

Abstract
Class I and class II molecules of the major histocompatibility complex present peptides to T cells. Class I molecules bind peptides that have been generated in the cytosol by proteasomes and delivered into the endoplasmic reticulum by the transporter associated with antigen presentation. In contrast, class II molecules are very efficient in the presentation of antigens that have been internalized and processed in endosomal/lysosomal compartments. In addition, class II molecules can present some cytosolic antigens by a TAP-independent pathway. To test whether this endogenous class II presentation pathway was linked to proteasome-mediated degradation of antigen in the cytosol, the N-end rule was utilized to produce two forms of the influenza virus matrix protein with different in vivo half-lives (10 min vs. 5 h) when expressed in human B cells. Whereas class I molecules presented both the short- and the long-lived matrix proteins, class II molecules presented exclusively the long-lived form of antigen. Thus, rapid degradation of matrix protein in the cytosol precluded its presentation by class II molecules. These data suggest that the turnover of long-lived cytosolic proteins, some of which is mediated by delivery into endosomal/lysosomal compartments, provides a mechanism for immune surveillance by CD4+ T cells.
Keywords: antigen processing, HLA-DR, N-end rule, protein degradation, ubiquitin
There are important distinctions between the antigen presentation pathways used by class I and class II molecules of the major histocompatibility complex (MHC) (for review, see ref. 1). MHC class I molecules, which present peptides to CD8+ T cells, acquire peptides in the endoplasmic reticulum. Except for some peptides that may be generated in the endoplasmic reticulum (2, 3), most of the peptides bound to MHC class I molecules are derived from proteins processed in the cytosol by the multicatalytic proteasomes (4). The resulting short peptides are delivered into the endoplasmic reticulum by a transporter associated with antigen presentation (TAP). This mechanism ensures that most of the peptides bound to MHC class I molecules are derived from endogenous proteins and provides a sensitive detection system for infected or transformed cells. Some cell types such as macrophages can engulf exogenous proteins that are subsequently released into the cytosol and processed by the proteasome- and TAP-dependent pathway for class I-restricted presentation (5–7).
In contrast, exogenous material taken up into endocytic compartments is the major source of antigens presented by MHC class II molecules to CD4+ T cells. The high efficiency of exogenous antigen presentation by MHC class II molecules provides the immune system with a sensitive detection system for the presence of foreign material. Internalized antigen is denatured by low pH and degraded by endosomal or lysosomal proteases, exposing peptide segments that bind to newly synthesized MHC class II molecules. Peptide loading onto class II molecules is facilitated by HLA-DM (reviewed in ref. 8). The resulting class II/peptide complexes are transported to the plasma membrane for detection by CD4+ T cells. An alternative presentation pathway, independent of HLA-DM but requiring internalization and recycling of cell surface class II molecules, is used for certain antigens (9).
In addition, ample evidence has accumulated for class II-restricted presentation of cytosolic antigens (10–13). The TAP-independence of class II-restricted presentation of cytosolic antigen established that this pathway was distinct from the class I pathway (13). The endogenous class II pathway shares features with the classical exogenous pathway: it is chloroquine-sensitive (10, 12) and requires a function(s) encoded in the class II region of the MHC, presumably HLA-DM (14). Processing of a cytosolic antigen for class II-restricted presentation took several hours, in contrast to a much more rapid presentation by MHC class I (11). The endogenous class II pathway may be inefficient or may operate selectively for certain antigens because MHC class II molecules have often failed to present cytosolic antigens (15–18).
The pathway by which some cytosolic antigens can be delivered into a class II compartment for loading onto class II molecules is still unknown. The chloroquine-sensitivity of this presentation does not imply processing of antigen in an endosomal/lysosomal compartment because chloroquine also blocks the prerequisite dissociation of the invariant chain from newly synthesized class II αβ/invariant chain complexes. Therefore, it is possible that cytosolic antigen presented by MHC class II undergoes proteasome-mediated degradation. Alternatively, cytosolic antigens may be delivered into class II compartments independently of proteasome-mediated processing.
To test whether degradation by proteasomes was required for class II-restricted presentation of cytosolic antigen, the N-end rule pathway of protein degradation (19, 20) was used to produce two forms of a cytosolic viral protein: one was targeted for rapid ubiquitin (Ub)-dependent degradation and the other remained stable in the cytosol. The matrix protein of influenza virus was chosen because of its inherent stability in the cytosol. In addition, it carries well-defined T-cell epitopes restricted by the human class I and class II MHC alleles HLA-A2 and HLA-DR1, respectively. Endogenous expression of M1 mediated by a recombinant vaccinia virus resulted in presentation of both class I and class II epitopes (10). The present study showed that whereas both the short- and long-lived forms of the antigen were presented efficiently by class I, only the long-lived form was presented by class II.
MATERIALS AND METHODS
Cells and Recombinant Viruses.
