

Abstract
It has recently been proposed that experimental autoimmune encephalomyelitis, once considered the classical Th1 disease, is predominantly Th17 driven. In this study we show that myelin-reactive Th1 preparations devoid of contaminating IL-17+ cells are highly pathogenic. In contrast, Th17 preparations lacking IFN-γ+ cells do not cause disease. Our key observation is that only Th1 cells can access the noninflamed CNS. Once Th1 cells establish the experimental autoimmune encephalomyelitis lesion, Th17 cells appear in the CNS. These data shed important new light on the ability of Th1 vs Th17 cells to access inflamed vs normal tissue. Because the IL-17-triggered release of chemokines by stromal cells could attract many other immune cells, allowing Th17 cells to access the tissues only under conditions of inflammation may be a key process limiting (auto)immune pathology. This has major implications for the design of therapeutic interventions, many of which are now aiming at Th17 rather than Th1 cells.
Experimental autoimmune encephalomyelitis (EAE)4 had long been considered the prototypic Th1-mediated autoimmune disease (1, 2) until recent findings suggested a primary role for Th17 cells in this model (3, 4). This paradigm shift has sparked a rapid and remarkable change in emphasis in the search for disease-modifying drugs away from the Th1 pathway toward the Th17 pathway (5-8). It is therefore important to have a clear understanding of the relevance of each of these pathways in autoimmune pathology. This requires an assessment of the ability of Th1 or Th17 cells to initiate pathology upon adoptive transfer in the absence of each other.
With the purity of the Th phenotype in mind, we have evaluated the EAE-initiating capacity of Th1 vs Th17 populations. Our results indicate that Th1 cells have a preferential ability to access the noninflamed CNS and that the pathology they cause promotes the subsequent infiltration of Th17 cells.
Materials and Methods
Mice, Ag, and tissue culture medium
C57BL/6 mice (CD45.2/CD90.2, CD45.1/CD90.2, or CD45.2/CD90.1), Tg4 × CD45.1 mice, and B10.PL mice were bred under specific pathogen-free conditions at the University of Edinburgh (Edinburgh, U.K.). Tg4 and B10.PL × RAG2-/- mice were bred under specific pathogen-free conditions at the University of Bristol (Bristol, U.K.). Experiments received University of Edinburgh/University of Bristol ethical approval and were performed under U.K. legislation. The myelin oligodendrocyte glycoprotein peptide 35-55 (pMOG; MEVGWYRSPFSRVVHLYRNGK) and myelin basic protein (MBP) acetylated (Ac) peptide Ac1-9 (Ac-ASQKRPSQR) were synthesized by the Advanced Biotechnology Centre, Imperial College (London, U.K.). The tissue culture medium was RPMI 1640 medium supplemented with 2 mM l-glutamine, 100 U/ml penicillin, 100 μg/ml streptomycin, and 5 × 10-5 M 2-ME (all from Invitrogen) and 5% FCS (Sigma-Aldrich).
Cell culture and passive EAE
C57BL/6 mice were immunized s.c. with 100 μg of pMOG in CFA. Ten days later, draining lymph node cells were recovered and cultured with 10 μg/ml pMOG for a total of 72 h. For Th1 polarization, 25 ng/ml rIL-12 (R & D Systems) and 25 ng/ml rIL-18 (MBL) were used. rIL-2 (0.5 ng/ml; R and D Systems) was added at the start of Th1-polarizing cultures, and the concentration of IL-2 was raised to 2.5 ng/ml after 48 h. For Th17-polarization, 20 ng/ml rIL-6, 20 ng/ml rIL-23, and 3 ng/ml rTGF-β (all from R&D Systems) were used. After 72 h, 4 × 106 blasts were injected i.v.
FACS-sorted CD4+CD62LhighCD25- Tg4 TCR transgenic cells were cultured for 72 h with irradiated B10.PL splenocytes and 10 μg/ml MBP Ac1-9 using the Th1- and Th17-polarizing conditions described above. After 72 h, 3 × 106 blasts were injected i.v. For transfer to B10.PL × RAG2-/- recipients, naive Tg4 splenic and lymph node populations were stimulated with 10 μg/ml Ac1-9 under either Th1-polarizing conditions (5 ng/ml IL-12, and 10 μg/ml anti-IL-4), or Th17-polarizing conditions (3 ng/ml TGF-β, 20 ng/ml IL-6, 20 ng/ml IL-23, and 3 μg/ml anti-IFN-γ) for 5 days. IL-2 (20 U/ml for Th1-polarization and 10 U/ml for Th17-polarization) was included from day 3. After 5 days of culture, 3 × 106 blasts were injected i.v. On the day of transfer, each mouse also received 200 ng of pertussis toxin. Clinical signs of EAE were assessed as described previously (9).
