

Abstract
Activating mutations in the Kit receptor tyrosine kinase have been identified in both rodent and human mast cell leukemia. One activating Kit mutation substitutes a valine for aspartic acid at codon 816 (D816V) and is frequently observed in human mastocytosis. Mutation at the equivalent position in the murine c-kit gene, involving a substitution of tyrosine for aspartic acid (D814Y), has been described in the mouse mastocytoma cell line P815. We have investigated the mechanism of oncogenic activation by this mutation. Expression of this mutant Kit receptor tyrosine kinase in a mast cell line led to the selective tyrosine phosphorylation of a 130-kDa protein and the degradation, through the ubiquitin-dependent proteolytic pathway, of a 65-kDa phosphoprotein. The 65-kDa protein was identified as the src homology domain 2 (SH2)-containing protein tyrosine phosphatase SHP-1, a negative regulator of signaling by Kit and other hematopoietic receptors, and the protein product of the murine motheaten locus. This mutation also altered the sites of receptor autophosphorylation and peptide substrate selectivity. Thus, this mutation activates the oncogenic potential of Kit by a novel mechanism involving an alteration in Kit substrate recognition and the degradation of SHP-1, an attenuator of the Kit signaling pathway.
Keywords: ubiquitin, signal transduction, oncogenes, mastocytosis
Receptor tyrosine kinases (RTKs) mediate cellular responses to a broad array of extracellular signals involved in the regulation of proliferation and differentiation. Ligand binding initiates a cascade of events, including receptor dimerization and autophosphorylation at specific tyrosine residues (1–3). Phosphotyrosine residues within specific sequence contexts then serve as high-affinity docking sites for intracellular proteins with Src homology 2 (SH2) domains (4, 5). The Kit RTK and its ligand Steel factor, that are encoded by the murine Dominant white spotting (W) and Steel (Sl) loci, respectively, play major roles in regulating at least four cell types, including primordial germ cells, hematopoietic cells, melanocytes, and the interstitial cells of Cajal (ICC) in the gut (6–9). Loss-of-function mutations at either the W or Sl locus result in sterility, anemia, mast cell deficiency, white coat color, and the absence of ICC and gut pacemaker activity. Gain of function mutations have also been described in the Kit RTK. These mutations have been found in both rodent and human mast-cell leukemic cell lines (10, 11) and in mast cells derived from patients with mastocytosis and urticaria pigmentosa (12, 13). Reintroduction of the cDNAs encoding these variant forms of Kit into the IC2 mast cell line leads to oncogenic transformation, consistent with the idea that these mutations cause the dysregulation of cell growth and differentiation, particularly in mast cell progenitors. One such activating mutation, located in the cytoplasmic kinase domain, results in the substitution of tyrosine for aspartic acid at codon 814 in mouse Kit (D814Y) or valine for aspartic acid at the equivalent position (D816V) in human KIT (14). These mutations lead to both ligand-independent Kit signaling and oncogenic activation, and selectively induce changes in morphology and gene expression in mast cells (15).
In this report, we demonstrate that the Kit D814Y mutation (KDY) alters the specificity of Kit for peptide substrates and results in the ubiquitin (Ub)-mediated degradation of SHP-1, a protein tyrosine phosphatase encoded by the murine motheaten (me) locus that negatively regulates Kit signaling (16). These observations suggest that the KDY mutation has both quantitative and qualitative effects on Kit signaling, altering the fidelity of signaling through the Kit receptor, and leading to the degradation of an important intracellular attenuator of extracellular signals.
MATERIALS AND METHODS
Cell Lines.
IC2 cells that express wild-type Kit or KDY mutant Kit were generated by retroviral infection. Details of the constructs, selection of expressing cells by surface fluorescence, and growth requirements for the cells are described in ref. 15.
Protein Analysis.
For analysis of phosphotyrosine-containing proteins, total cell lysates (10 mg) were resolved by SDS/PAGE, transferred to nitrocellulose, and probed with the 4G10 antibody to phosphotyrosine (Upstate Biotechnology). For analysis of SHP-1 protein, whole cell lysates (10 mg) were resolved by SDS/PAGE, transferred to nitrocellulose, and probed with the C-19 antibody to SHP-1 (Santa Cruz Technology), which is a polyclonal antibody directed against the C-terminal amino acids 576–595 of SHP-1. Alternatively, SHP-1 was immunoprecipitated from total cell lysates, followed by immunoblot analysis with anti-SHP-1 antiserum directed against the SH2 domain of SHP-1 (17) (generously provided by K. A. Siminovitch, Samuel Lunenfeld Research Institute). For analysis of ubiquitinated SHP-1, SHP-1 was immunoprecipitated from control and Steel-factor-stimulated IC2/Kit and IC2/KDY cells with anti-SHP-1 antiserum, as described (18). The immunoprecipitates were then resolved by SDS/PAGE and transferred to nitrocellulose membranes. The membrane was then autoclaved for 15 min to denature ubiquitinized proteins, before immunoblot analysis with a polyclonal antibody to Ub (Sigma).