The HLA-A2 and HLA-DR1 positive human B-cell line 721.45 has been described (21). Recombinant vaccinia viruses encoding the influenza virus A matrix protein M1 (Vac-M1), hemagglutinin protein H3 (Vac-H3), or the T7 RNA polymerase (Vac-T7) have been described (22–24). Fusion proteins between Ub and influenza virus matrix M1 were engineered that contained either a methionine or an arginine residue after the Ub cleavage site. An Ub cDNA (gift of A. Townsend, Oxford University) was subcloned in the plasmid pKS (Bluescript II KS+, Stratagene) as an EcoRI fragment. An Ub-Met fragment containing a Bsp120I site downstream of the Met codon was produced by PCR amplification using the forward Ub primer ACGTACGTGCGGCCGCGCCAACATGCAGATCTTCGTG with a NotI cloning site and the backward primer GTACGTGAATTCGGGCCCTTCATACCACCGCGCAGACGCAG with an EcoRI cloning site. An Ub-Arg fragment was similarly amplified using the backward primer GTACGTGAATTCGGGCCCTTTCGACCACCGCGCAGACGCAG. The 273-bp fragment was purified, digested with NotI and EcoRI, and ligated into NotI–EcoRI digested plasmid pKS downstream of the T7 polymerase promoter. The Bsp120I site in plasmid pKS was previously destroyed by a fill-in reaction and religation. The sequences of the Ub-Met and Ub-Arg inserts were verified by sequencing. A Bsp120I site was introduced upstream of the M1 coding sequence in plasmid pr8 ma (gift of P. Palese) (22) by PCR amplification with the forward primer ACGTACGTGGGCCCAATGAGTCTTCTAACGGAGG and the backward primer ACGTACGTGTCGACAGTAGAAACAAGGTAGTTTTTT that provided a SalI cloning site at the 3′ end of the M1 cDNA. The 1031-bp fragment was purified, digested with Bsp120I and SalI, and ligated to the plasmids pKS-Ub-Met and pKS-Ub-Arg digested with Bsp120I and SalI. In the resulting constructs the M1 coding region is preceded by Ub-Met-Lys-Gly-Pro or by Ub-Arg-Lys-Gly-Pro. Triple chimeric proteins containing the first 44 amino acids of the influenza virus A nucleoprotein (NP) between Ub and M1 were constructed as follows. Ub-Met-NP and Ub-Arg-NP fragments were amplified from plasmids ptac-85-Ub-Met-NP and ptac-85-Ub-Arg-NP (25) with the Ub forward primer and the backward primer ACGTACGTGAATTCGGGCCCTTGCACATTTGGATGTAGAATCGT that introduces a Bsp120I site after codon 44 of NP and provides an EcoRI cloning site. The 407-bp fragment was purified, digested with NotI and EcoRI, and ligated into plasmid pKS (lacking the Bsp120I site) digested with NotI and EcoRI. The M1 fragment amplified as described above was ligated into pKS-Ub-Met-NP and pKS-Ub-Arg-NP digested by Bsp120I and SalI. In the resulting constructs the M1 coding region is preceded by Ub-Met or Ub-Arg, and the next 43 NP codons fused to Lys-Gly-Pro codons. To produce recombinant vaccinia viruses, two independent NotI–SalI inserts from pKS-Ub-Met-NP-M1 and from pKS-Ub-Arg-NP-M1 were subcloned into a modified plasmid pSC11 containing NotI and SalI cloning sites (gift from I. Bacik, National Institute of Allergy and Infectious Diseases, National Institues of Health) (26). One clone for each construct was used to generate a recombinant vaccinia virus as described (27). The two independent Vac-Ub-Met-NP-M1 constructs gave identical results in multiple experiments, and so did the two Vac-Ub-Arg-NP-M1 constructs. Recombinant vaccinia viruses were purified on a sucrose gradient as described (27).
Vac-T7-Mediated Expression.
721.45 cells were infected with 10 plaque-forming units (pfu)/cell of Vac-T7 for 10 min in serum-free medium on ice followed by rotation for 30 min at 37°C. After removing the infection medium, 4 × 106 infected cells were transfected with 5 μg of plasmid using lipofectin (Life Technologies, Grand Island, NY) in 2 ml OptiMEM (Life Technologies) and incubated at 37°C for 2 h. The cells were then transferred to a T25 flask, 8 ml of complete medium was added, and the flask was incubated at 37°C for 2 h. Cells were starved in methionine-free medium for 30 min, labeled with [35S]methionine for 30 min and chased for 0 and 1 h in medium supplemented with 2 μM methionine.
Immunoprecipitations.
721.45 cells (2 × 106) were infected with 30 pfu/cell of vaccinia virus at 4°C for 20 min in 0.5 ml serum-free medium, and further incubated on a rotator for 1 h at 37°C. Infected cells were starved in methionine-free medium for 1 h, and labeled with 50 μCi (1 Ci = 37 GBq) [35S]methionine for 10 min (Vac-Ub-Met-NP-M1) or 5 min (Vac-Ub-Arg-NP-M1). Cells were resuspended in cold medium supplemented with 2 μM methionine and chased for up to 4 h at 37°C. Cells were harvested and lysed as described (3) except that the lysis buffer contained 1% Triton X-100 and 1 mg/ml BSA. Immunoprecipitation was performed with the M1-specific mAb M2/1C6 (gift of J. Yewdell, National Institute of Allergy and Infectious Diseases, National Institutes of Health) (28). In the calpain inhibitor I experiment, 2 × 106 721.45 cells were infected with 15 pfu/cell as described above. Cells were starved for 30 min in methionine-free medium, incubated for another 30 min in the same medium supplemented with 200 μM of calpain inhibitor I (Calbiochem), and labeled with [35S]methionine for 10 min in the presence of the inhibitor. Immunoprecipitates were analyzed by SDS/PAGE as described (3) and radioactive bands on the dried gels were quantitated using the AMBIS Radioanalytic Imaging system.
RNA Hybridization.