FACS
All Abs were from eBioscience except for anti-CD4-PerCP (BD Pharmingen) and anti-IL-17-allophycocyanin (BioLegend). Intracellular cytokine staining of peptide-stimulated cells was performed as previously described (9). For staining at the end of polarizing cultures, 1 μg/ml PMA, 50 ng/ml ionomycin, and 1 μl/ml brefeldin A was added for the last 4 h of culture.
Analysis of cytokine secretion
Ag-induced cytokine production by lymph node cells (6 × 105 per 200 μl per well) was assessed by ELISA. Polarized T cells (2 × 104 per 200 μl per well) were extensively washed before restimulation with irradiated splenocytes (2 × 105/well) and a dose range of Ag as indicated. After 72 h, cytokine concentration was determined by ELISA or cytometric bead array using an inflammation array (BD Pharmingen) or a Th1/Th2 10plex kit (Bender MedSystems) according to the manufacturer’s instructions.
Real-time PCR
RNA was extracted after 72 h of culture in the presence or absence of polarizing cytokines. Primer sequences used for ROR-γt and T-bet have been published previously (10). Expression was normalized to hypoxanthine phosphoribosyl-transferase and expressed as the fold induction over cells stimulated under neutral conditions.
Statistical analysis
Statistical analysis of results was performed using the Mann-Whitney U test and the two-tailed Student’s t test.
Results
Th1 cells initiate EAE upon passive transfer and promote the recruitment of Th17 cells to the CNS
Using draining lymph nodes from mice immunized with the pMOG peptide 35-55, our emphasis was to generate the cleanest possible Th1 and Th17 preparations. Stimulation with pMOG alone induced a mixed response dominated by IFN-γ production and a small proportion of cells producing IL-17 or coproducing IFN-γ and IL-17 (Fig. 1A). The addition of IL-12 and IL-18 maximized IFN-γ production and repressed IL-17 production, giving the highest ratio of IFN-γ to IL-17 producers (Fig. 1A). Although IL-23 induced the highest frequency of IL-17-producers (∼40% of cells), it did not reduce the overall proportion of IFN-γ producers (∼20% of cells made IFN-γ, either alone or in combination with IL-17). TGF-β was required to suppress IFN-γ production. The IL-6 plus IL-23 plus TGF-β combination therefore produced the most highly polarized Th17 population (Fig. 1A). As exemplified in Fig. 1B, administration of Th1 cells alone or Th1 together with Th17 cells led to clinical disease, whereas Th17 cells alone were not pathogenic. Similarly, populations generated in the presence of IL-6 and TGF-β (no IL-23) did not cause EAE alone and did not significantly modulate the course of disease when cotransferred with Th1 cells (Fig. 1C).
FIGURE 1.
