for homologous sequences in the C. briggsae genome using BLASTN (Altschul et al. 1997). True matches were defined as those having greater than 95% identity over greater than 95% of the query length. Fragmentary matches were defined as all other hits with p value of <0.001. We predicted all other ncRNA genes using the Rfam 3.0 library of covariance models (Griffiths-Jones et al. 2003) and the INFERNAL software suite (Eddy 2002) with Rfam family-specific score thresholds.
Repeat families
We used RepeatMasker (A. F. A. Smit and P. Green, unpublished data) to identify presumptive repetitive elements in C. briggsae. Two different repeat library files were used to run RepeatMasker. The first library file, elegans.lib, was obtained from RepBase (Jurka 2000). RepBase includes all previously identified C. elegans repeat elements, as well as repeat elements from a variety of vertebrate and invertebrate species.
When elegans.lib failed to identify the expected number of repeats in C. briggsae, we built a second repeat library file using RECON (Bao and Eddy 2002). For the initial all versus all pairwise comparison of genomic sequences, the result of which serves as input to RECON, we used WUBLAST (W. R. Gish, unpublished data; http://blast.wustl.edu/) with options “−kap E=0.00001 wordmask=dust wordmask=seg maskextra=20 −hspmax 5000 M=5 N=−11 Q=22 R=11.” All parameters of RECON were left at their default values. For RECON-defined families with more than ten copies, a consensus sequence for each was constructed by aligning the ten longest members of the family with DIALIGN2 (Morgenstern 1999), with its default options, then selecting a simple majority rule consensus residue for each column in the multialignment. For C. briggsae, 723 consensuses were built. A total of 554 consensus sequences were built for C. elegans following the same protocol.
RECON has a known artifact in which it identifies conserved protein family domain and multicopy noncoding genes such as tRNAs and rRNAs in addition to finding bona fide repeat family elements. We removed these cases from the C. elegans and C. briggsae RECON library files by applying a series of filters. Before filtering, C. elegans and C. briggsae RECON library files contained 554 and 723 entries, respectively. To remove known ncRNA species, we ran INFERNAL (Eddy 2002) together with Rfam (Griffiths-Jones et al. 2003). For C. briggsae, this step removed 22 tRNAs, two histone H3 genes, and one each of U1, U2, U5, and U6. For C. elegans, this step removed 20 tRNAs, three rRNAs, one histone H3 gene, and one each of U1, U2, U5, and U6.
Prior to removing gene family domains from the RECON libraries, it was necessary to remove transposons and other repetitive gene calls from the gene prediction sets. We used elegans.lib to identify “trusted repeats” in the RECON libraries, finding 188 trusted repeats in the RECON library for C. elegans and 72 trusted repeats in the RECON repeat library for C. briggsae. We then used BLAST (Altschul et al. 1997) to identify and remove trusted repeat sequences from the DNA sequences of the C. elegans and C. briggsae gene sets. A gene prediction was taken to be a transposable element if it had a TBLASTN hit of E-value <10−30 in the RepBase C. elegans library (version 8.2; Jurka 2000) or if its individual exons all had BLASTN hits of E-value <10−10 in the RECON repeat library for that species. This step removed 303 genes from C. elegans WS77 gene set, 434 genes from C. elegans hybrid gene set, and 627 genes from C. briggsae gene set.
We next used TBLASTN to match the library of RECON-identified repeat elements against the filtered C. elegans and C. briggsae hybrid gene sets as well as SwissProt, and we removed repeat families that matched proteins with an E-value <10−10. A total of 181 and 382 entries were removed from the C. elegans and C. briggsae RECON libraries, respectively. Finally, we manually examined those repeat entries with significant protein hits and added an additional eight entries to the trusted repeat library for C. briggsae. Manual examination did not yield more trusted repeats for C. elegans.
The final filtered RECON libraries contained 473 C. briggsae and 377 C. elegans entries.
The repeat family data can be found as Dataset S7.
Nucleotide-level sequence alignments
We used the WABA algorithm (Kent and Zahler 2002) to align the C. briggsae WGS assembly to the complete C. elegans genomic sequence. All WGS supercontigs were “cut” into 100 kbp pieces and individually aligned to the six C. elegans chromosomes. The coordinates of the regions of alignment were then transformed back into supercontig coordinates for further analysis.
To characterize raw WABA aligning segments, we partitioned the C. elegans genome into eight compartments corresponding to CDS, introns, 5′ and 3′ UTRs, upstream regions, downstream regions, and repeat regions. The remainder of the genome was considered to be intergenic. For consistency, upstream regions were considered to be 1,000 bp upstream of the translational start site, and downstream regions were considered to be 1,000 bp downstream of the translational stop, regardless of whether or not the UTRs of the gene were known. In the case of a region that could be scored in two or more ways, such as a region that is within the 1,000 bp downstream window of one gene and upstream of another, the region was assigned to one partition on the basis of left-to-right priority in the list above. For example, CDSs have priority over an intron. A WABA segment was scored as belonging to a partition if it shared at least one base overlap with a region in that partition.
For the purposes of comparison, we also repeated the whole-genome alignment with the BLASTZ algorithm (Schwarz et al. 2003). The results were highly comparable, but the coverage was slightly lower with BLASTZ (56% coverage with BLASTZ versus 65% coverage with WABA, not adjusted for overlaps).
Synteny block construction
To identify putative syntenic regions from the raw WABA alignment blocks, we merged adjacent “strong,” “weak,” and “coding” blocks into a smaller number of contiguous blocks. This reduced the 1.3 million raw WABA blocks to 104,097 contiguous blocks. From this set, we discarded blocks in which more than five C. elegans segments aligned to a segment of C. briggsae or vice versa. The remaining alignments were merged using the “simple merge” algorithm, which searches for and merges uninterrupted series of alignments that are monotonically increasing in both the C. elegans and C. briggsae genomes. This procedure yielded 26,231 merged blocks of alignment.
We then performed an additional round of merging using a dynamic programming algorithm to identify the longest monotonically increasing set of alignment blocks. Unlike the simple merge, this algorithm can jump over interrupted regions of colinearity. For each C. briggsae supercontig, the algorithm first finds the longest series of blocks then finds the next longest series using those left over from the first iteration. This continues until no blocks remain. During the process, the identification of monotonically increasing blocks is constrained so that no gap in the series can be greater than 100 kbp in either genome and so that no single gap can cause a relative expansion of greater than 5-fold in either the C. elegans or C. briggsae coordinates. This step yielded 13,467 merged blocks.