721.45 cells (10 × 106) were infected with 10 pfu/cell as described above. At the end of the incubation at 37°C in serum-free medium, FCS was added to a final concentration of 5% and the cells incubated for 3 h at 37°C. Cells were then washed in 10 mM Tris·HCl (pH 7.5) and 150 mM NaCl. RNA was extracted by the guanidine/acid/phenol technique (29). Ten micrograms total RNA was run on a 0.8% agarose/formaldehyde denaturing gel and transferred using a Stratagene pressure blotter to a Gene Screen Plus membrane (Dupont/NEN). The blot was probed overnight at 68°C with the NotI–SalI insert of Ub-Met-NP-M1 radiolabeled with [32P]dCTP using a Prime-It II kit from Stratagene. The blot was then washed with 2× standard saline citrate (SSC), 0.1% SDS at 42°C, 2× SSC/0.1% SDS at 68°C, 0.2× SSC/0.1% SDS at 68°C, and finally 0.1× SSC/0.1% SDS at 68°C. Each wash lasted 30 min.
Cytotoxicity Assays.
721.45 cells (5 × 105) were infected as described above. At the end of the incubation at 37°C in serum-free medium, fetal calf serum was added to a final concentration of 5% and the cells incubated for 2 h at 37°C. Cells were labeled with 50 μCi sodium [51Cr] chromate for 1 h in 0.2 ml, washed, counted and plated at 5 × 103 cells in V-bottom 96-well plates containing previously aliquoted effector T cells. The total time of vaccinia virus infection was 4 h. HLA-A2-restricted M1-specific T-cell lines (30) were a gift of W. Biddison (National Institute of Neurological Diseases and Stroke, National Institutes of Health). HLA-DR1-restricted M1-specific T-cell clones and 51Cr-release assays have been described (31).
RESULTS
A Fusion Protein of Ub and Influenza Virus Matrix Did Not Follow the N-End Rule.
Vectors encoding fusion proteins that consist of Ub followed by either methionine or arginine and the influenza virus matrix M1 protein were constructed (Fig. 1A). A similar strategy had been used by Townsend (25) to confer different in vivo half-lives on the influenza virus NP. According to the N-end rule, upon cleavage of the Ub, the protein with an N-terminal methionine should remain stable, whereas the one with an N-terminal arginine should be destabilized (19, 20, 32). Specific lysine residues in the N-terminal portion of the protein are also required for the degradation of a protein by the N-end rule pathway (20, 33). The first three lysine residues after cleavage of the N-terminal Ub extension of the Ub-Met-M1 and Ub-Arg-M1 fusion proteins are at positions 2, 26, and 40. The transient Vac-T7 polymerase expression system (24) was used to quickly test whether the Arg-M1 was rapidly degraded. The human B-cell line 721.45 was infected with Vac-T7 and transfected with plasmids encoding Ub-Met-M1 and Ub-Arg-M1 downstream of the T7 promoter. After labeling with [35S]methionine for 30 min, M1 proteins were immunoprecipitated (Fig. 1B). Comparable levels of Met-M1 and Arg-M1 proteins with the expected apparent molecular weight (28 kDa) were detected. Furthermore, Arg-M1 protein was still detectable after 1 h of chase (Fig. 1B).
Figure 1.
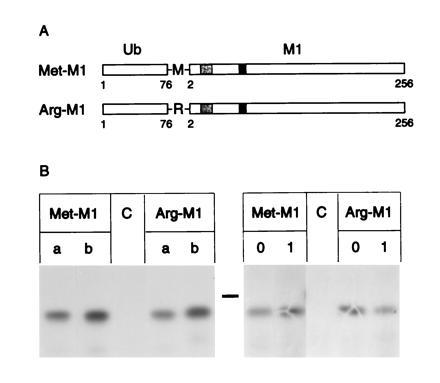
Biosynthesis of recombinant Ub-Met-M1 and Ub-Arg-M1 proteins. (A) Diagram of the Ub-Met-M1 and Ub-Arg-M1 proteins. The Ub protein (□) was fused to the matrix M1 protein (□ with shaded areas) with a methionine (Met-M1) or an arginine (Arg-M1) junction after the Ub cleavage site. The HLA-DR1-restricted epitope 18–31 and the HLA-A2-restricted epitope 58–66 are indicated by light and dark shading, respectively. (B) 721.45 cells infected with Vac-T7 and transfected with Ub-Met-M1, Ub-Arg-M1, or a control plasmid (C), as indicated, were labeled with [35S]methionine for 30 min. Lanes a and b represent independent transfections each with an independent recombinant construct. Lanes 0 and 1 represent cells chased for 0 and 1 h, respectively. Immunoprecipitated M1 molecules were revealed by SDS/PAGE and autoradiography. The bar indicates the position of a 30-kDa protein marker.
An N-Terminal Extension of the Matrix Protein Was Required to Obtain Half-Lives That Obey the N-End Rule.
As NP bearing the N-terminal arginine residue was degraded by the N-end rule pathway (25), we reasoned that the N-terminal portion of NP may carry appropriate signals (including lysine residues) that could be grafted onto the N-terminus of M1 and provide recognition of the NP-M1 fusion proteins by the N-end rule pathway. Thus, the first 44 amino acids of NP were fused to M1 with an additional Lys-Gly-Pro linker. This N-terminal fragment of NP was isolated from Ub-Met-NP and Ub-Arg-NP fusion constructs made previously (25). The first three lysine residues after cleavage of the N-terminal Ub extension of these Ub-Met-NP-M1 and Ub-Arg-NP-M1 fusion proteins (Fig. 2A) are at positions 7, 31, and 45. The Vac-T7 polymerase expression system revealed a lower amount of Arg-NP-M1 relative to that of Met-NP-M1 detectable after labeling with [35S]methionine for 30 min (Fig. 2B). To define more precisely the half-lives of these recombinant proteins two independent recombinant vaccinia viruses for each Ub-Met-NP-M1 and Ub-Arg-NP-M1 construct were generated and tested for protein stability after infection of 721.45 cells (Fig. 3). Both proteins were synthesized with the expected apparent molecular weight (33 kDa) but with very different half-lives: ≈5 h for Met-NP-M1 and ≈10 min for Arg-NP-M1. Half-lives were determined from two experiments by quantitation of bands in SDS/PAGE. The half-life of a wild-type M1 protein expressed from Vac-M1 was at least 6 h in several experiments (not shown). A short lag of 5–10 min was observed before degradation of Arg-NP-M1, presumably because the cells had been on ice before addition of the chase medium.