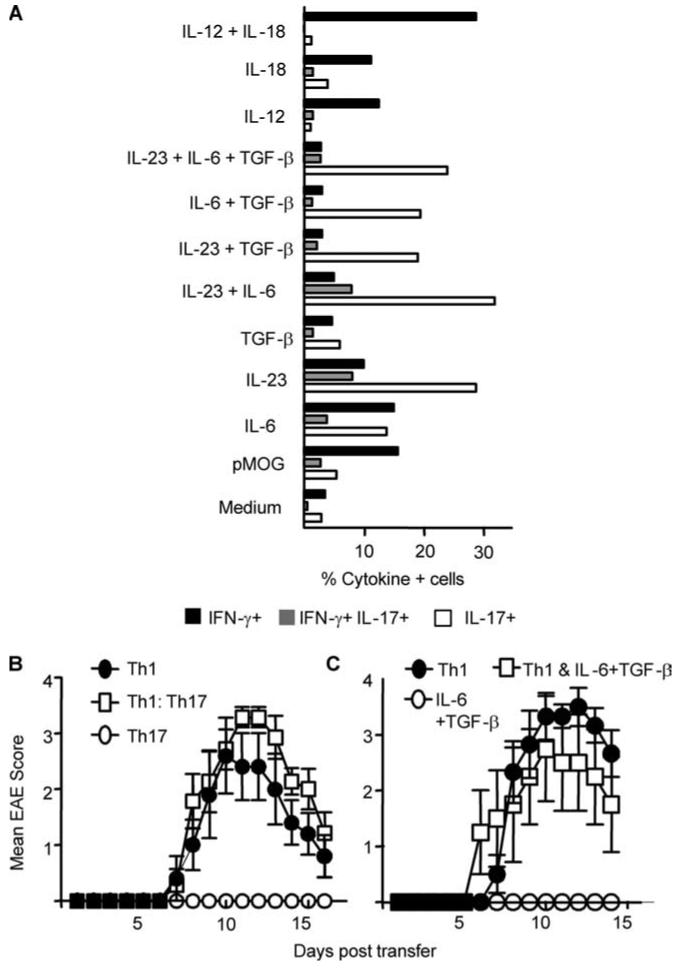
Polyclonal Th1 but not Th17 effectors initially activated in vivo induce disease upon passive transfer. Draining lymph node cells from pMOG-immunized mice were restimulated for 72 h with pMOG under Th1- or Th17-polarizing conditions. A, The proportion of CD4+ T cells capable of producing IFN-γ (black bars), IL-17 (open bars), or IFN-γ and IL-17 (gray bars) after 3 days of culture in the presence of pMOG and the indicated recombinant cytokines. B, EAE score in recipients of Th1- (closed circles), Th17- (open circles), or Th1- and Th17-polarized (open squares) cells. Data (n = 5-7 mice per group) are from one of three experiments giving consistent results. C, EAE score in recipients of Th1- (closed circles) and IL-6 plus TGF-β-polarized cells (open circles) or Th1- and IL-6 plus TGF-β-polarized cells (open squares) (n = 3-6 mice per group).
To assess their comparative capacities to home to the target organ, Th1-polarized and Th17-polarized cells were sourced from CD45.1/CD90.2 donors and CD45.2/CD90.1 donors, respectively, for injection into CD45.2 CD90.2 hosts. Both Th1 and Th17 populations were clearly identifiable in the spleen (Fig. 2A) with Th17 preparations at significantly higher frequencies than Th1 preparations when transferred either singly or in tandem (Fig. 2A). The picture from the CNS (Fig. 2D) was the reverse of that from the spleen. A strong Th1 infiltrate was clearly identifiable (∼100-fold enriched as a percentage of total CD4+ cells compared with the spleen). Th17 cells were not identifiable in the CNS if transferred alone but were present when transferred together with Th1 effectors (Fig. 2D). We conclude that Th1 lymphocytes have a superior capacity to home to the CNS and orchestrate inflammation and cellular recruitment, thereby facilitating the entry of Th17 cells.
FIGURE 2.
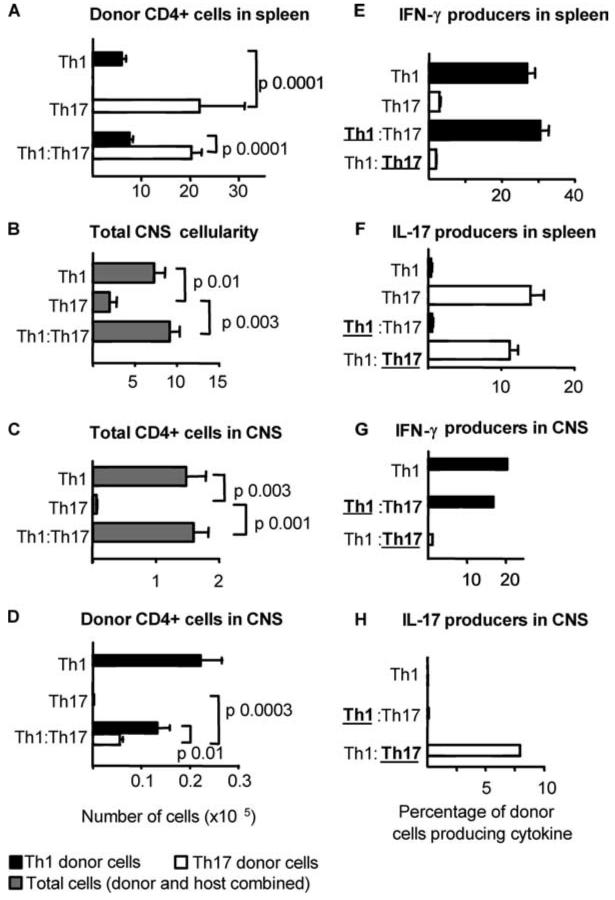
Th1- but not Th17-polarized effectors enter the CNS and initiate inflammation. Polyclonal Th1- (CD45.1/CD90.2) and Th17-polarized (CD45.2/CD90.1) cells generated from the draining lymph nodes of pMOG-immunized mice were transferred to CD45.2/CD90.2 hosts alone (Th1, Th17) or in combination at a 1:1 ratio (Th1:Th17). The distribution of donor cells on day 16 after transfer was assessed by FACS analysis. A, The number of Th1- (closed bars) and Th17-polarized (open bars) cells recovered from the spleen of recipient mice. B, Total number of mononuclear cells recovered from the CNS of recipient mice. C, Total number of CD4+ T cells recovered from the CNS (donor and host cells combined). D, Number of donor Th1 (closed bars) and Th17 (open bars) recovered from the CNS. E-H, Individual splenic populations (E and F) or pooled CNS mononuclear cells (G and H) sampled from recipient mice were stimulated overnight with pMOG, and cytokine production by donor cells originally polarized to Th1 cells (closed bars) and Th17 cells (open bars) was assayed by intracellular cytokine staining with IFN-γ (E and G) and IL-17 (F and H). Bars represent the mean ± SE of 5-7 recipient mice per group. In the cotransferred groups shown in panels E-H, the populations analyzed for cytokine production are identified with boldface type and are underlined.