[35S]Methionine Labeling.
Cells were starved of methionine for 5 h, and then pulse-labeled with [35S]methionine (1 mCi/ml; 1 Ci = 37 GBq) for 10 min. Newly synthesized proteins were chased for various times in the presence of nonradioactive methionine (150 mg/ml). Cell aliquots (1 × 107 cells per lane) were withdrawn and lysed, and the fate of radiolabeled SHP-1 was analyzed by immunoprecipitation of the lysates with the C-19 antibody to SHP-1 (Santa Cruz Technology), followed by SDS/PAGE and autoradiography. In some experiments, cells were incubated in the presence of 100 mM N-acetyl-leucinyl-leucinyl-norleucinyl-H (LLnL, Sigma), an inhibitor of the Ub-dependent proteosome, during the last hour of methionine starvation, and during the pulse and chase period.
Tryptic Phosphopeptide Mapping.
Cells were grown to 90% confluence, starved overnight in Dulbecco’s modified Eagle’s medium (DMEM)/20 mM Hepes, pH 7.2, containing fetal bovine serum (0.5%) and labeled for 4 h at 37°C in 5 ml of DMEM without phosphate/20 mM Hepes, pH 7.2/0.5% fetal calf serum, containing 10 mCi of [32P]orthophosphate per 10-cm dish. The Kit RTK from both control and Steel-factor-stimulated 3T3/Kit and 3T3/KDY cells was then immunoprecipitated (18) and proteins were resolved by SDS/PAGE on a 7.0% gel, transferred to Immobilon P, and visualized by autoradiography. Kit bands were excised, and digestion products were eluted from the membrane, after digestion with trypsin, and subjected to tryptic peptide mapping (19).
In Vitro Kinase Assay.
The Kit and KDY mutant proteins were immunoprecipitated from IC2/Kit and IC2/KDY cells, respectively (16), and used to phosphorylate the optimal peptide substrates for epidermal growth factor receptor (EGFR), Src, or Abl tyrosine kinase in the presence of [γ-32P]ATP (20). These substrates, AEEEEYFELVAKKKK, EAIYAAPFAKKKFEAKKK, and AEEEIYGEFEAKKK, were generously provided by L. C. Cantley and Z. Songyang (Harvard University). The in vitro kinase assay was perfomed as described (20)
Northern Blot Analysis.
Total cellular RNAs (10 mg) were resolved by electrophoresis on a 1% agarose gel, transferred to a nylon membrane, and probed with a 32P-labeled SHP-1 cDNA probe. A mouse ribosomal protein L32 cDNA probe was used as a loading control.
RESULTS
To address the mechanisms of oncogenic activation by the D814Y mutation, we introduced the wild-type (wt) or mutant KitD814Y (KDY) receptors into mouse IC2 cells (IC2/KDY), an immature mast cell line that does not express Kit. To avoid clonal variation, pools of G418-resistant cells were used. Expression of KDY, but not wt Kit, resulted in interleukin 3 independence and mastocytoma formation in vivo, confirming the transforming nature of this allele (15). Moreover, KDY induced IC2 differentiation, whereas wt Kit did not, even in the presence of Steel factor, suggesting that KDY delivers a qualitatively different signal to IC2 cells (15). To investigate further the differences in signaling between KDY and wt Kit, we first compared the spectrum of proteins phosphorylated on tyrosine in cells expressing either wt or activated Kit. KDY was constitutively tyrosine-phosphorylated, whereas the wt receptor was only tyrosine-phosphorylated after addition of Steel factor, the Kit ligand (Fig. 1A). In addition, IC2 cells expressing KDY exhibited two major differences in the spectrum of phosphotyrosine-containing proteins when compared with IC2/Kit cells stimulated with Steel factor. (i) A 130- to 140-kDa protein (p130) distinct from p145Kit that was heavily phosphorylated in IC2/KDY cells was poorly phosphorylated in IC2/Kit cells. (ii) A 65-kDa phosphotyrosine-containing protein, present in Steel factor-stimulated IC2/Kit cells and primary mast cells, was absent in IC2/KDY cells (Fig. 1A).
Figure 1.