Figure 2.
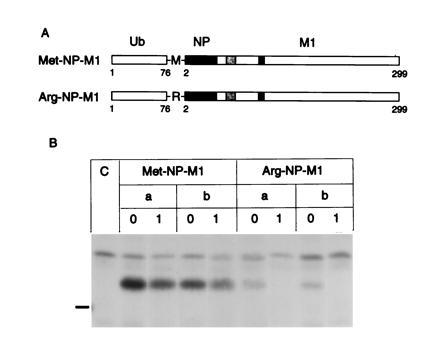
Biosynthesis of recombinant Ub-Met-NP-M1 and Ub-Arg-NP-M1 proteins. (A) Diagram of the proteins as in Fig. 1. (▪) Amino acids 2–44 of NP. (B) Pulse-chase analysis of recombinant proteins as in Fig. 1.
Figure 3.
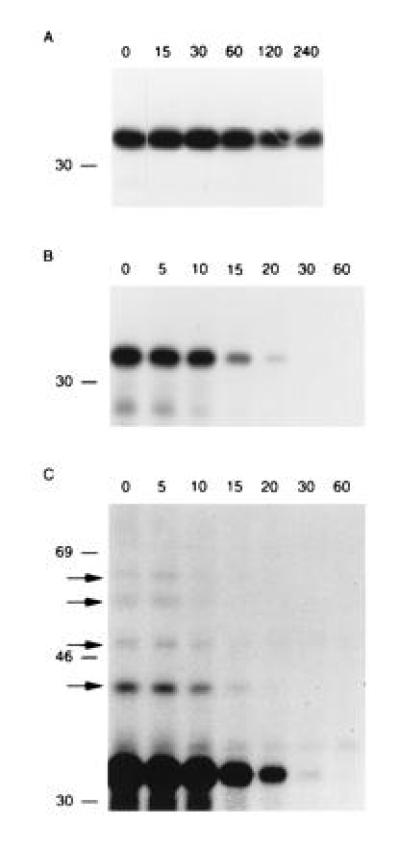
The Met-NP-M1 and Arg-NP-M1 fusion proteins follow the N-end rule. 721.45 cells were infected with Vac-Ub-Met-NP-M1 (A) or Vac-Ub-Arg-NP-M1 (B and C). After [35S]methionine labeling for 10 min (A) or 5 min (B and C), cells were chased for the indicated times in min. Immunoprecipitations of NP-M1 fusion proteins were revealed by SDS/PAGE and autoradiography. Exposure was 6 h in A and 3 days in (B). C is a longer exposure (11 days) of B. Arrows indicate slower migrating bands in C that were not visible in A even after a long exposure. The position of [14C]-labeled markers is indicated on the left.
A long exposure of the gel containing immunoprecipitated Arg-NP-M1 revealed additional bands corresponding to about 42, 49, 56, and 62 kDa (Fig. 3C). This ladder may well reflect multiubiquitination of the protein containing a destabilizing N-terminal amino acid and an appropriate lysine substrate (34). The bands may correspond to Arg-NP-M1 molecules with 1, 2, 3, and 4 Ub moieties. No such bands appeared in long exposures of the gel containing immunoprecipitated Met-NP-M1 (not shown).
Identical half-lives were determined for each pair of independent vaccinia viruses encoding Ub-Met-NP-M1 or Ub-Arg-NP-M1 proteins. We conclude that the Met-NP-M1 and Arg-NP-M1 proteins follow the N-end rule.
Equal Expression Levels from the Ub-Met-NP-M1 and Ub-Arg-NP-M1 Constructs.
The levels of transcription and translation from the Ub-Met-NP-M1 and Ub-Arg-NP-M1 recombinant genes in vaccinia viruses were expected to be very similar because these constructs differ only by the methionine or arginine codons. However, the amount of Arg-NP-M1 immunoprecipitated after a 5-min pulse with [35S]methionine was lower than that of Met-NP-M1 (not shown). This difference may be due to the very short half-life of Arg-NP-M1 or to a lower level of expression. To test whether expression levels of these two proteins differed, both RNA and protein levels were compared. RNA extracted from Vac-Ub-Met-NP-M1 and Vac-Ub-Arg-NP-M1 infected cells was separated by electrophoresis in a denaturing gel, transferred to a membrane and hybridized with a cDNA probe corresponding to full-length Ub-Met-NP-M1 (Fig. 4). Similar amounts of RNA of the expected size (≈1.5 kb) were detected.
Figure 4.