The polarization of pMOG-reactive donor populations is stable
The stability of the cytokine phenotype confirmed that the disease seen with Th1 populations was not because of residual IL-17 production and that the lack of disease with Th17 preparations did not reflect a loss of IL-17 production. Among the cells recovered from the spleen and the CNS, the phenotypes of both populations appeared stable (Fig. 2, E-H). It is important to emphasize that donor Th1 cells extracted from the CNS of diseased mice did not show any capacity for IL-17 production (Fig. 2H).
Th17-polarized, myelin-reactive TCR transgenic T cells can induce EAE, but only in the presence of IFN-γ-producing cells
The current understanding that IL-6 plus TGF-β is the key combination for driving Th17 differentiation of naive cells has come from either anti-CD3 stimulation of polyclonal populations or Ag stimulation of naive TCR transgenic cells. To date, the use of naive T cells differentiated in the presence of these cytokines has not been described in an EAE model. We made use of the Tg4 mouse that expresses a transgenic TCR recognizing the Ac1-9 peptide of MBP (11). We generated Th1- and Th17-polarized populations by primary in vitro stimulation of CD4+CD62LhighCD25- cells taken from Tg4 × CD45.1 mice for transfer into B10.PL (CD45.2) recipients (Fig. 3A). Th1-polarized cells (devoid of IL-17 producers) efficiently induced severe, often fatal EAE (Fig. 3B). The number of Th1 blasts transferred correlated well with the severity of EAE. EAE could be induced with as few as 1 × 106 Th1 blasts. Numbers beyond 4 × 106 led to high mortality (data not shown). In this model, Th17-polarized populations could occasionally induce some EAE, but this was a mild disease with delayed onset and reduced severity (Fig. 3B). Furthermore, despite stringent Th17 polarization pretransfer (Fig. 3A), those donor cells recovered from the CNS consistently displayed a capacity for IFN-γ production (Fig. 3C). There was a striking shift in the IL-17+:IFN-γ+ ratio in these populations from ∼200:1 at the time of transfer to ∼3:1 upon extraction from the CNS. In contrast, Th1 donor cells recovered from the CNS did not show any capacity for IL-17 production upon restimulation ex vivo (Fig. 3C). The Th1 phenotype was very stable during preclinical stages, through to the peak of disease (days 5-14; data not shown). In contrast. “Th17” cells recovered from the CNS had a propensity for IFN-γ production, suggesting a degree of instability upon reencounter with Ag (discussed below).
FIGURE 3.