Examination of the distribution of synteny block lengths showed a large asymmetric peak at 1,250 bp and a long tail of longer block lengths (data not shown). Most of these blocks involved a single unmerged WABA alignment and correlated poorly with the positions of previously identified orthologs. To exclude these small blocks from further analysis, we filtered the blocks for a lower size limit of 1,850 bp, which reduced the coverage of the C. elegans genome by 1.5% but excluded 64% of the merged blocks, leaving a final candidate list of 4,837 synteny blocks.
Each block was classified as an inversion, transposition, or reciprocal translocation by examining the breakpoint junctions a/b and c/d in C. briggsae:
A block was classified as an inversion if a was adjacent to c and b was adjacent to d from the perspective of C. elegans coordinates. A block was classified as a transposition if a and d were adjacent in C. elegans and a reciprocal event could not be identified. A block was classified as involving one or two reciprocal translocations if another breakpoint
could be found such that a was adjacent to f or e was adjacent to d in C. elegans.
Updating the C. elegans gene set
We used TBLASTN searches (Altschul et al. 1997) of the C. briggsae genome to see how many of the gene model changes suggested in the C. elegans hybrid gene set, compared to the WS77 gene set, were supported by C. briggsae similarity. We only considered new hybrid exons and extensions and truncations or deletions of existing WS77 exons where the extended or truncated region was greater than or equal to five amino acids (15 bp) long. If the C. elegans hybrid gene set predicted a new exon in an existing WS77 gene, we considered the new exon to be well-supported if the exon had a TBLASTN hit of E-value <10−3 in the C. briggsae genome that covered greater than or equal to 10 amino acids of the new exon. If the C. elegans hybrid gene set predicted an extension of an existing WS77 exon, we considered the extension to be well-supported if the extended exon had a TBLASTN hit of <10−3 in the C. briggsae genome which covered greater than or equal to 10 amino acids of the extended part. In contrast, if the C. elegans hybrid gene set predicted that an existing exon in a WS77 gene should be deleted, we considered the exon deletion to be well supported if the WS77 exon did not have any TBLASTN hit of E-value <0.1 that covered greater than or equal to five amino acids of the exon. Likewise, if the C. elegans hybrid gene set predicted that an existing exon in a WS77 gene should be truncated, we considered the exon truncation to be well-supported if the WS77 exon did not have any TBLASTN hit of E-value <0.1 that covered greater than ot equal to five amino acids of the truncated part.
Supporting Information
(64 MB GZ).
This directory comprises cb.hybrid.gff, a file of C. briggsae gene predictions using the “hybrid” method in GFF format (http://www.sanger.ac.uk/Software/formats/GFF/); cb.fa, file that contains conceptual translations of C. briggsae gene predictions in FASTA format; ws77.hybrid.gff, a file that contains C. elegans gene predictions using the “hybrid” method in GFF format; ws77.hybrid.fa, a file that contains C. elegans hybrid genes' conceptual translations in FASTA format; ws77.gff, a file that contains the canonical C. elegans gene predictions from Wormbase version WS77* (after filtering for transposase-containing genes and other artefacts); ws77.fa, a file that contains C. elegans WS77* genes' conceptual translations in FASTA format; gene2ip2go.briggsae, a file that contains InterPro annotations of predicted C. briggsae gene products, plus GO terms when available; and gene2ip2go.elegans, a file that contains InterPro annotations of predicted C. elegans gene products from WS77*, plus GO terms when available.
(20 MB ZIP).
This directory comprises orthologs.txt, a file that contains the list of C. briggsae/C. elegans ortholog pairs; orthologs-kaks.txt, a file that contains KA/KS values for ortholog pairs, as well as other information used to determine regional and functional variation in KA/KS; and cb_orphans and ce_orphans, files that contain lists of putative “orphan” genes that are found in one species but not in another.
(433 KB ZIP).
This directory comprises cb.waba_state.gff, a file that contains WABA alignments between C. briggsae and C. elegans; and cb.waba.gff, a file that contains WABA alignments in which transitions between adjacent WABA block types (“strong,” “weak” and “coding” ) have been merged.
(41.8 MB ZIP).
This directory comprises synteny_blocks.txt, a file that contains 4,837 synteny blocks identified between C. elegans and C. briggsae; and sorted_ultracontigs.txt, a file that contains 382 C. briggsae ultracontigs that have been ordered and oriented on the C. elegans genome relative to their largest synteny block.
(120 KB TXT).
This directory comprises tribe_cluster.txt, a file that contains TRIBE-MCL co-clusters of C. briggsae and C. elegans predicted proteins, with the clusters sorted with the largest first; tribe_cluster_classify_short.txt, a file that contains the Pfam and InterPro classification of co-clusters; and tribe_cluster_classify_long.txt, which is similar to the previous file, except that all InterPro/Pfam annotations are shown.
(360 KB ZIP).
This directory comprises Cb_repeat.lib, a FASTA-format file of RECON-predicted repeat elements from C. briggsae after removal of protein families; Ce_repeat.lib, a FASTA-format file of C. elegans predicted repeat elements; Cb_RECON_repeat.gff, a GFF-format file indicating the positions of predicted repeats in the C. briggsae genome; and Ce_RECON_repeat.gff, a GFF-format file indicating the positions of predicted repeats in the C. elegans genome.
(1.9 MB TXT).
The Genome Sequence of Caenorhabditis briggsae: A Platform for Comparative Genomics
The nematodes C. briggsae and C. elegans diverged 80–110 MYA, near to the time of divergence of human from mouse, but are almost indistinguishable morphologically. These figures demonstrate the high degree of morphological similarity between C. briggsae and C. elegans and show how the patterns of evolutionary conservation between the two species vary across the five autosomes (I–V) and sex chromosome (X) of C. elegans.
(50.6 MB PDF).
