

Abstract
Efficient 3′-end processing of cell cycle-regulated mammalian histone premessenger RNAs (pre-mRNAs) requires an upstream stem–loop and a histone downstream element (HDE) that base pairs with the U7 small ribonuclearprotein. Insertions between these elements have two effects: the site of cleavage moves in concert with the HDE and processing efficiency declines. We used Xenopus oocytes to ask whether compensatory length insertions in the human U7 RNA could restore the fidelity and efficiency of processing of mouse histone insertion pre-mRNAs. An insertion of 5 nt into U7 RNA that extends its complementary to the HDE compensated for both defects in processing of a 5-nt insertion substrate; a noncomplementary insertion into U7 did not. Yet, the noncomplementary insertion mutant U7 was shown to be active on insertion substrates further mutated to allow base pairing. Our results suggest that the histone pre-mRNA becomes rigidified upstream of its HDE, allowing the bound U7 small ribonucleoprotein to measure from the HDE to the cleavage site. Such a mechanism may be common to other RNA measuring systems. To our knowledge, this is the first demonstration of length suppression in an RNA processing system.
Keywords: U7 small nuclear ribonucleoprotein, histone gene expression
Cell cycle-regulated histone premessenger RNAs (pre-mRNAs) are unusual RNA polymerase II transcripts because they do not contain introns and are not polyadenylylated (for review, see refs. 1 and 2). Instead, their 3′ ends are formed by endonucleolytic cleavage. Two conserved elements on the histone pre-mRNA are required for efficient processing: a stem–loop structure found directly upstream of the site of cleavage and a purine-rich region located 9–17 nt (usually 11 ± 1 nt, see ref. 3) downstream of the cleavage site termed the histone downstream element (HDE) (Fig. 1). There are at least three trans-acting factors involved in histone mRNA maturation: a factor that binds the stem–loop element (7–11), an essential heat-labile factor whose role is not yet understood (12–14), and the low-abundance U7 small nuclear ribonucleoprotein (snRNP) (for reviews, see refs. 15 and 16), which contains at least two proteins of 50 kDa and 14 kDa (17–19) in addition to the core Sm proteins (20). Genetic suppression experiments established that the 5′ end of the U7 RNA base pairs with the purines of the HDE during histone pre-mRNA processing (6, 21) (see Fig. 1); the ability to form an small nuclear RNA (snRNA)–pre-mRNA duplex, rather than the exact sequence of the complementary regions, is critical for processing.
Figure 1.
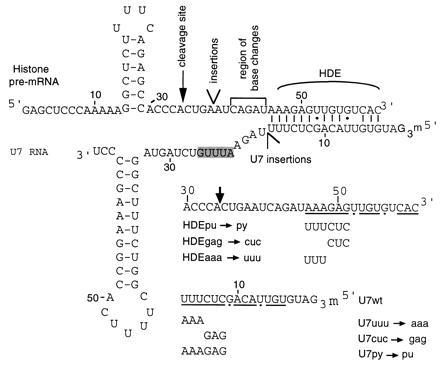
Diagram of mouse histone H2A pre-mRNA and human U7 RNA mutants. Partial sequence of the mouse histone H2A pre-mRNA (4), indicating the wild-type cleavage site, the site of insertions, the HDE residues that base pair to the U7 RNA, and the residues altered in the base substitution mutants. All histone pre-mRNA substrates contain an additional 14 nt of vector sequence at the 5′ end. The human U7 sequence (5) is shown with the Sm site shaded. Partial sequences of the base substitution HDE and U7 RNA suppressor mutants (6) are shown with altered residues dipicted below the wild-type sequence. Residues involved in base pairing to the U7 RNA or the histone pre-mRNA are underlined with G·U base pairs indicated by dots.
Previously, we reported the effects of systematically moving the HDE further downstream of the stem–loop in a mouse histone H2A pre-mRNA (22). The processed products generated in a HeLa cell in vitro system revealed that the site of 3′-end formation moves in concert with the movement of the HDE, remaining 11 ± 1 nt upstream. Decreases in processing efficiency were also observed as the HDE was distanced from the stem–loop. These results suggested that the U7 snRNP directs cleavage at a fixed distance from its binding site (termed the “molecular ruler” effect) and that a particular geometry of the processing complex is necessary for efficient cleavage.
Herein we have subjected the molecular ruler model to a critical test by asking whether compensatory length insertions in human U7 RNA can restore the fidelity and efficiency of processing of histone insertion mutant pre-mRNAs in the Xenopus oocyte. Our ability to obtain excellent length suppression of both the site and efficiency of cleavage confirms the existence of a measuring device in histone mRNA 3′-end maturation. We present a model for the processing complex in which U7 snRNP-associated protein(s) interact with the substrate in the region between the HDE and the cut site, as well as with the stem–loop, to correctly align the cleavage activity.
MATERIALS AND METHODS
Construction of Histone Pre-mRNA and U7 RNA Mutants.
A fragment of the mouse H2A-614 gene (4) was the wild-type substrate (HDEwt in ref. 6). Clones containing mutations were constructed using complementary deoxyoligonucleotides synthesized on an Applied Biosystems DNA synthesizer (W.M. Keck Biotechnology Resource Laboratory Oligonucleotide Synthesis Facility, Yale University). Template deoxyoligonucleotides for the histone pre-mRNA base substitution mutants had the sequences 5′-GCGCGTCTAGAACACTCTTACTTTTGGGGCAGGGCCAGGAAAGGCCAGGAAGACCGGCTACCGTGACACAACTCTTTXATGGGGGTCAGTGGGTGGCTCTGAAAGAGCCTTTTTGGGAGCT-3′ (where X is CCCTG, CCGTG, and CCGCC for 5C+2bp, 5C+3bp, 5C+5bp, respectively). The italicized guanylates produce an insertion of five cytidylate residues that is not present in the wild-type sequence. The primer used for extension by Taq polymerase (Perkin–Elmer) was 5′-CCCAAAAAGGCTCTTTTCAG-3′. The cloning procedure followed is described in ref. 22. HDEpu→py was described in ref. 6.