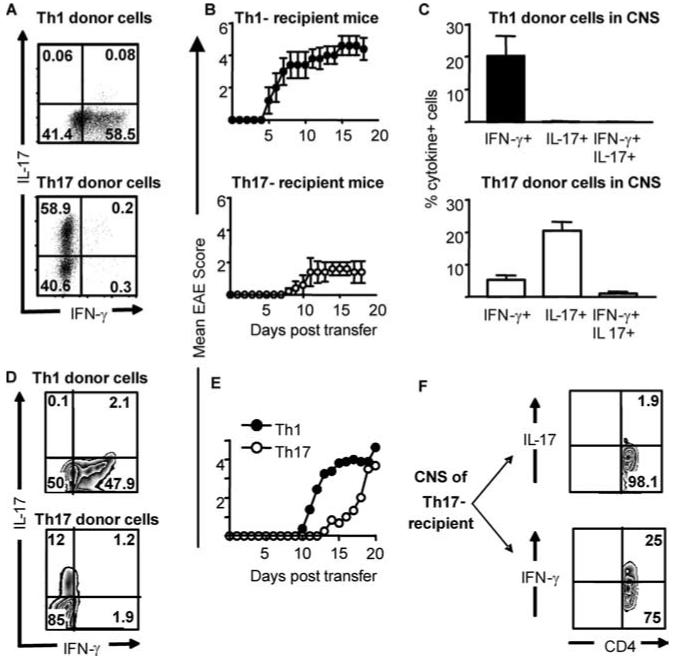
Th17-polarized cells from MBP-reactive TCR transgenic mice can induce mild/delayed EAE, correlating with the in vivo appearance of IFN-γ-producing cells. Th1- and Th17-polarized populations were generated from naive Tg4 mice and transferred into B10.PL mice (A-C) or B10.PL × RAG2-/- mice (D-F). A and D, Intracellular cytokine staining of Th1-polarized (top panels) and Th17-polarized (lower panels) Tg4 cells before transfer. B, Clinical course of EAE induced by the transfer of Th1 (upper panel) and Th17 (lower panel) cells, n = 5. C, Ac1-9 stimulated IFN-γ and IL-17 production by Th1 (upper panel) and Th17 (lower panel) donor cells recovered from the CNS as determined by intracellular cytokine staining. Graphs show the mean ± SD of cells recovered from three surviving Th1-transferred and five Th17-transferred mice. E, Th1 (closed symbols), or Th17-polarized (open symbols) Tg4 T cells (as shown in D) were transferred to B10.PL × RAG-/- recipients (three to four mice per group). F, Ac1-9-stimulated cytokine production by CD4+ T cells recovered from the CNS of a representative B10.PL × RAG2-/- recipient with a disease score of 3. Data are from two of seven experiments giving consistent results.
The data shown in Fig. 2 indicated that transferred IL-17 producing cells did not have an inherent fragility in vivo (they accumulated in large numbers in the spleen). Nevertheless, we sought to maximize their opportunity for clonal expansion and disease initiation by transfer into lymphopenic B10.PL × RAG2-/- hosts (Fig. 3, D-F). Th17 preparations could induce disease in RAG2-/- recipients (Fig. 3E). However, the disease induced with Th1 effectors was always more rapid, with higher mortality. Using the Th1 passive transfer system, we have recently reported that host-derived CD4+Foxp3+ cells are recruited to the inflamed CNS and play a key role in recovery from EAE (9). The absence of these cells in lymphopenic hosts therefore is probably an important factor contributing to this high mortality. Importantly, cells taken from the diseased CNS after transfer of Th17-polarized populations clearly showed the presence of IFN-γ-producing cells. In fact, as illustrated in Fig. 3F, IFN-γ-producing cells were found to markedly outnumber IL-17-producing cells in the CNS of these mice.
Signature cytokine production is more stable in Th1 than in Th17 populations
We assessed the stability of in vitro primed Th1 and Th17 populations following restimulation in the absence of exogenous polarizing cytokines. Both populations continued to produce their signature cytokine, but not the opposing cytokine, in the absence of secondary Ag stimulation (Fig. 4, A and B). We never observed IL-17 production by Th1 preparations, even at high Ac1-9 concentrations (Fig. 4A). In contrast high-dose Ac1-9 could provoke IFN-γ production by Th17 preparations (Fig. 4, B and C). Ag-induced TNF-α production was equivalent in Th1- and Th17-polarized cells (Fig. 4D), whereas the latter produced greater amounts of IL-6 (Fig. 4E).
FIGURE 4.