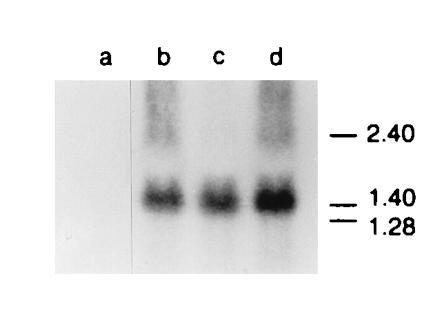
Vac-Ub-Met-NP-M1 and Vac-Ub-Arg-NP-M1 infected cells produce similar levels of RNA encoding M1 fusion proteins. (a) Uninfected 721.45 cells or (b) 721.45 cells infected with Vac-Ub-Met-NP-M1 (b) and Vac-Ub-Arg-NP-M1 (c and d). Total RNA was extracted from infected cells, separated on a denaturing gel, and transferred to a membrane. The membrane was hybridized with a Ub-Met-NP-M1 DNA probe and autoradiographed. The position of RNA markers in kb is indicated on the right.
To determine the level of protein synthesis independently of protein half-life, Vac-Ub-Met-NP-M1 and Vac-Ub-Arg-NP-M1 infected cells were labeled with [35S]methionine in the presence of calpain inhibitor I. This aldehyde inhibitor of serine and cysteine proteases inhibits the multicatalytic proteasome (35). Therefore, the use of this inhibitor prior to and during the labeling should block the degradation of the Arg-NP-M1 protein. Equivalent amounts of Met-NP-M1 and Arg-NP-M1 proteins were immunoprecipitated from Vac-Ub-Met-NP-M1 and Vac-Ub-Arg-NP-M1 infected cells using this protocol (Fig. 5). Quantitation of the protein amount (in arbitrary units) immunoprecipitated from cells infected with the two independent Vac-Met-NP-M1 constructs was 7300 and 8623 counts, and 6829 and 7330 counts from cells infected with the two independent Vac-Arg-NP-M1 constructs. We concluded that the expression levels of Ub-Met-NP-M1 and Ub-Arg-NP-M1 proteins were not significantly different.
Figure 5.
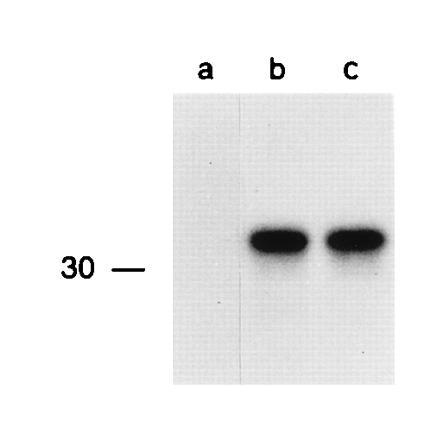
Vac-Ub-Met-NP-M1 and Vac-Ub-Arg-NP-M1 infected cells synthesize M1 fusion proteins at a similar rate. 721.45 cells were either uninfected (a), infected with Vac-Ub-Met-NP-M1 (b) or Vac-Ub-Arg-NP-M1 (c), treated with the calpain inhibitor I and labeled with [35S]methionine for 10 min in the continuing presence of the inhibitor. Immunoprecipitations of NP-M1 fusion proteins were revealed by SDS/PAGE and autoradiography. The position of a 14C-labeled marker is indicated on the left.
A Short Half-Life of M1 Eliminated Presentation to HLA-DR1-Restricted But Not to HLA-A2-Restricted T Cells.
The cytolytic M1-specific and HLA-DR1-restricted T-cell clone C3.5 killed 721.45 cells pulsed with synthetic peptide M1 18–31, 721.45 cells infected with Vac-M1, but not uninfected cells or cells infected with Vac-H3 (Fig. 6 a and b). Clone C3.5 also killed cells infected with the two Vac-Ub-Met-NP-M1 but not those infected with the two Vac-Ub-Arg-NP-M1 (Fig. 6 c and d). To test more quantitatively the difference in presentation of the short- and long-lived proteins and to control for the lack of presentation of Ub-Arg-NP-M1, cells were infected with different doses of vaccinia viruses and tested for lysis by both HLA-A2 and HLA-DR1-restricted T cells. As before, clone C3.5 lysed cells infected with Vac-Ub-Met-NP-M1 but not those infected with Vac-Ub-Arg-NP-M1 (Fig. 7). Recognition of cells infected with Vac-Ub-Met-NP-M1 was dose-dependent. Whereas some lysis still occurred with only 0.3 pfu/cell of Vac-Ub-Met-NP-M1, no lysis was detected with ≤30 pfu/cell of Vac-Ub-Arg-NP-M1 (Fig. 7 b and c). As expected, the M1-specific HLA-A2-restricted T-cell line Q157 lysed cells pulsed with peptide M1 58–66 but not untreated cells or cells infected with Vac-H3 (Fig. 7d). However, in contrast to the distinction between presentation of long- and short-lived proteins by HLA-DR1, both forms of antigen were efficiently presented by HLA-A2 molecules (Fig. 7 e and f).
Figure 6.
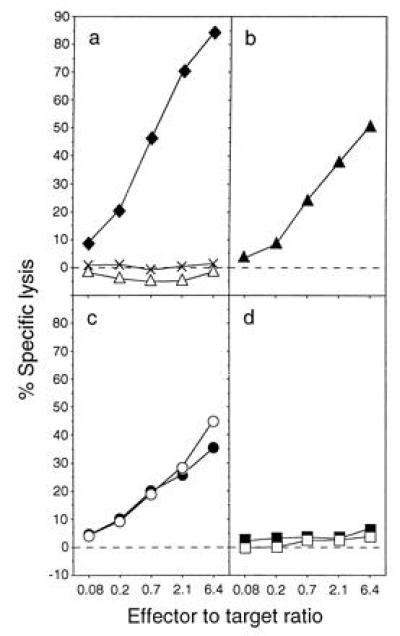
A short half-life precludes presentation of cytosolic M1 to HLA-DR1-restricted T cells. Lysis of 721.45 cells by the M1-specific T-cell clone C3.5. (a) Controls included untreated cells (×), cells incubated with 10 μg/ml synthetic peptide M1 18–31 (♦), and cells infected with Vac-H3 (▵). The other panels represent cells infected with Vac-M1 (b), two independent Vac-Ub-Met-NP-M1 (c), and two independent Vac-Ub-Arg-NP-M1 (d). Each infection was at 30 pfu/cell for 4 h.