Accession Numbers
The C. briggsae genomic sequence has been submitted to the GenBank WGS division as the 578 entries accessioned CAAC01000001 through CAAC01000578. The genes discussed in this paper can be found in the WormBase database: C06G3.7 (http://wormbase.org/db/gene/gene?name=C06G3.7); CBG05747 (http://wormbase.org/db/gene/gene?name=CBG05747); mir-35 (http://wormbase.org/db/gene/gene?name=mir-35); mir-41 (http://wormbase.org/db/gene/gene?name=mir-41); and snt-1 (http://wormbase.org/db/gene/gene?name=snt-1).
Acknowledgments
We thank Hugh Robertson for helpful discussions on the chemosensory receptor families and Paul Sternberg, Karin Kiontke, and Marie-Anne Felix for their insights into the anatomic differences among the Caenorhabditis species. We are grateful to Sean Eddy, Tom Jones, and Robbie Klein for providing us with the sequences of C. elegans RNAase P and U3 snoRNA. We thank David Baillie for discussions on the repetitive elements and for pointing us to key references. We thank Paula Kassos for proofreading the manuscript. We also are grateful to the three anonymous reviewers who reviewed this paper.
This work was supported by grants from the following funding agencies: National Institutes of Health, United States of America (P41 HG02223, 5P01 HG00956, 5U01 HG02042, T32 GM07754-22, RO1 GM42432); the National Science Foundation, United States of America (DBI-0132436); The Wellcome Trust, the United Kingdom; The United Kingdom's Medical Research Council.
J. E. Stajich was supported by National Institutes of Health training grant T32 GM07754-22. A. Coghlan was supported by a grant from Science Foundation Ireland.
A. Chinwalla, C. Clee, L. A. Fulton, R. E. Fulton, P. E. Kuwabara, T. L. Miner, P. Minx, R. W. Plumb, J. E. Schein, C. Wei, D. Willey, and R. K. Wilson performed the experiments.
L. D. Stein, Z. Bao, D. Blasiar, T. Blumenthal, M. R. Brent, N. Chen, L. Clarke, A. Coghlan, P. D'Eustachio, S. Griffiths-Jones, T. W. Harris, L. W. Hillier, R. Kamath, J. C. Mullikin, M. Sohrmann, J. Spieth, J. E. Stajich, R. Durbin, and R. H. Waterston analyzed the data.
A. Coulson, D. H. A. Fitch, and M. A. Marra contributed reagents/materials/analysis tools.
L. D. Stein, Z. Bao, T. Blumenthal, N. Chen, A. Coghlan, P. D'Eustachio, S. Griffiths-Jones, T. W. Harris, L. W. Hillier, J. C. Mullikin, M. Sohrmann, J. Spieth, J. E. Stajich, R. Durbin, and R. H. Waterston wrote the paper.
L. D. Stein and R. Durbin organized conference calls.
Abbreviations
- BAC
bacterial artificial chromosome
- CDS
coding sequence
- EGF
epidermal growth factor
- EST
expressed sequence tag
- FPC
FingerPrint Contig software
- GO
Gene Ontology
- GSC
Genome Sequencing Center
- KA
nonsynonymous substitution
- KS
synonymous substitution
- MCL
Markov clustering
- miRNA
microRNA
- ML
maximum likelihood
- mRNA
messenger RNA
- MY
million years
- MYA
million years ago
- ncRNA
noncoding RNA
- ORF
open reading frame
- OST
open reading frame sequence tag
- rDNA
ribosomal DNA
- RNAi
RNA inhibition
- rRNA
ribosomal RNA
- SD
standard deviation
- SECIS
selenocysteine insertion sequence
- snoRNA
small nucleolar RNA
- snRNA
small nuclear RNA
- tRNA
transfer RNA
- UTR
untranslated region
- WGS
whole-genome shotgun sequencing
Conflicts of Interest. The authors have declared that no conflicts of interest exist.
Author Contributions. L. W. Hillier, E. R. Mardis, J. C. Mullikin, J. Rogers, R. Durbin, and R. H. Waterston conceived and designed the experiments.
Academic Editor: Jonathan A. Eisen, The Institute for Genomic Research
References
- Aguinaldo AM, Turbeville JM, Linford LS, Rivera MC, Garey JR, et al. Evidence for a clade of nematodes, arthropods, and other moulting animals. Nature. 1997;387:489–493. doi: 10.1038/387489a0. [DOI] [PubMed] [Google Scholar]
- Altschul SF, Madden TL, Schaffer AA, Zhang J, Zhang A, et al. Gapped BLAST and PSI-BLAST: A new generation of protein database search programs. Nucleic Acids Res. 1997;25:3389–3402. doi: 10.1093/nar/25.17.3389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ambros V, Lee RC, Lavanway A, Williams PT, Jewell D. MicroRNAs and other tiny endogenous RNAs in C. elegans. Curr Biol. 2003;13:807–818. doi: 10.1016/s0960-9822(03)00287-2. [DOI] [PubMed] [Google Scholar]
- Bao Z, Eddy SR. Automated de novo identification of repeat sequence families in sequenced genomes. Genome Res. 2002;12:1269–1276. doi: 10.1101/gr.88502. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barnes TM, Kohara Y, Coulson A, Hekimi S. Meiotic recombination, noncoding DNA, and genomic organization in Caenorhabditis elegans. Genetics. 1995;141:159–179. doi: 10.1093/genetics/141.1.159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bateman A, Birney E, Cerruti L, Durbin R, Etwiller L, et al. The Pfam protein families database. Nucleic Acids Res. 2002;30:276–280. doi: 10.1093/nar/30.1.276. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blaxter M. Caenorhabditis elegans is a nematode. Science. 1998;282:2041–2046. doi: 10.1126/science.282.5396.2041. [DOI] [PubMed] [Google Scholar]
- Blumenthal T, Gleason KS. Caenorhabditis elegans operons: Form and function. Nat Rev Genet. 2003;4:112–120. doi: 10.1038/nrg995. [DOI] [PubMed] [Google Scholar]
- Boeckmann B, Bairoch A, Apweiler R, Blatter MC, Estreicher A, et al. The SWISS-PROT protein knowledgebase and its supplement TrEMBL in 2003. Nucleic Acids Res. 2003;31:365–370. doi: 10.1093/nar/gkg095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Britten R, Kohne D. Repeated sequences in DNA. Science. 1968;161:529–540. doi: 10.1126/science.161.3841.529. [DOI] [PubMed] [Google Scholar]