Human U7 RNA suppressor mutations were generated from overlapping deoxyoligonucleotides using standard cloning procedures (23). The following deoxyoligonucleotides were used: for mutants U7py→pu, U7cuc→gag, and U7 uuu→aaa, sense strand 5′-AGCTTAATACGACTCACTATAGGGTGTTACAGXTAGAATTTGTCTAGTAGGCTTTCTGGCTTTTCACCGGAAAGCCCC-3′ [where X is GAGAAA (U7py→pu), GAGTTT (U7cuc→gag), or CTCAAA (U7 uuu→aaa)], antisense strand 5′-GGGGCTTTCCGGTGAAAAGCCAGAAAGCCTACTAGACAAATTCTAXCTGTAACACCCTATAGTGACTCGTATTA-3′ [where X is TTTCTC (U7py→pu), AAACTC (U7cuc→gag), or TTTGAG (U7 uuu→aaa)]; for mutants U75bp, U75nobp, and U710, sense strand 5′-AGCTTAATACGACTCACTATAGGGTGTTACAGCTCTTTXTAGAATTTGTCTAGTAGGCTTTCTGGCTTTTCACCGGAAAGCCCC-3′ [where X is ATCTG (U75BP), CCGCC (U75nobp), or ATCTGCCCCC (U710)], antisense strand 5′-GGGGCTTTCCGGTGAAAAGCCAGAAAGCCTACTAGACAAATTCTAXAAAGAGCTGTAACACCCTATAGTGAGTCGTATTA-3′ [where X is CAGAT (U75bp), GGCGG (U75nobp), and GGGGGCAGAT (U710]. The double-stranded fragments were then cloned into a SmaI/HindIII-digested pSP65 vector (Promega). Mutants U7 uuu→aaa, U7cuc→gcg, and U7py→pu (6) were recloned as described above so that the correct 3′ end would be generated by in vitro transcription, using T7 RNA polymerase.
U7 transcription templates were generated by digestion with SmaI and T7 RNA polymerase run-off transcripts were primed with G(5′)ppp(5′)G. Histone transcription templates were generated by digestion with MaeIII and T3 RNA polymerase run-off transcripts were primed with G(5′)ppp(5′)G.
Nuclear Extracts and Processing Reactions.
Nuclear extracts were prepared as described (22) from HeLa cells obtained from the National Cell Culture Center (Coon Rapids, MN). In vitro processing was performed (22) for 1 h at 30°C. Tris·HCl (pH 7.5), EDTA, SDS and proteinase K were added to a final concentration of 10 mM, 25 mM, 0.5%, and 1 μg/μl, respectively, and incubated for 15 min at 65°C. Isolated RNAs were analyzed adjacent to RNAs isolated from oocytes on 8% polyacrylamide/8.3 M urea gels. MspI-digested pBR322 DNA provided size markers.
Oocyte Injections and RNA Isolation.
Oocytes for injection, stage 5 and early stage 6, were manually separated from Xenopus laevis ovaries in modified Barth’s saline (24, 25). Oocytes were cytoplasmically injected with 32.2 nl of antisense deoxyoligonucleotide (5′-AAGAGCTGTAACACTT-3′; 2 μg/μl), complementary to 16 nt at the 5′ end of Xenopus U7 RNA (26). After a 3- to 4-h incubation at 18°C, oocytes were cytoplasmically injected with 32.2 nl of various U7 RNAs at a concentration of 10 ng/μl and incubated at 18°C overnight (10–12 h). Then, 13.8 nl of various [α-32P]UTP-labeled histone transcripts at a concentration of 5–10 ng/μl were injected into the germinal vesicle (GV). Three to 4 h later, GVs were hand-isolated and RNA was recovered essentially as described (27), except that proteinase K at 400 μg/ml was added and tubes were incubated at 65°C for 5–15 min. Samples were extracted with phenol, phenol/chloroform/isoamyl alcohol, and precipitated with ethanol. Four to six GV equivalents of RNA were loaded per lane on an 8% polyacrylamide/8.3 M urea gel.
RESULTS
Processing of Insertion Mutant Histone Pre-mRNAs and U7 Suppression Activity in Xenopus Oocytes.
Xenopus laevis oocytes provide the opportunity to target a particular endogenous snRNP for destruction and then assess the activity and particle assembly of an in vitro transcribed snRNA introduced by microinjection (27). We have adopted this procedure for the study of histone pre-mRNA processing. The first step is a cytoplasmic injection of a deoxyoligonucleotide complementary to 16 nt at the 5′ end of Xenopus U7 RNA (26). After 3 h, sufficient for the antisense deoxyoligonucleotide to target degradation of the pool of Xenopus U7 by endogenous RNase H (28, 29) and for endogenous DNase to degrade the antisense deoxyoligonucleotide (data not shown), in vitro-transcribed human U7 RNA is microinjected into the oocyte cytoplasm. Following an overnight incubation, which allows U7 snRNP assembly and transport to the nucleus, [α-32P]UTP-labeled histone pre-mRNA derived from the mouse H2A-614 gene (4, 6) is injected into the GV (nucleus). Three to 4 h later the GVs are collected, and RNA is isolated and analyzed. Successful destruction of endogenous U7 RNA can be confirmed by the absence of a U7 primer extension product and by lack of processing of subsequently injected wild-type histone pre-mRNA.