Figure 7.
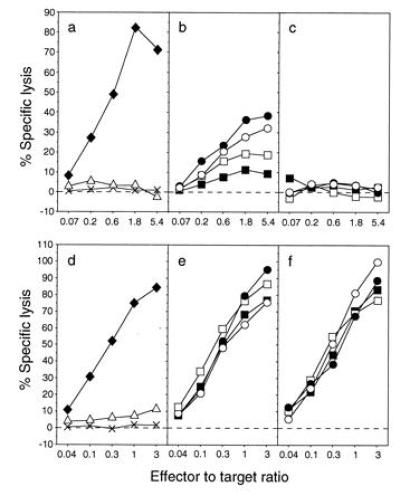
A long half-life is required for the presentation of M1 to HLA-DR1-restricted T cells but not to HLA-A2-restricted T cells. Lysis by the M1-specific HLA-DR1-restricted T-cell clone C3.5 (a–c) or by the M1-specific HLA-A2-restricted T-cell line Q157 (d–f) of 721.45 cells that were untreated (×), incubated with synthetic peptide (♦, M1 18–31 in a, M1 58–66 in d), or infected with Vac-H3 (▵), infected with Vac-Ub-Met-NP-M1 (b and e) or with Vac-Ub-Arg-NP-M1 (c and f). The doses of virus were 30 (○), 10 (•), 1 (□), and 0.3 (▪) pfu/cell and each infection lasted 4 h.
DISCUSSION
This study established that only a long-lived form of endogenous M1, the predominant cytosolic influenza virus antigen, was presented by MHC class II molecules to T cells. A short-lived form of the same M1 antigen was not presented despite similar expression level. Therefore, class II-restricted presentation of cytosolic M1 uses a different pathway from the rapid proteasome-mediated protein degradation. It is unlikely that the lack of presentation of the short-lived M1 was due to a failure of transport by TAP because expression of a minigene encoding the M1 18–31 epitope resulted in presentation by HLA-DR1 molecules in cells that did not express the invariant chain (36). Expression of the invariant chain in those cells blocked presentation of the minigene-encoded M1 epitope (36), suggesting that the short M1 epitope was loaded onto class II in the endoplasmic reticulum. In contrast, whole M1 antigen is most likely delivered to an endosomal/lysosomal compartment for processing because class II-restricted M1 presentation is independent of TAP (13), requires functions encoded in the MHC class II region (14), and, as shown here, is independent of rapid proteasome-mediated degradation.
Cells utilize several pathways for cytosolic protein turnover. Short-lived proteins are degraded mostly by proteasomes in the cytosol whereas the bulk of long-lived proteins is degraded by lysosomal proteases (37, 38). How long-lived proteins reach lysosomal compartments from the cytosol is not entirely clear but several mechanisms may account for their delivery to such class II processing compartments. Macro- and microautophagy are nonselective processes that result in the engulfment of cytosolic material into lysosomes (37). In addition, stress proteins of the 70-kDa heat shock protein family have been implicated in the selective and serum-regulated translocation of cytosolic proteins into lysosomes (39). The signal that mediates such translocation is found on many cytosolic proteins (38) and related sequence motifs are also found in both M1 and H3 antigens.
The Ub-M1 fusion protein first tested did not appear to follow the N-end rule pathway. Despite expression of molecules of the expected size that bore either the stabilizing methionine or the destabilizing arginine N-terminal residues after cleavage of Ub, both forms of M1 were long lived. Structural features of M1, that included an N-terminal Lys-Gly-Pro artificial extension, must be insufficient for targeting by the N-end rule pathway. On the other hand, addition of 44 amino acids derived from the N-terminal portion of influenza virus NP provided the necessary signals for degradation of M1 according to the N-end rule. In the yeast Saccharomyces cerevisiae, rules have been derived concerning the position of lysine residues that are critical for targeting by the Ub-dependent N-end rule pathway (20, 32). Similar rules appear to be relevant in higher eukaryotes (40), in that the context in which lysine residues are placed must also be important. The N-terminal region of NP carries the proper features for recognition by the N-end rule pathway, because NP itself follows the rule (25) and, as shown here, a 44-amino acid NP segment was sufficient to confer recognition of a protein that is otherwise not recognized. The Lys-Gly-Pro linker at the NP-M1 junction did not prevent the degradation of M1 by the N-end rule pathway.
The class I-restricted presentation of M1 was not affected by antigen half-life. This result may reflect the extreme sensitivity of the class I presentation pathway. Accelerated degradation of antigen has improved class I-restricted presentation in some circumstances. During the late phase of a vaccinia virus infection, a decrease in the half-life of NP improved its class I-restricted presentation (25). Furthermore, loading of a single dose of antigen into the cytosol by osmotic lysis of pinosomes resulted in class I-restricted presentation that was enhanced by rapid antigen degradation (41). Short-lived antigen was presented efficiently at early times, whereas long-lived antigen was presented after a delay of several hours (41). However, when antigens are synthesized within cells, as during virus infections, it is likely that class I-restricted presentation to T cells will function even with a slow protein degradation.