- Buettner C, Harney JW, Berry MJ. The Caenorhabditis elegans homologue of thioredoxin reductase contains a selenocysteine insertion sequence (SECIS) element that differs from mammalian SECIS elements but directs selenocysteine incorporation. J Biol Chem. 1999;274:21598–21602. doi: 10.1074/jbc.274.31.21598. [DOI] [PubMed] [Google Scholar]
- Butler MH, Wall SM, Luehrsen KR, Fox GE, Hecht RM. Molecular relationships between closely related strains and species of nematodes. J Mol Evol. 1981;18:18–23. doi: 10.1007/BF01733207. [DOI] [PubMed] [Google Scholar]
- C. elegans Sequencing Consortium. Genome sequence of the nematode C. elegans: A platform for investigating biology. Science. 1998;282:2012–2018. doi: 10.1126/science.282.5396.2012. [DOI] [PubMed] [Google Scholar]
- Castillo-Davis CI, Hartl DL. Genome evolution and developmental constraint in Caenorhabditis elegans. Mol Biol Evol. 2002;19:728–735. doi: 10.1093/oxfordjournals.molbev.a004131. [DOI] [PubMed] [Google Scholar]
- Civetta A, Singh RS. Sex-related genes, directional sexual selection, and speciation. Mol Biol Evol. 1998;15:901–909. doi: 10.1093/oxfordjournals.molbev.a025994. [DOI] [PubMed] [Google Scholar]
- Clamp M, Andrews D, Barker D, Bevan P, Cameron G, et al. Ensembl 2002: Accommodating comparative genomics. Nucleic Acids Res. 2003;31:38–42. doi: 10.1093/nar/gkg083. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cliften P, Sudarsanam P, Desikan A, Fulton L, Fulton B, et al. Finding functional features in Saccharomyces genomes by phylogenetic footprinting. Science. 2003;301:71–76. doi: 10.1126/science.1084337. [DOI] [PubMed] [Google Scholar]
- Coghlan A, Wolfe KH. Four-fold faster rate of genome rearrangement in nematodes than in Drosophila. Genome Res. 2002;12:857–867. doi: 10.1101/gr.172702. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cui M, Han M. cis-Regulatory requirements for vulval cell-specific expression of the Caenorhabditis elegans fibroBLAST growth factor gene egl-17. Dev Biol. 2003;257:104–116. doi: 10.1016/s0012-1606(03)00033-2. [DOI] [PubMed] [Google Scholar]
- Culetto E, Combes D, Fedon Y, Roig A, Toutant JP, et al. Structure and promoter activity of the 5′ flanking region of ace-1, the gene encoding acetylcholinesterase of class A in Caenorhabditis elegans. J Mol Biol. 1999;290:951–966. doi: 10.1006/jmbi.1999.2937. [DOI] [PubMed] [Google Scholar]
- Cutter AD, Payseur BA. Selection at linked sites in the partial selfer Caenorhabditis elegans. Mol Biol Evol. 2003;20:665–673. doi: 10.1093/molbev/msg072. [DOI] [PubMed] [Google Scholar]
- Eddy SR. A memory-efficient dynamic programming algorithm for optimal alignment of sequence to an RNA secondary structure. BMC Bioinformatics. 2002;2:18. doi: 10.1186/1471-2105-3-18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Edwards A, Voss H, Rice P, Civitello A, Stegemann J, et al. Automated DNA sequencing of the human HPRT locus. Genomics. 1990;6:593–608. doi: 10.1016/0888-7543(90)90493-e. [DOI] [PubMed] [Google Scholar]
- Ejima Y, Yang L. Trans-mobilization of genomic DNA as a mechanism for retrotransposon-mediated exon shuffling. Hum Mol Genet. 2003;12:1321–1328. doi: 10.1093/hmg/ddg138. [DOI] [PubMed] [Google Scholar]
- Ellis RE, Sulston JE, Coulson AR. The rDNA of C. elegans: Sequence and structure. Nucleic Acids Res. 1986;14:2345–2364. doi: 10.1093/nar/14.5.2345. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Enright AJ, Van Dongen S, Ouzounis CA. An efficient algorithm for large-scale detection of protein families. Nucleic Acids Res. 2002;30:1575–1584. doi: 10.1093/nar/30.7.1575. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ewing B, Green P. Base-calling of automated sequencer traces using PHRED. II. Error probabilities. Genome Res. 1998;8:186–194. [PubMed] [Google Scholar]
- Ewing B, Hillier L, Wendl MC, Green P. Base-calling of automated sequencer traces using PHRED. I. Accuracy assessment. Genome Res. 1998;8:175–185. doi: 10.1101/gr.8.3.175. [DOI] [PubMed] [Google Scholar]
- Evans D, Zorio D, MacMorris M, Winter CE, Lea K, et al. Operons and SL2 trans-splicing exist in nematodes outside the genus Caenorhabditis. Proc Natl Acad Sci U S A. 1997;94:9751–9756. doi: 10.1073/pnas.94.18.9751. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Felsenstein J. PHYLIP-phylogeny inference package (version 3.2) Cladistics. 1989;5:164–166. [Google Scholar]
- Fitch DHA. Evolution of male tail development in rhabditid nematodes related to Caenorhabditis elegans. Syst Biol. 1997;46:145–179. doi: 10.1093/sysbio/46.1.145. [DOI] [PubMed] [Google Scholar]
- Fitch DHA, Emmons SW. Variable cell positions and cell contacts underlie morphological evolution of the rays in the male tails of nematodes related to Caenorhabditis elegans. Dev Biol. 1995;170:564–582. doi: 10.1006/dbio.1995.1237. [DOI] [PubMed] [Google Scholar]
- Fraser AG, Kamath RS, Zipperlen P, Martinez-Campos M, Sohrmann M, et al. Functional genomic analysis of C. elegans chromosome I by systematic RNA interference. Nature. 2000;408:325–330. doi: 10.1038/35042517. [DOI] [PubMed] [Google Scholar]
- Fodor A, Riddle DL, Nelson FK, Golden JW. Comparison of a new wild-type Caenorhabditis briggsae with laboratory strains of C. briggsae and C. elegans. Nematologica. 1983;29:203–217. [Google Scholar]