To ask whether processing in the oocyte produces the same pattern of alternate cleavages of mouse histone pre-mRNA insertion mutants as had been documented in HeLa cell extracts in vitro (22), the identical mutant substrates (see Fig. 1) were injected into untreated oocytes or U7-depleted oocytes reconstituted with human U7 RNA (Fig. 2A). Untreated oocytes (lanes 11–20) yielded similar products to those obtained in the HeLa in vitro system (lanes 1–10). The only exception is the 5C insertion mutant, where the oocyte products (lane 15) migrate slightly faster than the in vitro product (lane 5). Lanes 31–40, oocytes depleted of endogenous U7 RNA, show that endogenous processing activity was almost completely abolished by the antisense deoxyoligonucleotide, although there is considerable nonspecific degradation of the substrates in the oocyte. In oocytes subsequently rescued with human U7 RNA prior to histone pre-mRNA addition (lanes 21–30), the patterns of processing of the histone insertion mutant substrates closely mimic those of in vitro processing (lanes 1–10) and untreated oocytes (lanes 11–20). Even though variability in injection efficiency is an inherent disadvantage of the oocyte system, the relative levels of products observed for the various insertion mutants reflect those detected in vitro. Processing efficiency in general decreases as the HDE is moved farther from the stem–loop; interestingly, the 5Cswap and 9C-A9 (which has an adenylate residue inserted upstream of the cleavage site) are not cleaved significantly better than 5C and 9C in the oocyte, as they are in the HeLa cell extract (compare lanes 6 and 9 with lanes 16, 19, 26, and 29). We conclude that human U7 RNA can assemble with Xenopus proteins to produce a snRNP that processes mouse histone insertion mutant pre-mRNAs equivalently to the Xenopus U7 snRNP and similarly to HeLa cell nuclear extract.
Figure 2.
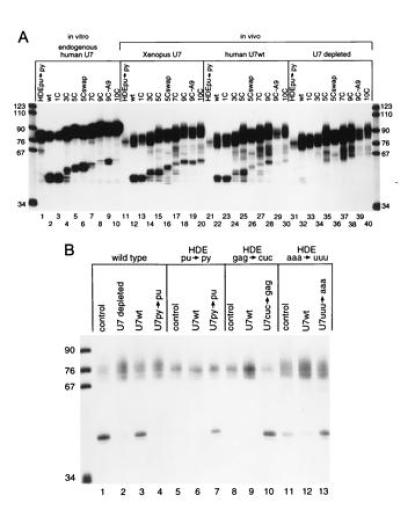
In vitro and oocyte processing of histone pre-mRNA insertion mutants and HDE base substitution mutants. (A) Internally labeled insertion substrates were processed in HeLa cell nuclear extract (lanes 1–10) or injected into the GV of untreated Xenopus oocytes (lanes 11–20), of oocytes where the endogenous Xenopus U7 RNA was degraded and subsequently rescued by injection of in vitro-transcribed human U7 RNA (lanes 21–30), or oocytes where the endogenous Xenopus U7 RNA had been degraded (lanes 31–40). Note that lane 29 was underloaded in this particular experiment. In at least six different experiments, progressive decreases in processing efficiency were observed with longer insertions. (B) Suppression of substitution mutations in the HDE in Xenopus oocytes. Wild-type histone pre-mRNA was injected into untreated oocytes (lane 1), oocytes depleted of U7 RNA (lane 2), depleted oocytes rescued with U7wt RNA (lane 3), or depleted oocytes rescued with U7py→pu (lane 4). In lanes 5–7, the HDEpu-py substrate was injected into untreated oocytes, U7wt rescued oocytes, or U7py→py rescued oocytes. In lanes 8–10, the HDEgag→cuc mutant was injected into untreated, U7wt rescued, or U7cuc→gag rescued oocytes. In lanes 11–13, the HDEaaa→uuu mutant was injected into untreated, U7wt rescued, or U7 uuu→aaa rescued oocytes.
To establish whether depleted Xenopus oocytes could be used for genetic suppression studies of mammalian histone pre-mRNA maturation, we next reproduced the suppression results of Bond et al. (6) in transfected HeLa cells (Fig. 2B). The wild-type substrate was cleaved in U7-depleted oocytes rescued with wild-type human U7 RNA (U7wt) (lane 3) but not with U7py→pu, where six of the pyrimidines that base pair with the HDE of the histone pre-mRNA are changed to purines (UUUCUC to AAAGAG, Fig. 1) (Fig. 2B, lane 4). The HDEpu→py histone pre-mRNA, where six purines of the HDE have been changed to pyrimidines (Fig. 1), was not detectably processed by U7wt (Fig. 2B, lane 6) but is very efficiently cleaved in the presence of the suppressor U7py→pu (lane 7), which restores the Watson–Crick base pairing potential between the HDE and the 5′ end of the U7 RNA (Fig. 1). Furthermore, mutation of the GAG of the HDE (HDEgag→cuc, Fig. 1) severely inhibited processing in both the oocyte and HeLa cells and was likewise well suppressed by U7cuc→gag in the oocyte (Fig. 2B, compare lanes 9 and 10). HDEaaa→uuu showed diminished processing relative to wild-type histone pre-mRNA in the presence of U7wt, as reported for HeLa cells (compare lanes 3 and 12). However, in the oocyte, the suppressor U7 uuu→aaa completely rescued processing of HDEaaa→uuu (compare lanes 12 and 13), whereas in HeLa cells suppression was only partial (6). Bond et al. (6) proposed that the endogenous (wild-type) U7 snRNP in HeLa cells might bind to the HDEaaa→uuu pre-mRNA to form inactive processing complexes, which would then interfere with the ability of U7 uuu→aaa to suppress. This explanation is consistent with our results: more efficient suppression resulted when the endogenous U7 RNA had been destroyed in the oocyte and could not compete with the injected snRNA being assessed for activity.