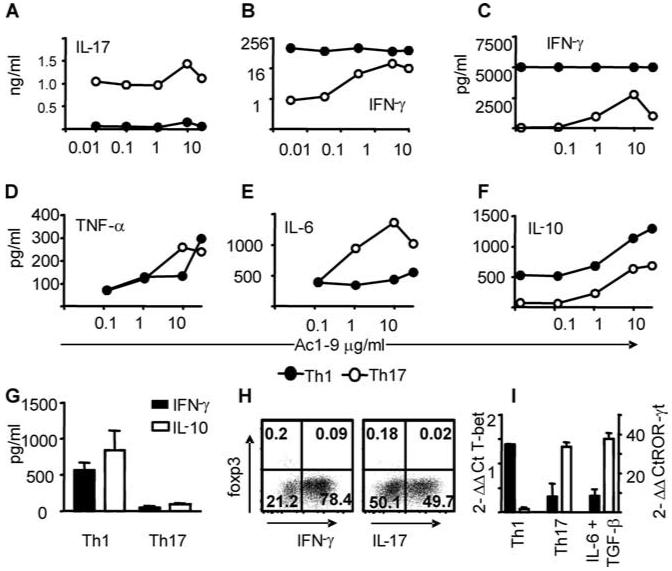
Th1-polarized cells are phenotypically stable. A-F, Th1-polarized (closed circles) and Th17-polarized (open circles) Tg4 cells were washed extensively and restimulated with graded concentrations of Ac1-9. Cytokine production after 72 h of culture was assessed by ELISA (A and B) or cytometric bead array (C-F). G, IFN-γ and IL-10 production by polyclonal Th1- and Th17-polarized cells derived from pMOG-primed mice as determined by ELISA. H, Signature cytokine production vs Foxp3 expression in populations of Th1 (left panel) and Th17 (right panel) Tg4 cells sampled on the day of transfer. I, Expression of T-bet (filled bars) and ROR-γt (open bars) at 72 h (expressed as fold induction over cells stimulated under neutral conditions).
Th17-polarized cells do not show increased IL-10 production or Foxp3 expression
It has recently been reported that, in addition to IL-17 production, TGF-β and IL-6 promote IL-10 production, which limits the pathogenic activity of T effectors (and may even confer suppressive activity), whereas IL-23 promotes the production of IL-17 alone, conferring pathogenicity (12). We consistently included IL-23 in our Th17-polarizing conditions alongside TGF-β and IL-6 to maximize pathogenic potential. Furthermore, upon restimulation, Th1-polarized Tg4 populations always produced higher levels of IL-10 than their Th17-polarized counterparts (Fig. 4F). Similarly, Th1-polarized populations from pMOG-primed mice produced IL-10, whereas this was negligible in Th17 preparations (Fig. 4G). Therefore IL-10 production by Th17-polarized cells does not account for their lack of pathogenicity in this instance. Our Th17 preparations were uniformly Foxp3-negative (Fig. 4H). Therefore, we have no evidence for a regulatory component that would inhibit EAE upon the transfer of Th17 preparations. This conclusion is supported by the fact that their inclusion did not suppress the pathogenic activity of Th1 populations (Fig. 1B). Populations polarized using only IL-6 and TGF-β did not significantly attenuate disease induced by Th1 effectors (Fig. 1C). Thus, our Th1-polarized populations were not sensitive to the suppressive activity of such cells, as reported by McGeachy et al. using IL-23-conditioned pathogenic populations (12). Expression of the lineage-specific transcription factors T-bet and ROR-γt in our pretransfer populations (following primary polarization) showed elevated T-bet expression in Th1 and elevated ROR-γt expression in Th17 preparations, respectively (Fig. 4I).
Discussion
Our data provide a picture of Th1 effectors preferentially infiltrating the noninflamed CNS to initiate inflammation that facilitates the recruitment of Th17 cells. These data are clearly at odds with the recent paradigm shift that has led to the role of Th1 cells in EAE being questioned. This has been based largely on active disease models using CFA immunization in gene knockout mice, in which the effects can be complex. For example, exaggerated EAE in IL-12(p35)-/- mice (3) is complicated by their lack of IL-35, which has a reported role in regulatory T cell function (13). To get a definitive picture of their comparative pathogenic potential, it is imperative to transfer disease with Th1 or Th17 cells, avoiding complications that might arise from the use of CFA in the host, and also to use populations that are as devoid of the opposite population as possible. The literature lacks clear evidence for a pure Th17 population, totally deficient in IFN-γ production, having any capacity to initiate CNS autoimmune disease. Previous studies (4) used IL-23-expanded, IL-17-producing preparations that clearly included contaminating IFN-γ+ cells (4, 12). Using such mixed donor populations, the contribution of IFN-γ- and IL-17-producing cells could not be determined. By using traceable populations with truly distinct cytokine secretion profiles, we have been able to dissect the comparative migratory potential and functional stability of Th1 vs Th17 cells with greater exactitude than previously achieved.