- Gene Ontology Consortium. Creating the gene ontology resource: Design and implementation. Genome Res. 2001;11:1425–1433. doi: 10.1101/gr.180801. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gilleard JS, Barry JD, Johnstone IL. cis-Regulatory requirements for hypodermal cell-specific expression of the Caenorhabditis elegans cuticle collagen gene dpy-7. Mol Cell Biol. 1997;17:2301–2311. doi: 10.1128/mcb.17.4.2301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goff SA, Ricke D, Lan TH, Presting G, Wang R, et al. A draft sequence of the rice genome (Oryza sativa L. ssp. japonica) Science. 2002;296:92–100. doi: 10.1126/science.1068275. [DOI] [PubMed] [Google Scholar]
- Gonczy P, Echeverri C, Oegema K, Coulson A, Jones SJ, et al. Functional genomic analysis of cell division in C. elegans using RNAi of genes on chromosome III. Nature. 2000;408:331–336. doi: 10.1038/35042526. [DOI] [PubMed] [Google Scholar]
- Gordon D, Abajian C, Green P. CONSED: A graphical tool for sequence finishing. Genome Res. 1998;8:195–202. doi: 10.1101/gr.8.3.195. [DOI] [PubMed] [Google Scholar]
- Gower NJ, Temple GR, Schein JE, Marra M, Walker DS, et al. Dissection of the promoter region of the inositol 1,4,5-trisphosphate receptor gene, itr-1 in C. elegans: A molecular basis for cell-specific expression of IP3R isoforms. J Mol Biol. 2001;306:145–157. doi: 10.1006/jmbi.2000.4388. [DOI] [PubMed] [Google Scholar]
- Grad Y, Aach J, Hayes GD, Reinhart BJ, Church GM, et al. Computational and experimental identification of C. elegans microRNAs. Mol Cell. 2003;11:1253–1263. doi: 10.1016/s1097-2765(03)00153-9. [DOI] [PubMed] [Google Scholar]
- Gray YH. It takes two transposons to tango: Transposable-element-mediated chromosomal rearrangements. Trends Genet. 2000;16:461–468. doi: 10.1016/s0168-9525(00)02104-1. [DOI] [PubMed] [Google Scholar]
- Griffiths-Jones S, Bateman A, Marshall M, Khanna A, Eddy SR. Rfam: An RNA family database. Nucleic Acids Res. 2003;31:439–441. doi: 10.1093/nar/gkg006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gu X, Zhang J. A simple method for estimating the parameter of substitution rate variation among sites. Mol Biol Evol. 1997;14:1106–1113. doi: 10.1093/oxfordjournals.molbev.a025720. [DOI] [PubMed] [Google Scholar]
- Gu X, Wang Y, Gu J. Age distribution of human gene families shows significant roles of both large- and small-scale duplications in vertebrate evolution. Nat Genet. 2002;31:205–209. doi: 10.1038/ng902. [DOI] [PubMed] [Google Scholar]
- Guthrie C, Abelson J. Organization and expression of tRNA genes in Saccharomyces cerevisiae. In: Strathern J, Broach J, editors. The molecular biology of the yeast Saccharomyces: Metabolism and gene expression. Cold Spring Harbor, New York: Cold Spring Harbor Laboratory Press; 1982. pp. 487–528. [Google Scholar]
- Haag ES, Wang S, Kimble J. Rapid coevolution of the nematode sex-determining genes fem-3 and tra-2. Curr Biol. 2002;12:2035–2041. doi: 10.1016/s0960-9822(02)01333-7. [DOI] [PubMed] [Google Scholar]
- Harris LJ, Prasad S, Rose AM. Isolation and sequence analysis of Caenorhabditis briggsae repetitive elements related to the Caenorhabditis elegans transposon Tc1. J Mol Evol. 1990;30:359–369. doi: 10.1007/BF02101890. [DOI] [PubMed] [Google Scholar]
- Hope IA, Mounsey A, Bauer P, Aslam S. The forkhead gene family of Caenorhabditis elegans. Gene. 2003;304:43–55. doi: 10.1016/s0378-1119(02)01175-7. [DOI] [PubMed] [Google Scholar]
- Hubbard T, Barker D, Birney E, Cameron G, Chen Y, et al. The Ensembl genome database project. Nucleic Acids Res. 2002;30:38–41. doi: 10.1093/nar/30.1.38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hughes AL, Friedman R. 2R or not 2R: Testing hypotheses of genome duplication in early vertebrates. J Struct Funct Genomics. 2003;3:85–93. [PubMed] [Google Scholar]
- Ihaka R, Gentleman R. R: A language for data analysis and graphics. J Comp Graph Stat. 1996;5:299–314. [Google Scholar]
- International Human Genome Sequencing Consortium. Initial sequencing and analysis of the human genome. Nature. 2001;409:860–921. doi: 10.1038/35057062. [DOI] [PubMed] [Google Scholar]
- Jordan IK, Rogozin IB, Wolf YI, Koonin EV. Microevolutionary genomics of bacteria. Theor Popul Biol. 2002;61:435–447. doi: 10.1006/tpbi.2002.1588. [DOI] [PubMed] [Google Scholar]
- Jovelin R, Ajie BC, Phillips PC. Molecular evolution and quantitative variation for chemosensory behavior in the nematode genus Caenorhabditis. Mol Ecol. 2003;12:1325–1337. doi: 10.1046/j.1365-294x.2003.01805.x. [DOI] [PubMed] [Google Scholar]
- Jurka J. RepBase update: A database and an electronic journal of repetitive elements. Trends Genet. 2000;16:418–420. doi: 10.1016/s0168-9525(00)02093-x. [DOI] [PubMed] [Google Scholar]
- Kamath RS, Fraser AG, Dong Y, Poulin G, Durbin R, et al. Systematic functional analysis of the Caenorhabditis elegans genome using RNAi. Nature. 2003;421:231–237. doi: 10.1038/nature01278. [DOI] [PubMed] [Google Scholar]
- Kellis M, Patterson N, Endrizzi M, Birren B, Lander ES. Sequencing and comparison of yeast species to identify genes and regulatory elements. Nature. 2003;423:241–254. doi: 10.1038/nature01644. [DOI] [PubMed] [Google Scholar]
- Kent WJ, Zahler AM. Conservation, regulation, synteny, and introns in a large-scale C. briggsae–C. elegans genomic alignment. Genome Res. 2000;8:1115–1125. doi: 10.1101/gr.10.8.1115. [DOI] [PubMed] [Google Scholar]
- Kirouac M, Sternberg PW. cis-Regulatory control of three cell fate-specific genes in vulval organogenesis of Caenorhabditis elegans and C. briggsae. Dev Biol. 2003;257:85–103. doi: 10.1016/s0012-1606(03)00032-0. [DOI] [PubMed] [Google Scholar]