A 5C Insertion in Histone Pre-mRNA Is Rescued by Inclusion of Five Complementary Nucleotides in U7 RNA.
We then asked whether insertions into the human U7 RNA could restore the wild-type site and level of cleavage of histone H2A insertion mutant pre-mRNAs. Two different 5-nt insertions (Fig. 3A) were made directly 3′ to the U7 nucleotides that base pair to the HDE of the histone substrate (between nt 17 and 18) upstream of the Sm binding site (shaded in Fig. 1A). One U7 insertion mutation, called U75bp, contains 5 nt complementary to the five residues immediately upstream of the HDE of the H2A substrate, while U75nobp contains five inserted nucleotides without potential to base pair with the pre-mRNA.
Figure 3.
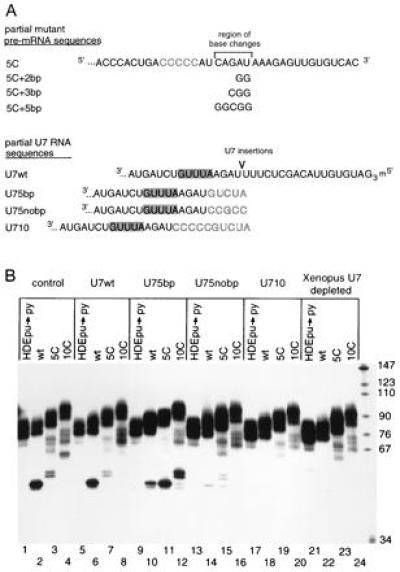
Processing of histone insertion mutant pre-mRNAs in the presence of length suppressor U7 RNAs. (A) Partial sequences of histone insertion substitution mutants and U7 RNA length suppressor mutants. The 5C insertion in the histone pre-mRNA is indicated by block letters while further base substitutions in this substrate are listed below. The point of insertion into U7 RNA is marked, with the inserted residues in blocked residues and the Sm site shaded. (B) In lanes 1–4, histone pre-mRNAs HDEpu→py, wild-type, 5C, and 10C were injected into untreated oocytes. The same histone pre-mRNAs were injected into depleted oocytes rescued with the following material. Lanes: 5–8, in vitro-transcribed U7wt RNA; 9–12, U75bp RNA; 13–16, U75nobp RNA; 17–20, U710 RNA; 21–24, not rescued.
When depleted oocytes (Fig. 3B, lanes 21–24) were complemented with U75bp (lanes 9–12), processing of the 5C insertion mutant was rescued both with respect to the site of cleavage and the efficiency of cleavage (compare lane 11 to lanes 2, 3, 6, and 7). The 10C insertion substrate (lane 12) gave similar results: it was cleaved to a much greater extent (compare with lanes 4 and 8) and its products migrated faster than those generated by U7wt. The sizes of the 10C cleavage products (lane 12) in the presence of U75bp were comparable to those of the processed 5C histone substrate in the presence of U7wt (lane 7), as expected. We conclude that both the fidelity and efficiency of processing of these histone insertion mutant pre-mRNAs can be rescued by a length suppressor U7 snRNA.
In contrast, U75nobp, whose 5-nt insertion is not complementary to the histone pre-mRNA sequence upstream of the HDE, was severely compromised in its ability to promote processing (Fig. 3B, lanes 13–16). Yet, it assembled into an anti-Sm precipitable particle, contained a hypermethylated cap, and was imported into the nucleus (data not shown). Two possible explanations for its lack of function were considered: (i) the increased stability of interaction between the pre-mRNA and the U75bp snRNP compared with the U75nobp particle is essential for length suppression or (ii) the particular nucleotides that have been inserted into U75nobp nonspecifically disrupt snRNP function.
Functionality of the U75nobp snRNP Can Be Demonstrated with Mutant 5C Histone Pre-mRNA Substrates.
To determine whether the difference in the ability of U75bp and U75nobp to rescue processing of the 5C substrate was due simply to the relative stability of their base pairing interactions with the substrate, U710 was generated (Fig. 3A). This construct contains a 10-nt insertion immediately 3′ to the U7 region that base pairs to the HDE; the 5′-most nucleotides of the insertion are identical to those inserted into U75bp, while the next 5 nt are identical to those inserted into U75nobp, except that the guanosine residue is replaced by cytosine to prevent potential base pairing (see Fig. 3A). Despite the extended base pairing interaction with the substrate expected for this mutant U7 RNA, U710 was incapable of rescuing the processing of either the 5C or 10C pre-mRNA (Fig. 3B, lanes 17–20). This suggests that the inability of U75nobp to suppress processing of the 5C substrate originates from some property of the inserted nucleotides: either their inability to base pair with the pre-mRNA substrate or some nonspecific deleterious effect on the reconstituted U7 snRNP particle.
To rule out a nonspecific effect of the particular nucleotides inserted into U75nobp, three additional base substitutions were made in the 5C histone pre-mRNA substrate. 5C+2bp, 5C+3bp, and 5C+5bp pre-mRNAs allow two, three, and five contiguous base pairs, respectively, to form with the 5-nt insertion of U75nobp, thereby stepwise extending the HDE duplex (Fig. 3A). Each new histone pre-mRNA substrate was first tested for processing in the HeLa in vitro extract (Fig. 4, lanes 1–6). The 5C+2bp and 5C+3bp substrates yielded products and levels of processing comparable to those of 5C (compare lanes 3 with lanes 4 and 5), as expected, while 5C+5bp was surprisingly not cleaved (lane 6). In the oocyte, similar patterns for the former two base substitution mutants were observed when processing was promoted by either the endogenous Xenopus (lanes 9–11) or complementing wild-type human U7 snRNA (lanes 15–17). 5C+5bp showed low levels of product in vivo in the presence of Xenopus or human U7 RNA (lanes 12 and 18) (see below).