Three recent reports of actively induced EAE show that the majority of IL-17-producing CD4+ cells in the CNS also produce IFN-γ (10, 14, 15). We also see these cells after culture with IL-23 alone (Fig. 1A), but not after culture with IL-23, IL-6, and TGF-β. However, whenever we saw disease after Th17 transfer, IFN-γ-producing cells were always over-represented in the CNS compared with their frequency at the time of transfer (Fig. 3). The ability of Th17-polarized populations to gain IFN-γ-producing capacity upon subsequent antigenic stimulation (Fig. 4) may lead to the organ-specific enrichment of IFN-γ+ and IFN-γ+IL-17+ cells in the CNS as has been seen following the transfer of Th17-polarized populations (12). A recent study reporting a pathogenic role for Th17-polarized populations in autoimmune disease of the eye also indicated that their ability to produce IL-17 waned with continued stimulation (16). Consistent with our data, that study showed the induction of disease by a Th1 cell line in the complete absence of IL-17 production (16). Clearly, there is a need for better understanding of the stability of the Th17 phenotype in terms of the production of IL-17 vs other cytokines thought to be more associated with a Th1 response. It may be that either cell can cause pathology under the appropriate conditions and that this may not require the production of the Th1 or Th17 signature cytokines. For example, both cell types can produce significant amounts of TNF-α (Fig. 4). Our data indicate that Th1 cells enter the CNS and modify conditions therein, making it both attractive and accessible to Th17 cells. Understanding the basis for the differential migratory potential and cytokine stability of these polarized populations might allow the development of therapeutic strategies to modulate their distribution and function in vivo.
Footnotes
This work was supported by grants form the Medical Research Council (U.K.) and the Wellcome Trust. C.A.S. is supported by the Batchworth Trust. S.M.A. is a Medical Research Council Senior Research Fellow and a Research Councils U.K. Fellow in Translational Medicine.
- EAE
- experimental autoimmune encephalomyelitis
- Ac
- acetylated
- MBP
- myelin basic protein
- pMOG
- myelin oligodendrocyte glycoprotein peptide.
Publisher's Disclaimer: The costs of publication of this article were defrayed in part by the payment of page charges. This article must therefore be hereby marked advertisement in accordance with 18 U.S.C. Section 1734 solely to indicate this fact.
Disclosures
The authors have no financial conflict of interest.
References
- 1.Merrill JE, Kono DH, Clayton J, Ando DG, Hinton DR, Hofman FM. Inflammatory leukocytes and cytokines in the peptide-induced disease of experimental allergic encephalomyelitis in SJL and B10.PL mice. Proc. Natl. Acad. Sci. USA. 1992;89:574–578. doi: 10.1073/pnas.89.2.574. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Ando DG, Clayton J, Kono D, Urban JL, Sercarz EE. Encephalitogenic T cells in the B10.PL model of experimental allergic encephalomyelitis (EAE) are of the Th-1 lymphokine subtype. Cell Immunol. 1989;124:132–143. doi: 10.1016/0008-8749(89)90117-2. [DOI] [PubMed] [Google Scholar]
- 3.Cua DJ, Sherlock J, Chen Y, Murphy CA, Joyce B, Seymour B, Lucian L, To W, Kwan S, Churakova T, et al. Interleukin-23 rather than interleukin-12 is the critical cytokine for autoimmune inflammation of the brain. Nature. 2003;421:744–748. doi: 10.1038/nature01355. [DOI] [PubMed] [Google Scholar]