- Korf I, Flicek P, Duan D, Brent MR. Integrating genomic homology into gene structure prediction. 2001;(Bioinformatics 17 Suppl 1: S140–S148) doi: 10.1093/bioinformatics/17.suppl_1.s140. [DOI] [PubMed] [Google Scholar]
- Krause M, Harrison SW, Xu SQ, Chen L, Fire A. Elements regulating cell- and stage-specific expression of the C. elegans MyoD family homolog hlh-1. Dev Biol. 1994;166:133–148. doi: 10.1006/dbio.1994.1302. [DOI] [PubMed] [Google Scholar]
- Lau NC, Lim LP, Weinstein EG, Bartel DP. An abundant class of tiny RNAs with probable regulatory role in Caenorhabditis elegans. Science. 2001;294:858–862. doi: 10.1126/science.1065062. [DOI] [PubMed] [Google Scholar]
- Lee RC, Ambros V. An extensive class of small RNAs in Caenorhabditis elegans. Science. 2001;294:862–864. doi: 10.1126/science.1065329. [DOI] [PubMed] [Google Scholar]
- Lev-Maor G, Sorek R, Shomron N, Ast G. The birth of an alternatively spliced exon: 3′ Splice-site selection in Alu exons. Science. 2003;300:1288–1291. doi: 10.1126/science.1082588. [DOI] [PubMed] [Google Scholar]
- Lim LP, Lau NC, Weinstein EG, Abdelhakim A, Yekta S, et al. The microRNAs of Caenorhabditis elegans. Genes Dev. 2003;17:991–1008. doi: 10.1101/gad.1074403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lowe TM, Eddy SR. tRNAscan-SE: A program for improved detection of transfer RNA genes in genomic sequence. Nucleic Acids Res. 1997;25:955–964. doi: 10.1093/nar/25.5.955. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Long M. Evolution of novel genes. Curr Opin Genet Dev. 2001;11:673–680. doi: 10.1016/s0959-437x(00)00252-5. [DOI] [PubMed] [Google Scholar]
- Maeda I, Kohara Y, Yamamoto M, Sugimoto A. Large-scale analysis of gene function in Caenorhabditis elegans by high-throughput RNAi. Curr Biol. 2001;11:171–176. doi: 10.1016/s0960-9822(01)00052-5. [DOI] [PubMed] [Google Scholar]
- Marra MA, Kucaba TA, Dietrich NL, Green ED, Brownstein B, et al. High-throughput fingerprint analysis of large-insert clones. Genome Res. 1997;7:1072–1084. doi: 10.1101/gr.7.11.1072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marshall SD, McGhee JD. Coordination of ges-1 expression between the Caenorhabditis pharynx and intestine. Dev Biol. 2001;239:350–363. doi: 10.1006/dbio.2001.0442. [DOI] [PubMed] [Google Scholar]
- Mewes HW, Albermann K, Bahr M, Frishman D, Gleissner A, et al. Overview of the yeast genome. 1997;(Nature 387 Suppl: 7–65) doi: 10.1038/42755. [DOI] [PubMed] [Google Scholar]
- Morgenstern B. DIALIGN2: Improvement of the segment-to-segment approach to multiple sequence alignment. Bioinformatics. 1999;15:211–218. doi: 10.1093/bioinformatics/15.3.211. [DOI] [PubMed] [Google Scholar]
- Mullikin JC, Ning Z. The Phusion assembler. Genome Res. 2003;13:81–90. doi: 10.1101/gr.731003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Myers EW, Sutton GG, Delcher AL, Dew IM, Fasulo DP, et al. A whole-genome assembly of Drosophila. Science. 2000;287:2196–2204. doi: 10.1126/science.287.5461.2196. [DOI] [PubMed] [Google Scholar]
- Nei M, Gojobori T. Simple methods for estimating the numbers of synonymous and nonsynonymous nucleotide substitutions. Mol Biol Evol. 1986;3:418–426. doi: 10.1093/oxfordjournals.molbev.a040410. [DOI] [PubMed] [Google Scholar]
- Nelson DW, Honda BM. Two highly conserved transcribed regions in the 5S DNA repeats of the nematodes Caenorhabditis elegans and Caenorhabditis briggsae. Nucleic Acids Res. 1989;17:8657–8667. doi: 10.1093/nar/17.21.8657. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nigon V, Dougherty EC. Reproductive patterns and attempts at reciprocal crossing of Rhabditis elegans Maupas, 1900, and Rhabditis briggsae Dougherty & Nigon, 1949 (Nematoda: Rhabditidae) J Exp Zool. 1949;112:485–503. doi: 10.1002/jez.1401120307. [DOI] [PubMed] [Google Scholar]
- Ning Z, Cox AJ, Mullikin JC. SSAHA: A fast search method for large DNA databases. Genome Res. 2001;11:1725–1729. doi: 10.1101/gr.194201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Notredame C, Higgins DG, Heringa J. T-COFFEE: A novel method for fast and accurate multiple sequence alignment. J Mol Biol. 2000;302:205–217. doi: 10.1006/jmbi.2000.4042. [DOI] [PubMed] [Google Scholar]
- Pearson WR. Searching protein sequence libraries: Comparison of the sensitivity and selectivity of the Smith–Waterman and FASTA algorithms. Genomics. 1991;11:635–650. doi: 10.1016/0888-7543(91)90071-l. [DOI] [PubMed] [Google Scholar]
- Piano F, Gunsalus K. RNAi-based functional genomics in Caenorhabditis elegans. Curr Genomics. 2002;3:69–81. [Google Scholar]
- Plasterk RHA, von Luenen HGAM. Transposons. In: Riddle DL, Meyer BJ, Priess JR, editors. C. elegans II. Cold Spring Harbor, New York: Cold Spring Harbor Laboratory Press; 1997. pp. 97–116. [PubMed] [Google Scholar]
- Prasad SS, Harris LJ, Baillie DL, Rose AM. Evolutionarily conserved regions in Caenorhabditis transposable elements deduced by sequence comparison. Genome. 1991;34:6–12. doi: 10.1139/g91-002. [DOI] [PubMed] [Google Scholar]
- Ranz JM, Casals F, Ruiz A. How malleable is the eukaryotic genome?: Extreme rate of chromosomal rearrangement in the genus Drosophila. Genome Res. 2001;11:230–239. doi: 10.1101/gr.162901. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reboul J, Vaglio P, Rual JF, Lamesch P, Martinez M, et al. C. elegans ORFeome version 1.1: Experimental verification of the genome annotation and resource for proteome-scale protein expression. Nat Genet. 2003;34:35–41. doi: 10.1038/ng1140. [DOI] [PubMed] [Google Scholar]