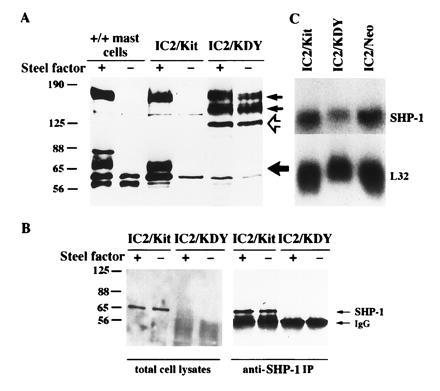
Reduced SHP-1 protein in mast cells expressing the mutant KDY. (A) Anti-phosphotyrosine blots of whole cell lysates obtained from control and Steel-factor-stimulated mast cells, IC2/Kit, and IC2/KDY cells. The small arrows indicate the positions of the mature (upper arrow) and immature (lower arrow) forms of p145Kit, the open arrow indicates the phosphorylated p130 protein, and the large solid arrow indicates the 65-kDa protein. (B) Detection of SHP-1 protein in cells expressing Kit or KDY. (Left) Immunoblot of SHP-1 with C-19 antibody to SHP-1 on total cell lysates. (Right) SHP-1 was immunoprecipitated with the C-19 anti-SHP-1 antibodies from control and Steel-factor-stimulated IC2/Kit and IC2/KDY cells. Western blots of the immunoprecipitates were probed with anti-SHP-1 antiserum directed against the SH2 domain of SHP-1 (17). (C) Expression of SHP-1 transcripts in cells expressing Kit or KDY. Total cellular RNA was separated by gel electrophoresis, and the blot was hybridized to cDNA probe for SHP-1 and mouse ribosomal protein L32.
Several proteins are phosphorylated by Kit in response to Steel factor (22–29). One of these proteins SHP-1 [formerly called protein tyrosine phosphatase 1C (PTP1C), hematopoietic cell phosphatase (HCP), or SH2-containing protein tyrosine phosphatase 1 (SHPTP1)] is a 65-kDa protein tyrosine phosphatase that negatively regulates signaling through a number of receptors, including Kit (16, 29–34). Mice with mutations at the motheaten (me) locus, the gene that encodes SHP-1, exhibit multiple hematologic abnormalities (35), consistent with the deregulated activation of multiple signaling pathways. To test whether the p65 missing from IC2/KDY cells was SHP-1, we analyzed SHP-1 expression in IC2 cells expressing either the wt Kit or KDY. SHP-1 was abundantly expressed in IC2/Kit cells (Fig. 1B) and phosphorylated on tyrosine after stimulation with Steel factor (data not shown). In contrast, SHP-1 was undetectable in IC2/KDY cells (Fig. 1B), despite the presence of SHP-1 mRNA in these cells (Fig. 1C).
To test whether the SHP-1 transcripts were translated in IC2/KDY cells, we labeled cells with [35S]methionine, followed by immunoprecipitation and analysis with anti-SHP-1 antibody. 35S-labeled SHP-1 produced in IC2/Kit cells was not significantly degraded after the 15-min chase (97.9% remained after 15 min). In contrast, only 49% of the labeled SHP-1 produced IC2/KDY cells remained after 15 min. This analysis revealed that SHP-1 was made in IC2/KDY cells but rapidly degradated (Fig. 2A).
Figure 2.
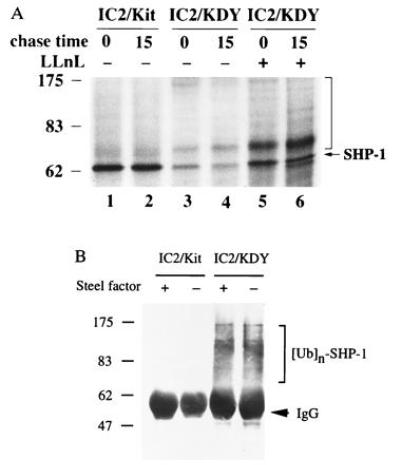
(A) Rapid turnover of SHP-1 in cells expressing KDY. After a 10-min pulse with [35S]methionine, 35S-labeled SHP-1 was chased for 15 min. The vertical bar indicates the multiple protein bands larger than 65 kDa immunoprecipitated by anti-SHP-1 antibody C-19. (B) Ubiquitination of SHP-1 in cells expressing KDY. SHP-1 immunoprecipitates from control and Steel-factor-stimulated IC2/Kit and IC2/KDY cells were examined by immunoblot analysis with an antibody to Ub.
Ubiquitin-Mediated SHP-1 Degradation.