The data presented here established a selectivity in the presentation of cytosolic antigens by MHC class II. The requirement for a long-lived form of antigen provides an explanation for the lack of class II-restricted presentation of several antigens, such as engineered cytosolic forms of lysozyme (15) and influenza virus (H2 subtype) hemagglutinin (17) that were both short-lived. High expression levels of the short-lived cytosolic lysozyme did result in class II-restricted presentation (12), possibly via the unusual TAP-dependent pathway sometimes used by class II (13). It is interesting to note that a cytosolic form of hemagglutinin from another influenza virus subtype (H3) with a relatively long half-life (>2.5 h) was presented by class II in a TAP-independent manner (13). The selectivity in presentation of cytosolic antigen by MHC class II should alleviate some of the confusion in the field (18). It is also worth noting that presentation of cytosolic M1 by class II was not a consequence of cytopathic effects due to viral infections because the experiments, including those reported earlier (10, 13, 14), were carried out during the early phase of vaccinia virus infection that precedes the cytopathic effects caused by the late phase of infection. In addition, identification of class II-associated peptides derived from cytosolic proteins in normal cells has provided the best evidence that MHC class II molecules acquire peptides from cytosolic proteins even in the absence of virus infection. Such peptides have been identified by several groups, from several class II molecules, and from different types of class II-expressing cells (42–47). Our data suggest that long-lived cytosolic proteins may contribute peptides for recognition by CD4+ T cells, and that this endogenous class II pathway may participate in the immune surveillance.
Acknowledgments
We thank B. Moss and G. Smith for vaccinia viruses, I. Bacik, P. Palese and A. Townsend for plasmids, W. Biddison for T cells, J. Yewdell for antibodies, A. Brooks and J. Yewdell for comments on the manuscript, and A. Varshavsky for useful discussions.
Footnotes
Abbreviations: H3, H3 hemagglutinin protein of influenza virus A; M1, M1 matrix protein of influenza virus A; MHC, major histocompatibility complex; NP, nucleoprotein of influenza virus A; TAP, transporter associated with antigen presentation; Ub, ubiquitin.
References
- 1.Germain R N. Cell. 1994;76:287–299. doi: 10.1016/0092-8674(94)90336-0. [DOI] [PubMed] [Google Scholar]
- 2.Hammond S A, Bollinger R C, Tobery T W, Siliciano R F. Nature (London) 1993;364:158–161. doi: 10.1038/364158a0. [DOI] [PubMed] [Google Scholar]
- 3.Guéguen M, Biddison W E, Long E O. J Exp Med. 1994;180:1989–1994. doi: 10.1084/jem.180.5.1989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Rock K L, Gramm C, Rothstein L, Clark K, Stein R, Dick L, Hwang D, Goldberg A L. Cell. 1994;78:761–771. doi: 10.1016/s0092-8674(94)90462-6. [DOI] [PubMed] [Google Scholar]
- 5.Kovacsovics-Bankowski M, Rock K L. Science. 1995;267:243–246. doi: 10.1126/science.7809629. [DOI] [PubMed] [Google Scholar]
- 6.Sousa C R E, Germain R N. J Exp Med. 1995;182:841–851. doi: 10.1084/jem.182.3.841. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Norbury C C, Hewlett L J, Prescott A R, Shastri N, Watts C. Immunity. 1995;3:783–791. doi: 10.1016/1074-7613(95)90067-5. [DOI] [PubMed] [Google Scholar]
- 8.Roche P A. Immunity. 1995;3:259–262. doi: 10.1016/1074-7613(95)90111-6. [DOI] [PubMed] [Google Scholar]
- 9.Pinet V, Vergelli M, Martin R, Bakke O, Long E O. Nature (London) 1995;375:603–606. doi: 10.1038/375603a0. [DOI] [PubMed] [Google Scholar]
- 10.Jaraquemada D, Marti M, Long E O. J Exp Med. 1990;172:947–954. doi: 10.1084/jem.172.3.947. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Hackett C J, Yewdell J W, Bennink J R, Wysocka M. J Immunol. 1991;146:2944–2951. [PubMed] [Google Scholar]
- 12.Brooks A G, McCluskey J. J Immunol. 1993;150:3690–3697. [PubMed] [Google Scholar]
- 13.Malnati M S, Marti M, LaVaute T, Jaraquemada D, Biddison W, DeMars R, Long E O. Nature (London) 1992;357:702–704. doi: 10.1038/357702a0. [DOI] [PubMed] [Google Scholar]
- 14.Malnati M S, Ceman S, Weston M, DeMars R, Long E O. J Immunol. 1993;151:6751–6756. [PubMed] [Google Scholar]
- 15.Calin-Laurens V, Forquet F, Mottez E, Kanellopoulos J, Godeau F, Kourilsky P, Gerlier D, Rabourdin-Combe C. Eur J Immunol. 1991;21:761–769. doi: 10.1002/eji.1830210332. [DOI] [PubMed] [Google Scholar]
- 16.Weiss S, Bogen B. Cell. 1991;64:767–776. doi: 10.1016/0092-8674(91)90506-t. [DOI] [PubMed] [Google Scholar]