- Reese MG, Hartzell G, Harris NL, Ohler U, Abril JF, et al. Genome annotation assessment in Drosophila melanogaster. Genome Res. 2000;10:483–501. doi: 10.1101/gr.10.4.483. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rice P, Longden I, Bleasby A. The European molecular biology open source suite. Trends Genet. 2000;16:276–277. doi: 10.1016/s0168-9525(00)02024-2. [DOI] [PubMed] [Google Scholar]
- Robertson HM. Updating the str and srj (stl) families of chemoreceptors in Caenorhabditis nematodes reveals frequent gene movement within and between chromosomes. Chem Senses. 2001;26:151–159. doi: 10.1093/chemse/26.2.151. [DOI] [PubMed] [Google Scholar]
- Rogic S, Ouellette BF, Mackworth AK. Improving gene recognition accuracy by combining predictions from two gene-finding programs. Bioinformatics. 2002;18:1034–1045. doi: 10.1093/bioinformatics/18.8.1034. [DOI] [PubMed] [Google Scholar]
- Roy SW, Fedorov A, Gilbert W. Large-scale comparison of intron positions in mammalian genes shows intron loss but no gain. Proc Natl Acad Sci U S A. 2003;100:7158–7162. doi: 10.1073/pnas.1232297100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Salamov AA, Solovyev VV. Ab initio gene finding in Drosophila genomic DNA. Genome Res. 2000;10:516–522. doi: 10.1101/gr.10.4.516. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sambrook J, Fritsch EF, Maniatis T. 2nd ed. Cold Spring Harbor, New York: Cold Spring Harbor Laboratory Press; 1989. Molecular cloning: A laboratory manual; pp. 1–659. [Google Scholar]
- Sanford C, Perry MD. Asymmetrically distributed oligonucleotide repeats in the Caenorhabditis elegans genome sequence that map to regions important for meiotic chromosome segregation. Nucleic Acids Res. 2001;29:2920–2926. doi: 10.1093/nar/29.14.2920. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sankoff D. Comparative mapping and genome rearrangement. In: Dekkers JCM, Lamont SJ, Rothschild MF, editors. From Jay Lush to genomics: Visions for animal breeding and genetics. Ames, Iowa: Iowa State University; 1999. pp. 124–134. [Google Scholar]
- Schwartz S, Kent WJ, Smit A, Zhang Z, Baertsch R, et al. Human–mouse alignments with BLASTZ. Genome Res. 2003;13:103–107. doi: 10.1101/gr.809403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shabalina SA, Kondrashov AS. Pattern of selective constraint in C. elegans and C. briggsae genomes. Genet Res. 1999;74:23–30. doi: 10.1017/s0016672399003821. [DOI] [PubMed] [Google Scholar]
- Sluder AE, Mathews SW, Hough D, Yin VP, Maina CV. The nuclear receptor superfamily has undergone extensive proliferation and diversification in nematodes. Genome Res. 1999;9:103–120. [PubMed] [Google Scholar]
- Smith TF, Waterman MS. Identification of common molecular subsequences. J Mol Biol. 1981;147:195–197. doi: 10.1016/0022-2836(81)90087-5. [DOI] [PubMed] [Google Scholar]
- Soderlund C, Longden I, Mott R. FPC: A system for building contigs from restriction fingerprinted clones. Comput Appl Biosci. 1997;13:523–535. doi: 10.1093/bioinformatics/13.5.523. [DOI] [PubMed] [Google Scholar]
- Soderlund C, Humphray S, Dunham A, French L. Contigs built with fingerprints, markers and FPCV4.7. Genome Res. 2000;10:1772–1787. doi: 10.1101/gr.gr-1375r. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stajich JE, Block D, Boulez K, Brenner SE, Chervitz SA, et al. The Bioperl toolkit: Perl modules for the life sciences. Genome Res. 2002;12:1611–1618. doi: 10.1101/gr.361602. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stankiewicz P, Lupski Genome architecture, rearrangements, and genomic disorders. Trends Genet. 2002;18:74–82. doi: 10.1016/s0168-9525(02)02592-1. [DOI] [PubMed] [Google Scholar]
- Stein L, Sternberg P, Durbin R, Thierry-Mieg J, Spieth J. WormBase: Network access to the genome and biology of Caenorhabditis elegans. Nucleic Acids Res. 2001;29:82–86. doi: 10.1093/nar/29.1.82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stothard P, Pilgrim D. Sex-determination gene and pathway evolution in nematodes. Bioessay. 2003;25:221–231. doi: 10.1002/bies.10239. [DOI] [PubMed] [Google Scholar]
- Sulston J, Mallett F, Staden R, Durbin R, Horsnell T, et al. Software for genome mapping by fingerprinting techniques. Comput Appl Biosci. 1988;4:125–132. doi: 10.1093/bioinformatics/4.1.125. [DOI] [PubMed] [Google Scholar]
- Tabata S, Kaneko T, Nakamura Y, Kotani H, Kato T, et al. Sequence and analysis of chromosome 5 of the plant Arabidopsis thaliana. Nature. 2000;408:823–826. doi: 10.1038/35048507. [DOI] [PubMed] [Google Scholar]
- Takezaki N, Rzhetsky A, Nei M. Phylogenetic test of the molecular clock and linearized trees. Mol Biol Evol. 1995;12:823–833. doi: 10.1093/oxfordjournals.molbev.a040259. [DOI] [PubMed] [Google Scholar]
- Thompson JD, Higgins DG, Gibson TJ. CLUSTAL W: Improving the sensitivity of progressive multiple sequence alignment through sequence weighting, positions-specific gap penalties, and weight matrix choice. Nucleic Acids Res. 1994;22:4673–4680. doi: 10.1093/nar/22.22.4673. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Van Dongen S. Performance criteria for graph clustering and Markov cluster experiments. In: Technical Report INS-R0012. National Research Institute for Mathematics and Computer Science in the Netherlands, Amsterdam. 2000 Available: http://ftp.cwi.nl//CWIreports/INS/INS-R0012.pdf. [Google Scholar]