- 4.Langrish CL, Chen Y, Blumenschein WM, Mattson J, Basham B, Sedgwick JD, McClanahan T, Kastelein RA, Cua DJ. IL-23 drives a pathogenic T cell population that induces autoimmune inflammation. J. Exp. Med. 2005;201:233–240. doi: 10.1084/jem.20041257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Kleinschek MA, Owyang AM, Joyce-Shaikh B, Langrish CL, Chen Y, Gorman DM, Blumenschein WM, McClanahan T, Brombacher F, Hurst SD, et al. IL-25 regulates Th17 function in autoimmune inflammation. J. Exp. Med. 2007;204:161–170. doi: 10.1084/jem.20061738. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Chen Y, Langrish CL, McKenzie B, Joyce-Shaikh B, Stumhofer JS, McClanahan T, Blumenschein W, Churakovsa T, Low J, Presta L, et al. Anti-IL-23 therapy inhibits multiple inflammatory pathways and ameliorates autoimmune encephalomyelitis. J. Clin. Invest. 2006;116:1317–1326. doi: 10.1172/JCI25308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Uyttenhove C, Van Snick J. Development of an anti-IL-17A auto-vaccine that prevents experimental auto-immune encephalomyelitis. Eur. J. Immunol. 2006;36:2868–2874. doi: 10.1002/eji.200636662. [DOI] [PubMed] [Google Scholar]
- 8.Kikly K, Liu L, Na S, Sedgwick JD. The IL-23/Th(17) axis: therapeutic targets for autoimmune inflammation. Curr. Opin. Immunol. 2006;18:670–675. doi: 10.1016/j.coi.2006.09.008. [DOI] [PubMed] [Google Scholar]
- 9.O’Connor R,A, Malpass KH, Anderton SM. The inflamed central nervous system drives the activation and rapid proliferation of Foxp3+ regulatory T cells. J. Immunol. 2007;179:958–966. doi: 10.4049/jimmunol.179.2.958. [DOI] [PubMed] [Google Scholar]
- 10.Ivanov II, McKenzie BS, Zhou L, Tadokoro CE, Lepelley A, Lafaille JJ, Cua DJ, Littman DR. The orphan nuclear receptor RORγt directs the differentiation program of proinflammatory IL-17+ T helper cells. Cell. 2006;126:1121–1133. doi: 10.1016/j.cell.2006.07.035. [DOI] [PubMed] [Google Scholar]
- 11.Liu GY, Fairchild PJ, Smith RM, Prowle JR, Kioussis D, Wraith DC. Low avidity recognition of self-antigen by T cells permits escape from central tolerance. Immunity. 1995;3:407–415. doi: 10.1016/1074-7613(95)90170-1. [DOI] [PubMed] [Google Scholar]
- 12.McGeachy MJ, Bak-Jensen KS, Chen Y, Tato CM, Blumenschein W, McClanahan T, Cua DJ. TGF-β and IL-6 drive the production of IL-17 and IL-10 by T cells and restrain TH-17 cell-mediated pathology. Nat. Immunol. 2007;8:1390–1397. doi: 10.1038/ni1539. [DOI] [PubMed] [Google Scholar]
- 13.Collison LW, Workman CJ, Kuo TT, Boyd K, Wang Y, Vignali KM, Cross R, Sehy D, Blumberg RS, Vignali DA. The inhibitory cytokine IL-35 contributes to regulatory T-cell function. Nature. 2007;450:566–569. doi: 10.1038/nature06306. [DOI] [PubMed] [Google Scholar]
- 14.Suryani S, Sutton I. An interferon-γ-producing Th1 subset is the major source of IL-17 in experimental autoimmune encephalitis. J. Neuroimmunol. 2007;183:96–103. doi: 10.1016/j.jneuroim.2006.11.023. [DOI] [PubMed] [Google Scholar]
- 15.Axtell RC, Xu L, Barnum SR, Raman C. CD5-CK2 binding/activation-deficient mice are resistant to experimental autoimmune encephalomyelitis: protection is associated with diminished populations of IL-17-expressing T cells in the central nervous system. J. Immunol. 2006;177:8542–8549. doi: 10.4049/jimmunol.177.12.8542. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Luger D, Silver PB, Tang J, Cua D, Chen Z, Iwakura Y, Bowman EP, Sgambellone NM, Chan C-C, Caspi RR. Either a Th17 or a Th1 effector response can drive autoimmunity: conditions of disease induction affect dominant effector category. J. Exp. Med. 2008;205:799–810. doi: 10.1084/jem.20071258. [DOI] [PMC free article] [PubMed] [Google Scholar]