Figure 4.
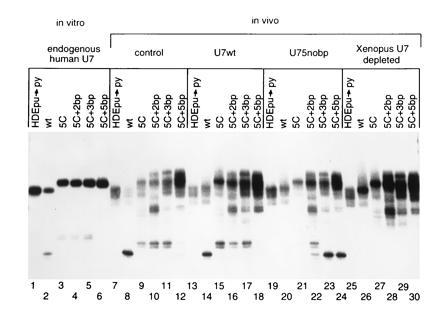
Processing of 5C insertion/base substitution mutants in the presence of U75nobp. Lanes 1–6 contain in vitro processing of pre-mRNAs HDEpu→py, wild-type, 5C, 5C+2bp, 5C+3bp, and 5C+5bp, respectively. The same substrates were injected into the following oocytes. Lanes: 7–12, untreated Xenopus oocytes; 13–18, oocytes depleted of Xenopus U7 RNA and rescued with U7wt RNA; 19–24, depleted oocytes rescued with U75nobp RNA; 25–30, depleted oocytes.
Processing of the base substitution 5C substrates in the oocyte then revealed that U75nobp does assemble into an active snRNP particle (Fig. 4, lanes 22–24). When only two of the five inserted nucleotides of U75nobp were complementary to the histone pre-mRNA (lane 22), partial suppression corresponding to processing at both the 5C site and the wild-type site was observed. When either three or all five of the inserted residues in U75nobp could base pair to the substrate, both the site and level of processing were restored to normal (lanes 23 and 24). This complete suppression demonstrates that the functionality of U75nobp can be observed by assaying it with the correct substrates. These experiments also suggest that at least three out of five of the inserted nucleotides must base pair to give complete length suppression. It is important to note that 5C+5bp was fully suppressed by U75nobp (lane 24), demonstrating that it can be an active substrate even though it was poorly cleaved by the U7wt (lanes 12 and 18).
DISCUSSION
We have confirmed the validity of the “molecular ruler” model for 3′-end processing of histone pre-mRNAs by demonstrating that the deleterious effects of insertions into the mouse H2A substrate can be suppressed by comparable length insertions into human U7 RNA. We observed length suppression of two histone pre-mRNA insertion mutants, 5C and 10C (Fig. 3B). With the U75bp mutant, which lengthens the U7–HDE duplex by 5 bp, the cleavage site moved 5 nt upstream and the efficiency increased significantly for both substrates. With the U75nobp mutant, suppression was optimal if the insertion could form 3 or 5 bp with the substrate (Figs. 3B and 5); 2 bp generated partial suppression, while no base pairs showed little processing activity and no length suppression (Fig. 4). The U7 insertion mutants all showed severely diminished processing on the wild-type (normal length) substrate (Figs. 3B and 4). We discuss a model for the processing complex that includes rigidification of the nonconserved histone pre-mRNA residues upstream of the HDE and targeting of the cleavage factor to a specific site by the bound U7 snRNP stimulated by interactions with the stem–loop.
Figure 5.
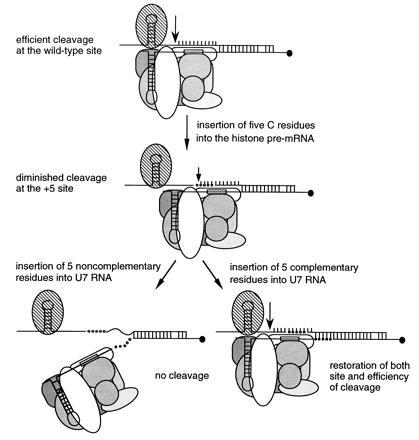
Models of histone pre-mRNA processing complexes. The wild-type situation is compared with complexes formed on the 5C insertion substrate with the wild-type U7 snRNP, U75nobp snRNP, and the U75bp snRNP. Arrows show the sites of cleavage with levels indicated by arrow size. SLBP is shown as a striped oval, core Sm proteins bound to the Sm binding site (boxed) are different shades of gray, the two known U7 snRNP specific proteins are white ovals, and the hypermethylated cap on the 5′ end of U7 RNA is a solid circle. Base pairing between the U7 RNAs and the substrate HDE sequences is indicated. Insertions into the histone pre-mRNA and the U7 RNAs are illustrated as open and solid circles, respectively. Rigidification of the residues upstream of the HDE is diagrammed as involving protein–backbone contacts (see text).
Interactions Critical for Efficient Histone Pre-mRNA Processing.
Based on results presented herein and previously, we propose that three critical interactions between the histone pre-mRNA substrate and the processing machinery dictate the site of cleavage and the efficiency of processing (Fig. 5). The first is base pairing between the U7 RNA and the HDE of the histone pre-mRNA (6, 20–22, 30–32). The second is contacts between the U7 snRNP and the stem–loop with its associated factors. An increase in the distance between these two interactions can be tolerated, but only with certain constraints. As revealed by our length suppression experiments, rigidification of the substrate between the stem–loop and the snRNP binding site contributes a third set of critical interactions.
We observed that simply increasing the length (and hence the stability) of the base pairing interaction with the U7 snRNP is not sufficient to overcome the negative effects of insertions into the histone pre-mRNA: the 10C insertion was not rescued by U710, which can form the same extended U7–HDE duplex as the 5C insertion coupled with U75bp (Fig. 3B). Apparently, the five non-base-pairing nucleotides in U710 are deleterious. This suggests that a required geometry of the processing complex is disrupted by insertions into U7 RNA that allow too much conformational flexibility of the snRNP particle in the region upstream of its Sm site (see Fig. 5).