- 17.Kittlesen D J, Brown L R, Braciale V L, Sambrook J P, Gething M J, Braciale T J. J Exp Med. 1993;177:1021–1030. doi: 10.1084/jem.177.4.1021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Oxenius A, Bachmann M F, Ashton-Rickardt P G, Tonegawa S, Zinkernagel R M, Hengartner H. Eur J Immunol. 1995;25:3402–3411. doi: 10.1002/eji.1830251230. [DOI] [PubMed] [Google Scholar]
- 19.Bachmair A, Finley D, Varshavsky A. Science. 1986;234:179–186. doi: 10.1126/science.3018930. [DOI] [PubMed] [Google Scholar]
- 20.Varshavsky A. Cold Spring Harbor Symp Quant Biol. 1996;60:461–478. doi: 10.1101/sqb.1995.060.01.051. [DOI] [PubMed] [Google Scholar]
- 21.Kavathas P, Bach F H, DeMars R. Proc Natl Acad Sci USA. 1980;77:4251–4255. doi: 10.1073/pnas.77.7.4251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Smith G L, Levin J Z, Palese P, Moss B. Virology. 1987;160:336–345. doi: 10.1016/0042-6822(87)90004-3. [DOI] [PubMed] [Google Scholar]
- 23.Gould K G, Scotney H, Townsend A R, Bastin J, Brownlee G G. J Exp Med. 1987;166:693–701. doi: 10.1084/jem.166.3.693. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Fuerst T R, Niles E G, Studier F W, Moss B. Proc Natl Acad Sci USA. 1986;83:8122–8126. doi: 10.1073/pnas.83.21.8122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Townsend A, Bastin J, Gould K, Brownlee G, Andrew M, Coupar B, Boyle D, Chan S, Smith G. J Exp Med. 1988;168:1211–1224. doi: 10.1084/jem.168.4.1211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Eisenlohr L C, Bacik I, Bennink J R, Bernstein K, Yewdell J W. Cell. 1992;71:963–972. doi: 10.1016/0092-8674(92)90392-p. [DOI] [PubMed] [Google Scholar]
- 27.Earl P L, Moss B. In: Current Protocols in Molecular Biology. Ausubel F M, Brent R, Kingston R E, Moore D D, Seidman J G, Smith J A, Struhl K, editors. New York: Wiley; 1988. pp. 16.17.1–16.17.16. [Google Scholar]
- 28.Yewdell J W, Frank E, Gerhard W. J Immunol. 1981;126:1814–1819. [PubMed] [Google Scholar]
- 29.Chomczynski P, Sacchi N. Anal Biochem. 1987;162:156–159. doi: 10.1006/abio.1987.9999. [DOI] [PubMed] [Google Scholar]
- 30.Shimojo N, Cowan E P, Engelhard V H, Maloy W L, Coligan J E, Biddison W E. J Immunol. 1989;143:558–564. [PubMed] [Google Scholar]
- 31.Karp D R, Long E O. J Exp Med. 1992;175:415–424. doi: 10.1084/jem.175.2.415. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Bachmair A, Varshavsky A. Cell. 1989;56:1019–1032. doi: 10.1016/0092-8674(89)90635-1. [DOI] [PubMed] [Google Scholar]
- 33.Baldi L, Brown K, Franzoso G, Siebenlist U. J Biol Chem. 1996;271:376–379. doi: 10.1074/jbc.271.1.376. [DOI] [PubMed] [Google Scholar]
- 34.Chau V, Tobias J W, Bachmair A, Marriott D, Ecker D J, Gonda D K, Varshavsky A. Science. 1989;243:1576–1583. doi: 10.1126/science.2538923. [DOI] [PubMed] [Google Scholar]
- 35.Vinitsky A, Michaud C, Powers J C, Orlowski M. Biochemistry. 1992;31:9421–9428. doi: 10.1021/bi00154a014. [DOI] [PubMed] [Google Scholar]
- 36.Long E O, LaVaute T, Pinet V, Jaraquemada D. J Immunol. 1994;153:1487–1494. [PubMed] [Google Scholar]
- 37.Marzella L, Glaumann H. In: Lysosomes: Their Role in Protein Breakdown. Glaumann H, Ballard F J, editors. London: Academic; 1987. pp. 319–367. [Google Scholar]
- 38.Chiang H L, Dice J F. J Biol Chem. 1988;263:6797–6805. [PubMed] [Google Scholar]
- 39.Chiang H L, Terlecky S R, Plant C P, Dice J F. Science. 1989;246:382–385. doi: 10.1126/science.2799391. [DOI] [PubMed] [Google Scholar]
- 40.Levy F, Johnsson N, Rumenapf T, Varshavsky A. Proc Natl Acad Sci USA. 1996;93:4907–4912. doi: 10.1073/pnas.93.10.4907. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Grant E P, Michalek M T, Goldberg A L, Rock K L. J Immunol. 1995;155:3750–3758. [PubMed] [Google Scholar]
- 42.Nelson C A, Roof R W, McCourt D W, Unanue E R. Proc Natl Acad Sci USA. 1992;89:7380–7383. doi: 10.1073/pnas.89.16.7380. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Newcomb J R, Cresswell P. J Immunol. 1993;150:499–507. [PubMed] [Google Scholar]
- 44.Chicz R M, Urban R G, Gorga J C, Vignali D A, Lane W S, Strominger J L. J Exp Med. 1993;178:27–47. doi: 10.1084/jem.178.1.27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Freed J H, Marrack P. Chem Immunol. 1993;57:88–112. [PubMed] [Google Scholar]
- 46.Gordon R D, Young J A, Rayner S, Luke R W, Crowther M L, Wordsworth P, Bell J, Hassall G, Evans J, Hinchliffe S A. Eur J Immunol. 1995;25:1473–1476. doi: 10.1002/eji.1830250553. [DOI] [PubMed] [Google Scholar]
- 47.Kirschmann D A, Duffin K L, Smith C E, Welply J K, Howard S C, Schwartz B D, Woulfe S L. J Immunol. 1995;155:5655–5662. [PubMed] [Google Scholar]