The Ub-dependent proteolytic pathway plays an important role in the degradation of key regulatory proteins (36, 37). We therefore examined whether the rapid turnover of SHP-1 in IC2/KDY cells might be mediated by a Ub-dependent degradation pathway. First, we examined whether the peptide aldehyde N-acetyl-leucinyl-leucinyl-norleucinyl-H (LLnL, also called MG101 or calpain inhibitor I), a potent inhibitor of the proteolytic activity that mediates degradation of Ub-conjugated proteins (38, 39), prevented SHP-1 degradation. As shown in Fig. 2A, the addition of LLnL stabilized SHP-1, suggesting that SHP-1 is degraded via the Ub-dependent pathway. We also observed multiple protein bands larger than 65 kDa in IC2/KDY cells after immunoprecipitation with anti-SHP-1 antibody. To confirm that these novel bands were multiply ubiquitinated SHP-1, SHP-1 was immunoprecipitated from IC2/Kit and IC2/KDY cells and examined by immunoblot analysis with an anti-Ub antibody. Ubiquitinated SHP-1 was not detected in either control or Steel-factor-stimulated IC2/Kit cells; in contrast, multiply ubiquitinated SHP-1 was present in IC2/KDY cells (Fig. 2B). Thus, SHP-1 is ubiquitinated and degraded in cells expressing KDY but not in cells expressing the wt Kit RTK stimulated with Steel factor.
Autophosphorylation of the wt Kit and KDY RTK.
The D814Y substitution in Kit lies in a region of the cytoplasmic kinase domain implicated in substrate recognition by Ser/Thr protein kinases (40, 41). Furthermore, mutations in the RET RTK in patients with multiple endocrine neoplasia type 2B, located 20 residues C-terminal of the KDY mutation, alter substrate specificity (20, 42). The mutations in RET and Kit both change a residue conserved in RTKs to a residue typical of nonreceptor tyrosine kinases (NRTKs). RTK autophosphorylation sites normally associate with proteins containing group III SH2 domains, whereas NRTKs, including Src and Abl, typically phosphorylate motifs that bind group I SH2 domains (20, 43, 44). Thus, the KDY mutation might also result in an alteration in substrate specificity and ligand-independent activation. To test this hypothesis, 32P-labeled Kit and KDY receptors were isolated from control and Steel-factor-stimulated cells and subjected to tryptic phosphopeptide mapping (Fig. 3) and phosphoamino acid analysis. All the phosphopeptides present in maps of unstimulated Kit receptors (Fig. 3, schematic map, peptides 1 to 7) contain phosphoserine (data not shown). The intensity of the phosphoserine-containing peptides 1, 2, 3, 6, and 7 in Fig. 3 decreased while others (peptides 4 and 5 in Fig. 3) increased after stimulation with Steel factor. Steel factor also induced the appearance of several new phosphopeptides (peptides a to d, Fig. 3) that likely represent autophosphorylation sites. The phosphopeptide maps of the mutant receptor from control or Steel-factor-stimulated cells were identical, consistent with the notion that KDY is constitutively activated (Fig. 3). While phosphopeptides a and b were phosphorylated to levels comparable to those observed in the wt receptor, phosphopeptides c and d were undetectable (Fig. 3). Thus, the D814Y mutation leads to a marked change in autophosphorylation site selection.
Figure 3.
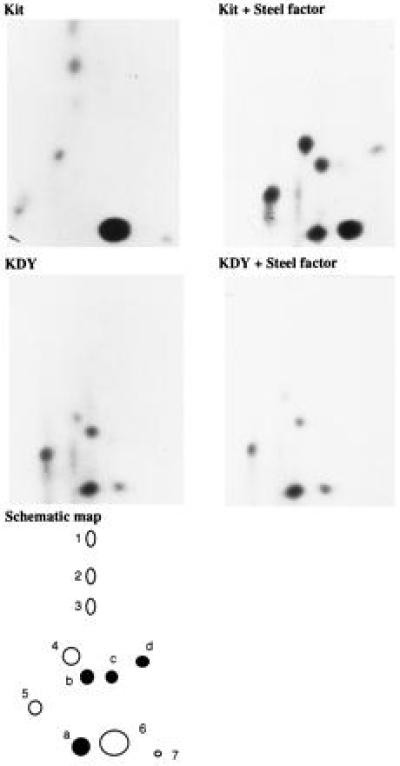
Tryptic phosphopeptide mapping of the Kit and KDY receptors. 32P-labeled Kit and KDY receptors isolated from control and Steel-factor-stimulated cells were digested with trypsin and peptides were resolved by separation in two dimensions on TLC plates. The results are summarized in a schematic phosphopeptide map. The phosphopeptides present in the unstimulated wt Kit receptor, which all contain phosphoserine, are shown as open circles (peptides 1 to 7); phosphopeptides that appear in the wt Kit receptor after stimulation with Steel factor, representing presumptive autophosphorylation sites, are shown as solid circles (peptides a to d).