Much circumstantial evidence supports essential interactions between the U7 snRNP and the conserved stem–loop element of histone pre-mRNAs. Deletion of the stem–loop causes nearly complete loss of substrate activity (8, 33, 34), whereas in the absence of a downstream HDE the stem–loop alone does not promote 3′-end formation (33, 35–37). Interactions are apparently mediated by a bound protein (stem–loop binding protein) since competition with oligonucleotides that mimic the stem–loop reduce processing to the same extent (3, 10) as the introduction of mutations into the conserved loop sequence that is essential for binding of this 45-kDa protein (9, 38–40). Such contacts between the stem–loop and its factors and the U7 snRNP could contribute to efficient cleavage by properly orienting the cleavage factor or by creating a binding site for a cleavage stimulatory factor.
If independent interactions are made by the U7 snRNP with the HDE, on the one hand, and the stem–loop, on the other, one might expect that insertions made into the intervening region of the histone pre-mRNA, which is nonconserved and presumably single stranded, could simply loop out to allow efficient cleavage at the wild-type site. This does not occur, arguing instead that in the processing complex the pre-mRNA becomes rigidified upstream of the duplex it forms with the U7 snRNP; since this sequence is not conserved, nonspecific backbone interactions are predicted (see Fig. 5).
Nature of the “Molecular Ruler.”
The component(s) that rigidify the histone pre-mRNA through nonspecific backbone interactions could be either a tightly bound U7 snRNP protein (see Fig. 5) or a non-snRNP component assembled by the snRNP–substrate interaction. The U7-specific Sm site may play a role in directing binding of such a protein(s) since substitution of the U7 Sm site with a U1 Sm site disrupts U7 snRNP’s ability to participate in histone pre-mRNA 3′-end processing (41). On the other hand, the assembly of the molecular measuring device is not dependent on the hypermethylated cap; replacement of the GpppG cap of U7 with ApppG does not alter suppression activity (unpublished observations). Likewise, we can conclude that measurement does not occur strictly from the proximal end of the duplex formed by U7–HDE base pairing. First, our results with U75nobp and base substitution mutant substrates (Fig. 4) demonstrate that variation in the distance from the end of the duplex to the cleavage site is allowed. Second, the HDEaaa→uuu mutant (Fig. 1) is processed at the normal site in both transfected HeLa cells (6) and the oocyte (Fig. 2B, lanes 11–13), despite the putative disruption of three proximal base pairs of the duplex formed with wild-type U7 RNA. Since uracil residues are known to form non-Watson–Crick interactions, we tested processing of the wild-type histone pre-mRNA in the presence of U7 uuu→aaa, which would form three less stable A·A base pairs. Even with this snRNA–substrate combination, in which the end of the U7 HDE duplex should be moved 3 nt downstream, the wild-type cleavage site was used.
The minimum length of histone pre-mRNA substrate requiring rigidification would be ≈35 Å if the nonconserved 11 nt between the cut site and the HDE become stacked, as in a helix. This distance could be spanned by a protein α-helix of 24 residues, as visualized in the binding of tRNASer to its aminoacyl-tRNA synthetase (42), or by interactions with β-sheet residues, as in certain RNA viruses (43). Since the mammalian U7 RNA in its snRNP is well protected from micrococcal nuclease digestion beyond 21 nt from its 5′ end (44, 45), the protecting protein(s) is appropriately located and could itself be the rigidifying component.
Our model (Fig. 5) would explain why insertions into the U7 RNA close to the nuclease protection boundary, if they are not conformationally restricted by base pairing to the substrate, could interfere with either assembly of the rigidifying component or effective interactions between the U7 snRNP containing the rigidifying component and the pre-mRNA. Crosslinking experiments aimed at identifying the cleavage factor and the rigidifying component (presumably a protein) should establish their relationship to the U7 snRNP.
Measuring Devices in RNA Processing.
There are several other RNA processing events where catalysis occurs a fixed distance from a recognition element in the substrate. In tRNA splicing, the endonuclease cuts at sites measured along the folded tRNA (46–48), an A-form helix. Also, in 5′-end maturation of tRNAs by RNase P, the cleavage site in most cases appears to be selected by measuring along the length of the acceptor duplex, probably including the T-stem (for reviews, see refs. 49–51). In apolipoprotein B editing, the distance between a mooring sequence and the editing site is critical for efficient editing (52), suggesting that the processing proteins (53–56) specifically structure the intervening single-stranded RNA. Likewise, the length of the poly(A) tail added to most mRNAs is intriguingly restricted to about 200 nt (57, 58); but how measurement occurs is not known. Ribose methylation of preribosomal RNA requires small nucleolar RNAs (snoRNAs) (59), which exhibit extensive complementarity (greater than 10 bp) to a region of 18S or 28S rRNA and target the rRNA nucleotide positioned five residues upstream of a cis-element (box D or D′) on the bound snoRNA, clearly implicating a measuring device.
Classically, length suppression was achieved in a translation system by a frameshift suppressor tRNA with an extra nucleotide in the anticodon loop, which restored the reading frame by causing a 4-nt translocation of the mRNA on the ribosome (60). No length suppression experiments have yet been reported for any other of the above RNA processing systems but may reveal commonalities among these systems and provide further insights into the functioning of RNA-based measuring devices.
Acknowledgments
We thank V. Myer, T. McConnell, and the rest of the Steitz lab for stimulating discussions and advice. Special thanks go to B. Peculis and M.-D. Shu for sharing their Xenopus oocyte expertise, and to T. Chang, T. McConnell, J. Mermoud, A. Parker, and B. Peculis for critical reading of this manuscript. This research was supported by Grant GM26514 from the National Institutes of Health.