- Waterston RH, Lindblad-Toh K, Birney E, Rogers J, Abril JF, et al. Initial sequencing and comparative analysis of the mouse genome. Nature. 2002;420:520–562. doi: 10.1038/nature01262. [DOI] [PubMed] [Google Scholar]
- Wang X, Chamberlin HM. Multiple regulatory changes contribute to the evolution of the Caenorhabditis lin-48 ovo gene. Genes Dev. 2002;16:2345–2349. doi: 10.1101/gad.996302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wendl M, Dear S, Hodgson D, Hillier L. Automated sequence preprocessing in a large-scale sequencing environment. Genome Res. 1998;8:975–984. doi: 10.1101/gr.8.9.975. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wootton JC, Federhen S. Analysis of compositionally biased regions in sequence databases. Methods Enzymol. 1996;266:554–571. doi: 10.1016/s0076-6879(96)66035-2. [DOI] [PubMed] [Google Scholar]
- Wuchty S, Fontana W, Hofacker IL, Schuster P. Complete suboptimal folding of RNA and the stability of secondary structures. Biopolymers. 1999;49:145–165. doi: 10.1002/(SICI)1097-0282(199902)49:2<145::AID-BIP4>3.0.CO;2-G. [DOI] [PubMed] [Google Scholar]
- Yang Z. PAML: A program package for phylogenetic analysis by maximum likelihood. Comput Appl Biosci. 1997;13:555–556. doi: 10.1093/bioinformatics/13.5.555. [DOI] [PubMed] [Google Scholar]
- Yang Z, Nielsen R, Golman N, Pederson AK. Codon-substitution models for heterogeneous selection pressure at amino-acid sites. Genetics. 2000;155:431–449. doi: 10.1093/genetics/155.1.431. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zdobnov EM, Apweiler R. InterProScan: An integration platform for the signature-recognition methods in InterPro. Bioinformatics. 2001;17:847–848. doi: 10.1093/bioinformatics/17.9.847. [DOI] [PubMed] [Google Scholar]
- Zdobnov EM, von Mering C, Letunic I, Torrents D, Suyama M, et al. Comparative genome and proteome analysis of Anopheles gambiae and Drosophila melanogaster. Science. 2002;298:149–159. doi: 10.1126/science.1077061. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
(64 MB GZ).
This directory comprises cb.hybrid.gff, a file of C. briggsae gene predictions using the “hybrid” method in GFF format (http://www.sanger.ac.uk/Software/formats/GFF/); cb.fa, file that contains conceptual translations of C. briggsae gene predictions in FASTA format; ws77.hybrid.gff, a file that contains C. elegans gene predictions using the “hybrid” method in GFF format; ws77.hybrid.fa, a file that contains C. elegans hybrid genes' conceptual translations in FASTA format; ws77.gff, a file that contains the canonical C. elegans gene predictions from Wormbase version WS77* (after filtering for transposase-containing genes and other artefacts); ws77.fa, a file that contains C. elegans WS77* genes' conceptual translations in FASTA format; gene2ip2go.briggsae, a file that contains InterPro annotations of predicted C. briggsae gene products, plus GO terms when available; and gene2ip2go.elegans, a file that contains InterPro annotations of predicted C. elegans gene products from WS77*, plus GO terms when available.
(20 MB ZIP).
This directory comprises orthologs.txt, a file that contains the list of C. briggsae/C. elegans ortholog pairs; orthologs-kaks.txt, a file that contains KA/KS values for ortholog pairs, as well as other information used to determine regional and functional variation in KA/KS; and cb_orphans and ce_orphans, files that contain lists of putative “orphan” genes that are found in one species but not in another.
(433 KB ZIP).
This directory comprises cb.waba_state.gff, a file that contains WABA alignments between C. briggsae and C. elegans; and cb.waba.gff, a file that contains WABA alignments in which transitions between adjacent WABA block types (“strong,” “weak” and “coding” ) have been merged.
(41.8 MB ZIP).
This directory comprises synteny_blocks.txt, a file that contains 4,837 synteny blocks identified between C. elegans and C. briggsae; and sorted_ultracontigs.txt, a file that contains 382 C. briggsae ultracontigs that have been ordered and oriented on the C. elegans genome relative to their largest synteny block.
(120 KB TXT).
This directory comprises tribe_cluster.txt, a file that contains TRIBE-MCL co-clusters of C. briggsae and C. elegans predicted proteins, with the clusters sorted with the largest first; tribe_cluster_classify_short.txt, a file that contains the Pfam and InterPro classification of co-clusters; and tribe_cluster_classify_long.txt, which is similar to the previous file, except that all InterPro/Pfam annotations are shown.
(360 KB ZIP).
This directory comprises Cb_repeat.lib, a FASTA-format file of RECON-predicted repeat elements from C. briggsae after removal of protein families; Ce_repeat.lib, a FASTA-format file of C. elegans predicted repeat elements; Cb_RECON_repeat.gff, a GFF-format file indicating the positions of predicted repeats in the C. briggsae genome; and Ce_RECON_repeat.gff, a GFF-format file indicating the positions of predicted repeats in the C. elegans genome.
(1.9 MB TXT).
The Genome Sequence of Caenorhabditis briggsae: A Platform for Comparative Genomics
The nematodes C. briggsae and C. elegans diverged 80–110 MYA, near to the time of divergence of human from mouse, but are almost indistinguishable morphologically. These figures demonstrate the high degree of morphological similarity between C. briggsae and C. elegans and show how the patterns of evolutionary conservation between the two species vary across the five autosomes (I–V) and sex chromosome (X) of C. elegans.
(50.6 MB PDF).