Peptide Substrate Selectivity of Kit and KDY.
We next assayed both the wt and mutant KDY receptors in vitro for protein kinase activity using optimal peptide substrates for the EGF receptor (EGFR) and the Abl and Src kinases (20, 45). As expected, Kit preferentially phosphorylated the optimal peptide substrate for the EGFR. KDY also phosphorylated this substrate, but, in contrast to wt Kit, phosphorylated the Abl peptide substrate more efficiently than the EGFR substrate (Fig. 4). Neither receptor significantly phosphorylated the Src peptide substrate (Fig. 4). Thus these data suggest that the codon 814 mutation in KDY markedly altered substrate specificity.
Figure 4.
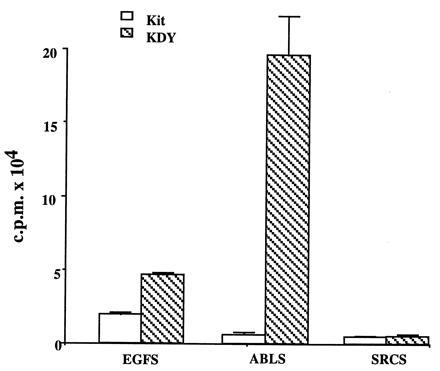
Substrate specificity of the wt Kit and mutant KDY receptors. Receptors were immunoprecipitated from IC2/Kit and IC2/KDY cells, respectively, with rabbit anti-Kit antiserum and used to phosphorylate the EGFR optimal substrate (EGFS) AEEEEYFELVAKKKK (10 mM), Abl optimal substrate (ABLS) EAIYAAPFAKKKFEAKKK (10 mM), or Src optimal substrate (SRCS) AEEEIYGEFEAKKK (10 mM).
DISCUSSION
In this paper, we have analyzed the biochemical mechanism of action of the codon 814 mutation in the murine Kit receptor (KDY). This analysis has revealed that the KDY mutation results in two profound changes. (i) There is a marked alteration in substrate recognition by the mutant receptor, as determinded by examining the pattern of receptor autophosphorylation sites, the selectivity of the receptor for model peptide substrates optimal for receptor as opposed to nonreceptor tyrosine kinases (45), and the pattern of tyrosine-phophorylated proteins in cells expressing either the wt or mutant Kit receptors. (ii) Expression of KDY in mast cells was associated with the selective tyrosine phosphorylation of a 130-kDa protein and the absence of a 65-kDa phosphoprotein present in cells expressing wt Kit. We have identified this 65-kDa protein as SHP-1, a SH2-containing protein tyrosine phosphatase, encoded by the murine me locus (46, 47). The absence of SHP-1 in cells expressing KDY was due to its selective degradation by Ub-mediated proteolysis.
SHP-1 negatively regulates signaling through a number of hematopoietic receptors, including the interleukin 3 (31), erythropoietin (32), immunoglobulin (34), and Kit receptors (16), consistent with the observations the multiple hematopoietic abnormalities observed in me mice. We have also recently demonstrated that me mutant mice have increased numbers of mast cells in the skin and that loss of SHP-1 activity in mice homozygous for mutations at both the me and W loci suppresses the mast cell deficiency associated with W mutations (16). Thus, these biochemical and genetic data suggest that SHP-1 is an important attenuator of signals emanating from the Kit receptor. Thus SHP-1 degradation in cells expressing the mutant Kit receptor would deregulate Kit receptor signaling. Furthermore, since SHP-1 regulates signaling from a number of hematopoietic receptors, the loss of SHP-1 in cells expressing KDY might also deregulate signaling through a number of cytokine receptors.
The phenotypes of W and me mice demonstrate the importance of the Kit receptor signaling pathway in normal mast cell development. Thus, the activation of the Kit pathway by the constitutive kinase activity of the KDY mutant receptor, coupled with the degradation of SHP-1 would explain the massive expansion of mast cells in patients with urticaria pigmentosa, mastocytosis, and mast cell leukemia. Moreover, Steel factor can synergize with a number of hematopoietic growth factors to promote proliferation and differentiation of hematopoietic progenitors (48, 49), which may explain the other hematopoietic abnormalities associated with certain cases of mastocytosis (50, 51).