Footnotes
Abbreviations: snRNP, small nuclear ribonucleoprotein; snRNA, small nuclear RNA; pre-mRNA, premessenger RNA; GV, germinal vesicle; HDE, histone downstream element.
References
- 1.Marzluff W F. Gene Expression. 1992;2:93–97. [PMC free article] [PubMed] [Google Scholar]
- 2.Wittop K T, Schümperli D. Eur J Biochem. 1994;219:25–42. doi: 10.1007/978-3-642-79502-2_3. [DOI] [PubMed] [Google Scholar]
- 3.Cho D C, Scharl E C, Steitz J A. RNA. 1995;1:905–914. [PMC free article] [PubMed] [Google Scholar]
- 4.Hurt M M, Chodchoy N, Marzluff W F. Nucleic Acids Res. 1989;17:8876. doi: 10.1093/nar/17.21.8876. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Mowry K L, Steitz J A. Science. 1987b;238:1682–1687. doi: 10.1126/science.2825355. [DOI] [PubMed] [Google Scholar]
- 6.Bond U M, Yario T A, Steitz J A. Genes Dev. 1991;5:1709–1722. doi: 10.1101/gad.5.9.1709. [DOI] [PubMed] [Google Scholar]
- 7.Mowry K L, Steitz J A. Mol Cell Biol. 1987a;7:1663–1672. doi: 10.1128/mcb.7.5.1663. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Vasserot A P, Schaufele F J, Birnstiel M L. Proc Natl Acad Sci USA. 1989;86:4345–4349. doi: 10.1073/pnas.86.12.4345. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Pandey N B, Sun J H, Marzluff W F. Nucleic Acids Res. 1991;19:5653–5659. doi: 10.1093/nar/19.20.5653. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Dominski Z, Sumerel J, Hanson R J, Marzluff W F. RNA. 1995;1:915–923. [PMC free article] [PubMed] [Google Scholar]
- 11.Hanson R J, Sun J H, Willis D G, Marzluff W F. Biochemistry. 1996;35:2146–2156. doi: 10.1021/bi9521856. [DOI] [PubMed] [Google Scholar]
- 12.Gick O, Kramer A, Vasserot A, Birnstiel M L. Proc Natl Acad Sci USA. 1987;84:8937–8940. doi: 10.1073/pnas.84.24.8937. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Lüscher B, Schümperli D. EMBO J. 1987;6:1721–1726. doi: 10.1002/j.1460-2075.1987.tb02423.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Larson D E, Hoffmann I, Zahradka P, Birnstiel M L, Sells B H. Biochim Biophys Acta. 1992;1131:139–144. doi: 10.1016/0167-4781(92)90068-b. [DOI] [PubMed] [Google Scholar]
- 15.Birnstiel M L, Schaufele F J. Structure and Function of Minor snRNPs. Berlin: Springer; 1988. [Google Scholar]
- 16.Mowry K L, Steitz J A. Trends Biochem Sci. 1988;13:447–451. doi: 10.1016/0968-0004(88)90220-4. [DOI] [PubMed] [Google Scholar]
- 17.Smith H O, Tabiti K, Schaffner G, Soldati D, Albrecht U, Birnstiel M L. Proc Natl Acad Sci USA. 1991;88:9784–9788. doi: 10.1073/pnas.88.21.9784. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Mital R, Albrecht U, Schümperli D. Nucleic Acids Res. 1993;21:1049–1050. doi: 10.1093/nar/21.4.1049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Stefanovic B, Hackl W, Lührmann R, Schümperli D. Nucleic Acids Res. 1995;23:3141–3151. doi: 10.1093/nar/23.16.3141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Strub K, Birnstiel M L. EMBO J. 1986;5:1675–1682. doi: 10.1002/j.1460-2075.1986.tb04411.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Schaufele F, Gilmartin G M, Bannwarth W, Birnstiel M L. Nature (London) 1986;323:777–781. doi: 10.1038/323777a0. [DOI] [PubMed] [Google Scholar]
- 22.Scharl E C, Steitz J A. EMBO J. 1994;13:2432–2440. doi: 10.1002/j.1460-2075.1994.tb06528.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Maniatis T, Fritsch E F, Sambrook J. Molecular Cloning: A Laboratory Manual. Cold Spring Harbor, NY: Cold Spring Harbor Lab. Press; 1982. [Google Scholar]
- 24.Peng H B. Methods Cell Biol. 1991;36:657–662. [PubMed] [Google Scholar]
- 25.Smith L D, Xu W, Varnold R L. Methods Cell Biol. 1991;36:45–60. doi: 10.1016/s0091-679x(08)60272-1. [DOI] [PubMed] [Google Scholar]
- 26.Phillips S C, Birnstiel M L. Biochim Biophys Acta. 1992;1131:95–98. doi: 10.1016/0167-4781(92)90104-8. [DOI] [PubMed] [Google Scholar]
- 27.Peculis B A, Steitz J A. Cell. 1993;73:1233–1245. doi: 10.1016/0092-8674(93)90651-6. [DOI] [PubMed] [Google Scholar]
- 28.Hamm J, Dathan N A, Mattaj I W. Cell. 1989;59:159–169. doi: 10.1016/0092-8674(89)90878-7. [DOI] [PubMed] [Google Scholar]
- 29.Prives C, Foukal D. Methods Cell Biol. 1991;36:185–210. doi: 10.1016/s0091-679x(08)60278-2. [DOI] [PubMed] [Google Scholar]