It is interesting to note the parallels between the Kit KDY mutation and the mutation at position 918 of the RET RTK observed in patients with multiple endocrine neoplasia type 2B. This latter mutation converts RET into a transforming oncogene and alters its substrate specificity (20, 42). The mutations in KIT and RET are located 20 amino acids apart, in subdomains VII and VIII of the kinase domain, respectively (40, 41). Furthermore, both mutations change a residue conserved in RTKs to a residue typical of NRTKs. RTKs normally associate with signaling molecules containing group III SH2 domains, whereas NRTKs, including Src and Abl, preferentially interact with proteins containing group I SH2 domains (20, 45). Therefore, the 814/816 and the 918 mutations in the Kit and RET receptors, respectively, may alter their conformation, such that they now interact preferentially with proteins with group I SH2 domains. Although the relevant substrates affected by the RET multiple endocrine neoplasia type 2B mutation are not yet known, our data suggest that the D814Y mutation in the Kit RTK alters the fidelity of Kit signaling, resulting in changes in autophosphorylation and in the preferential phosphorylation of a 130-kDa protein (p130). The selective tyrosine phosphorylation of p130 in cells expressing the oncogenic KDY suggests that it might be a positive effector of cell growth. It clearly will be of interest to characterize p130 further and to determine whether the degradation of SHP-1 is a direct consequence of this alteration in the fidelity of the Kit receptor signaling pathway.
In summary, our results suggest a novel mechanism for oncogenic activation of a RTK involving a change in substrate specificity coupled with the degradation of a protein tyrosine phosphatase. These data suggest that signals emanating from a tyrosine kinase can target specific proteins for proteolysis. Participation of protein kinases in protein turnover has been shown for IkB, which is degraded after serine-threonine phosphorylation (52). Finally, these experiments illustrate the importance of the events downstream of a RTK to the signals and responses perceived by the cell. The KDY mutation appears to induce profound quantitative and qualitative changes in the Kit signaling pathway, leading to factor-independent growth, differentiation, and oncogenic transformation.
Acknowledgments
We are grateful to S. I. Yahara and D. Baltimore for providing cell lines IC2 and BOSC 23, respectively; Shin-Ichi Nishikawa for the gift of the Ack2 antibody; K. A. Siminovitch for providing anti-PTP1C antiserum; and L. C. Cantley and Z. Songyang for generously providing the peptide substrates for EGFR, Abl, and Src. Work described herein was supported by grants from the Medical Research Council of Canada and the National Cancer Institute of Canada. R.P. was supported by a David C. Rae Memorial Fellowship from the Leukemia Research Fund of Canada. A.B and T.P. are International Research Scholars of the Howard Hughes Medical Institute.
Footnotes
Abbreviations: RTK, receptor tyrosine kinase; KDY, Kit receptor with a D814Y mutation; wt, wild type; SH2, src homology domain 2; EGFR, epidermal growth factor receptor; UB, ubiquitin; NRTK, nonreceptor tyrosine kinase.
References
- 1.van der Geer P, Hunter T, Lindberg R A. Annu Rev Cell Biol. 1994;10:251–337. doi: 10.1146/annurev.cb.10.110194.001343. [DOI] [PubMed] [Google Scholar]
- 2.Schlessinger J, Ullrich A. Neuron. 1992;9:383–391. doi: 10.1016/0896-6273(92)90177-f. [DOI] [PubMed] [Google Scholar]
- 3.Heldin C H. Cell. 1995;80:213–223. doi: 10.1016/0092-8674(95)90404-2. [DOI] [PubMed] [Google Scholar]
- 4.Koch C A, Anderson D, Moran M F, Ellis C, Pawson T. Science. 1991;252:668–674. doi: 10.1126/science.1708916. [DOI] [PubMed] [Google Scholar]