- 30.Cotton M, Gick O, Vasserot A, Schaffner G, Birnstiel M L. EMBO J. 1988;7:801–808. doi: 10.1002/j.1460-2075.1988.tb02878.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Melin L, Soldati D, Mital R, Streit A, Schümperli D. EMBO J. 1992;11:691–697. doi: 10.1002/j.1460-2075.1992.tb05101.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Spycher C, Streit A, Stefanovic B, Albrecht D, Koning T H, Schümperli D. Nucleic Acids Res. 1994;22:4023–4030. doi: 10.1093/nar/22.20.4023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Birchmeier C, Grosschedl R, Birnstiel M L. Cell. 1982;28:739–745. doi: 10.1016/0092-8674(82)90053-8. [DOI] [PubMed] [Google Scholar]
- 34.Mowry K L, Oh R, Steitz J A. Mol Cell Biol. 1989;9:3105–3108. doi: 10.1128/mcb.9.7.3105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Birchmeier C, Folk W, Birnstiel M L. Cell. 1983;35:433–440. doi: 10.1016/0092-8674(83)90176-9. [DOI] [PubMed] [Google Scholar]
- 36.Georgiev O, Birnstiel M L. EMBO J. 1985;4:481–489. doi: 10.1002/j.1460-2075.1985.tb03654.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Chodchoy N, Pandey N B, Marzluff W F. Mol Cell Biol. 1991;11:497–509. doi: 10.1128/mcb.11.1.497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Pandey N B, Williams A S, Sun J H, Brown V D, Bond U, Marzluff W F. Mol Cell Biol. 1994;14:1709–1720. doi: 10.1128/mcb.14.3.1709. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Williams A S, Ingledue T C, III, Kay B K, Marzluff W F. Nucleic Acids Res. 1994;22:4660–4666. doi: 10.1093/nar/22.22.4660. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Williams A S, Marzluff W F. Nucleic Acids Res. 1995;23:654–662. doi: 10.1093/nar/23.4.654. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Grimm C, Stefanovic B, Schümperli D. EMBO J. 1993;12:1229–1238. doi: 10.1002/j.1460-2075.1993.tb05764.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Biou V, Yaremchuk A, Tukalo M, Cusack S. Science. 1994;263:1404–1410. doi: 10.1126/science.8128220. [DOI] [PubMed] [Google Scholar]
- 43.Chen Z, Stauffacher C, Li Y, Schmidt T, Bomu W, Kamer G, Shanks M, Lomonossoff G, Johnson J E. Science. 1989;245:154–159. doi: 10.1126/science.2749253. [DOI] [PubMed] [Google Scholar]
- 44.Cotten M, Schaffner G, Birnstiel M L. Mol Cell Biol. 1989;9:4479–4487. doi: 10.1128/mcb.9.10.4479. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Hoffmann I, Birnstiel M L. Nature (London) 1990;346:665–668. doi: 10.1038/346665a0. [DOI] [PubMed] [Google Scholar]
- 46.Mattoccia E, Baldi I M, Gandini-Attardi D, Ciafre S, Tocchini-Valentini G P. Cell. 1988;55:731–738. doi: 10.1016/0092-8674(88)90231-0. [DOI] [PubMed] [Google Scholar]
- 47.Reyes V M, Abelson J. Cell. 1988;55:719–730. doi: 10.1016/0092-8674(88)90230-9. [DOI] [PubMed] [Google Scholar]
- 48.Baldi M I, Mattoccia E, Bufardeci E, Fabbri S, Tocchini-Valentini G P. Science. 1992;255:1404–1408. doi: 10.1126/science.1542788. [DOI] [PubMed] [Google Scholar]
- 49.Altman S, Kirsebom L, Talbot S. FASEB J. 1993;7:7–14. doi: 10.1096/fasebj.7.1.7916700. [DOI] [PubMed] [Google Scholar]
- 50.Kirsebom L A. Mol Microbiol. 1995;17:411–420. doi: 10.1111/j.1365-2958.1995.mmi_17030411.x. [DOI] [PubMed] [Google Scholar]
- 51.Pace N R, Brown J W. J Bacteriol. 1995;177:1919–1928. doi: 10.1128/jb.177.8.1919-1928.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Backus J W, Smith H C. Nucleic Acids Res. 1992;20:6007–6014. doi: 10.1093/nar/20.22.6007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Teng B B, Burant C F, Davidson N O. Science. 1993;260:1816–1819. doi: 10.1126/science.8511591. [DOI] [PubMed] [Google Scholar]
- 54.Lau P P, Zhu H-J, Baldini A, Charnsangavej C, Chan L. Proc Natl Acad Sci USA. 1994;91:8522–8526. doi: 10.1073/pnas.91.18.8522. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Schock D, Kuo S R, Steinburg M F, Bolognino M, Sparks C E, Smith H C. Proc Natl Acad Sci USA. 1996;93:1097–1102. doi: 10.1073/pnas.93.3.1097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Yang Y, Smith H C. Biochem Biophys Res Commun. 1996;218:797–801. doi: 10.1006/bbrc.1996.0142. [DOI] [PubMed] [Google Scholar]
- 57.Wahle E, Keller W. Annu Rev Biochem. 1992;61:419–440. doi: 10.1146/annurev.bi.61.070192.002223. [DOI] [PubMed] [Google Scholar]
- 58.Wahle E. Biochim Biophys Acta. 1995;1261:183–194. doi: 10.1016/0167-4781(94)00248-2. [DOI] [PubMed] [Google Scholar]
- 59.Kiss-Laszlo Z, Henry Y, Bachellerie J P, Caizergues-Ferrer M, Kiss T. Cell. 1996;85:1077–1088. doi: 10.1016/s0092-8674(00)81308-2. [DOI] [PubMed] [Google Scholar]
- 60.Riddle D L, Carbon J. Nature (London) New Biol. 1973;242:230–234. doi: 10.1038/newbio242230a0. [DOI] [PubMed] [Google Scholar]