- 5.Pawson T. Nature (London) 1995;373:573–580. doi: 10.1038/373573a0. [DOI] [PubMed] [Google Scholar]
- 6.Dubreuil P, Rottapel R, Reith A D, Forrester L, Bernstein A. Ann NY Acad Sci. 1990;599:58–65. doi: 10.1111/j.1749-6632.1990.tb42364.x. [DOI] [PubMed] [Google Scholar]
- 7.Ward S, Burns A, Toriheshi S, Sanders K. J Physiol (London) 1994;480:91–97. doi: 10.1113/jphysiol.1994.sp020343. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Huizinga J D, Thuneberg L, Kluppel M, Malysz J, Middelsen H B, Bernstein A. Nature (London) 1995;373:347–349. doi: 10.1038/373347a0. [DOI] [PubMed] [Google Scholar]
- 9.Russell E S. Adv Genet. 1979;20:357–459. [PubMed] [Google Scholar]
- 10.Furitsu T, Tsujimura T, Tono T, Ikeda H, Kitayama H, Koshimizu U, Sugahara H, Butterfield J H, Ashman L K, Kanayama Y, Matsuzawa Y, Kitamura Y, Kanakura Y. J Clin Invest. 1993;92:1736–1744. doi: 10.1172/JCI116761. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Tsujimura T, Furitsu T, Morimoto M, Isozaki K, Nomura S, Matsuzawa Y, Kitamura Y, Kanakura Y. Blood. 1994;83:2619–2626. [PubMed] [Google Scholar]
- 12.Longley B, Tyrrell L, Lu S-Z, Ma Y-S, Langley K, Ding T-G, Duffy T, Jacobs P, Tang L, Modlin I. Nat Genet. 1996;12:312–314. doi: 10.1038/ng0396-312. [DOI] [PubMed] [Google Scholar]
- 13.Nagata H, Worobec A S, Oh C D, Chowdhury B A, Tannenbaum S, Suzuki Y, Metcalfe D D. Proc Natl Acad Sci USA. 1995;92:10560–10564. doi: 10.1073/pnas.92.23.10560. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Kitayama H, Kanakura Y, Furitsu T, Tsujimura T, Oritani K, Ikeda H, Sugahara H, Mitsui H, Kanayama Y, Kitamura Y. Blood. 1995;85:790–798. [PubMed] [Google Scholar]
- 15.Piao X, Bernstein A. Blood. 1996;87:3117–3123. [PubMed] [Google Scholar]
- 16.Paulson R F, Vesely S, Siminovitch K, Bernstein A. Nat Genet. 1996;13:309–315. doi: 10.1038/ng0796-309. [DOI] [PubMed] [Google Scholar]
- 17.Kozlowski M, Mlinaric-Rascan I, Feng G, Shen R, Pawson A, Simnovitch K. J Exp Med. 1993;178:2157–2163. doi: 10.1084/jem.178.6.2157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Rottapel R, Reedijk M, Williams D E, Lyman S D, Anderson D M, Pawson T, Bernstein A. Mol Cell Biol. 1991;11:3043–3051. doi: 10.1128/mcb.11.6.3043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.van der Geer P, Hunter T. Electrophoresis. 1994;15:544–554. doi: 10.1002/elps.1150150173. [DOI] [PubMed] [Google Scholar]
- 20.Songyang Z, Carraway K L, III, Eck M J, Harrison S C, Feldman R A, Mohammadi M, Schlessinger J, Hubbard S R, Smith D P, Eng C, Lorenzo M J, Ponder B A J, Mayer B J, Cantley L C. Nature (London) 1995;373:536–539. doi: 10.1038/373536a0. [DOI] [PubMed] [Google Scholar]
- 21.Reith A D, Rottapel R, Giddens E, Brady C, Forrester L, Bernstein A. Genes Dev. 1990;4:390–400. doi: 10.1101/gad.4.3.390. [DOI] [PubMed] [Google Scholar]
- 22.Lev S, Givol D, Yarden Y. EMBO J. 1991;10:647–654. doi: 10.1002/j.1460-2075.1991.tb07993.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Lev S, Givol D, Yarden Y. Proc Natl Acad Sci USA. 1992;89:678–682. doi: 10.1073/pnas.89.2.678. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Duronio V, Welham M J, Abraham S, Dryden P, Schrader J W. Proc Natl Acad Sci USA. 1992;89:1587–1591. doi: 10.1073/pnas.89.5.1587. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Alai M, Mui A L, Cutler R L, Bustelo X R, Barbacid M, Krystal G. J Biol Chem. 1992;267:18021–18025. [PubMed] [Google Scholar]
- 26.Welham M J, Schrader J W. J Immunol. 1992;149:2772–2783. [PubMed] [Google Scholar]
- 27.Serve H, Hsu Y C, Besmer P. J Biol Chem. 1994;269:6026–6030. [PubMed] [Google Scholar]
- 28.Cutler R L, Liu L, Damen J E, Krystal G. J Biol Chem. 1993;268:21463–21465. [PubMed] [Google Scholar]
- 29.Yi T, Ihle J N. Mol Cell Biol. 1993;13:3350–3358. doi: 10.1128/mcb.13.6.3350. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Yeung Y G, Berg K L, Pixley F J, Angeletti R H, Stanley E R. J Biol Chem. 1992;267:23447–23450. [PubMed] [Google Scholar]
- 31.Yi T, Mui A L, Krystal G, Ihle J N. Mol Cell Biol. 1993;13:7577–7586. doi: 10.1128/mcb.13.12.7577. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Klingmuller U, Lorenz U, Cantley L C, Neel B G, Lodish H F. Cell. 1995;80:729–738. doi: 10.1016/0092-8674(95)90351-8. [DOI] [PubMed] [Google Scholar]
- 33.Lorenz U, Ravichandran K S, Pei D, Walsh C T, Burakoff S J, Neel B G. Mol Cell Biol. 1994;14:1824–1834. doi: 10.1128/mcb.14.3.1824. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Pani G, Kozlowski M, Cambier J, Mills G, Siminovitch K. J Exp Med. 1995;181:2077–2084. doi: 10.1084/jem.181.6.2077. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Shultz L D. Curr Top Microbiol Immunol. 1988;137:216–222. doi: 10.1007/978-3-642-50059-6_32. [DOI] [PubMed] [Google Scholar]
- 36.Scheffner M, Werness B A, Huibregtse J M, Levine A J, Howley P M. Cell. 1990;63:1129–1136. doi: 10.1016/0092-8674(90)90409-8. [DOI] [PubMed] [Google Scholar]
- 37.Ciechanover A. Cell. 1994;79:13–21. doi: 10.1016/0092-8674(94)90396-4. [DOI] [PubMed] [Google Scholar]
- 38.Palombella V J, Rando O J, Goldberg A L, Maniatis T. Cell. 1994;78:773–785. doi: 10.1016/s0092-8674(94)90482-0. [DOI] [PubMed] [Google Scholar]
- 39.Rock K, Gramm C, Rothstein L, Clark K, Stein R, Dick L, Hwang D, Goldberg A. Cell. 1994;78:761–771. doi: 10.1016/s0092-8674(94)90462-6. [DOI] [PubMed] [Google Scholar]
- 40.Hanks S K, Quinn A M, Hunter T. Science. 1988;241:42–52. doi: 10.1126/science.3291115. [DOI] [PubMed] [Google Scholar]
- 41.Knighton D R, Zheng J H, Ten E L, Xuong N H, Taylor S S, Sowadski J M. Science. 1991;253:414–420. doi: 10.1126/science.1862343. [DOI] [PubMed] [Google Scholar]
- 42.Santoro M, Carlomagno F, Romano A, Bottaro D P, Dathan N A, Grieco M, Fusco A, Vecchio G, Matoskova B, Kraus M H, DiFiore P P. Science. 1995;267:381–383. doi: 10.1126/science.7824936. [DOI] [PubMed] [Google Scholar]
- 43.Songyang Z, Shoelson S E, McGlade J, Olivier P, Pawson T, Bustelo X R, Barbacid M, Sabe H, Hanafusa H, Yi T, Ren R, Baltimore D, Ratnofsky S, Feldman R, Catnley L. Mol Cell Biol. 1994;14:2777–2785. doi: 10.1128/mcb.14.4.2777. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Songyang Z, Shoelson S E, Chaudhuri M, Gish G, Pawson T, Haser W G, King F, Roberts T, Ratnofsky S, Lechleider R J, Neel B, Birge R, Fajoudo J, Chou M, Hanafusa H, Schaffhausen B, Cantley L. Cell. 1993;72:767–778. doi: 10.1016/0092-8674(93)90404-e. [DOI] [PubMed] [Google Scholar]
- 45.Songyang Z, Blechner S, Hoagland N, Hoekstra M F, Piwnica W H, Cantley L C. Curr Biol. 1994;4:973–982. doi: 10.1016/s0960-9822(00)00221-9. [DOI] [PubMed] [Google Scholar]
- 46.Tsui H W, Siminovitch K A, de Souza L, Tsui F W. Nat Genet. 1993;4:124–129. doi: 10.1038/ng0693-124. [DOI] [PubMed] [Google Scholar]
- 47.Shultz L D, Schweitzer P A, Rajan T V, Yi T, Ihle J N, Matthews R J, Thomas M L, Beier D R. Cell. 1993;73:1445–1454. doi: 10.1016/0092-8674(93)90369-2. [DOI] [PubMed] [Google Scholar]
- 48.McNiece I, Langley K, Zsebo K. Exp Hematol. 1991;19:226–231. [PubMed] [Google Scholar]
- 49.McNiece I, Langlay K, Zsebo K. J Immunol. 1991;146:3785–3790. [PubMed] [Google Scholar]
- 50.Longley B, Duffy T, Kohn S. J Am Acad Dermatol. 1995;32:545–561. doi: 10.1016/0190-9622(95)90336-4. [DOI] [PubMed] [Google Scholar]
- 51.Metcalfe D. J Invest Dermatol. 1991;96:2S–4S. [PubMed] [Google Scholar]
- 52.Chen Z, Hagler J, Palombella V, Melandri F, Schere D, Ballard D, Maniatis T. Genes Dev. 1995;9:1586–1597. doi: 10.1101/gad.9.13.1586. [DOI] [PubMed] [Google Scholar]