

Abstract
A cytotoxic T lymphocyte (CTL) clone generated in vitro from the peripheral blood of a healthy HLA-A2-positive individual against a synthetic p53 protein-derived wild-type peptide (L9V) was shown to kill squamous carcinoma cell lines derived from two head and neck carcinomas, which expressed mutant p53 genes, in a L9V/HLA-A2 specific and restricted fashion. Thus, the normal tolerance against endogenously processed p53 protein-derived self-epitopes can be broken by peptide-specific in vitro priming. p53 protein-derived wild-type peptides might thus represent tumor associated target molecules for immunotherapeutical approaches.
Keywords: tumor immunology, tumor antigens, T cells, cytotoxicity, HLA restriction
The molecular and genetic basis for the development of neoplasia is an abnormal expression of genes and their encoded DNA regulatory proteins important for cellular control of growth, differentiation, and cell death (1–3). Neoplastic development may result in the presentation of tumor-associated peptides derived from regulatory intracellular proteins by major histocompatibility complex class I (MHC-I) molecules on the surface of the malignant cell (4, 5). In healthy individuals, as well as in cancer patients, such tumor-relevant peptide–MHC-I complexes may serve as potential targets for specific HLA-restricted cytotoxic CD8+ T cells. Mutations of the p53 gene are present in more than 50% of all human malignancies and more than 65% of oral squamous cell carcinomas (6–8).
MHC-I associated peptides derived from cytosolic proteins encoded by regulatory genes such as protooncogene and tumor suppressor genes, might be of particular relevance for the immunological surveillance and immunotherapy of tumors. If mutated, these proteins may be overexpressed in a mutant non-functioning form in the cytosol of the tumor cells. Potentially, this could lead to an increased intracellular proteolysis and subsequent MHC-I processing of protein derived peptides. During the last few years, in vitro priming procedures in human lymphocyte culture systems have led to the generation of cytotoxic T lymphocytes (CTLs) with specificity for peptides derived from proteins encoded by various protooncogenes and tumor suppressor genes such as ras, HER-2/neu, bcr/abl, and p53 (9–11). With a few exceptions (12–14), none of the CTLs generated were reported to lyse tumor cells endogenously expressing the particular oncogenic protein. Thus, it is still uncertain whether CTL recognition of peptides derived from DNA regulatory proteins play any part in anti-tumor reactivity in vivo.
In contrast, recent data from studies of human malignant melanomas have shown that self-peptides derived from intracellular enzymes, structural proteins, and differentiation antigens such as tyrosinase, gp100, BAGE, and the MART-1 differentiation antigens can serve as targets for MHC-I-restricted CTL reactivity (15–18).
We quantitated the T cells infiltrating squamous cell carcinomas of head and neck (SCCHN) cancer patients (19, 20). T-cell infiltration was found to be strongly correlated with the capability to propagate tumor infiltrating T lymphocytes in vitro. Some of these propagated tumor infiltrating T lymphocyte cultures displayed a CD3+CD8+ phenotype and were capable of killing autologous tumor cells in an MHC-I-restricted fashion (20). Immunohistological examinations showed that 23 of 44 SCCHN overexpressed the p53 protein (19). Thus, we found it of importance to clarify whether HLA-A2+ SCCHN cells, which overexpress p53 protein and are sensitive to killing by autologous tumor infiltrating T lymphocytes, are also sensitive to killing by a HLA-A2-restricted CTL clone, 3C5, generated in vitro (21) against the p53-derived, HLA-A*0201 binding wild-type peptide, LLGRNSFEV (21, 22).
MATERIALS AND METHODS
Cells.
The 3C5 CTL clone was generated from a HLA-A*0201-positive healthy donor by limiting dilution culture of peripheral blood T cells incubated with transporter associated with antiqeu processing (TAP)-deficient T2 cells (23) pulsed with synthetic p53264–272 peptide (21). This peptide displays a binding motif for HLA-A*0201 (22), the affinity constant (Kd) being approximately 6 nM (S. Buus, personal communication).
Conditions for removing, phenotyping, and culturing squamous carcinoma cells from head and neck cancer patients have been described (20). In brief, small pieces of carcinoma tissue were plated in culture medium. Immediately following plating, strongly adherent tumor cells grew out. At confluency, the tumor cells were separated from the plastic by trypsin/EDTA and passaged. SCC-4 and SCC-9 cells were purchased from American Type Culture Collection and cultured in ordinary RPMI 1640 culture medium containing 10% fetal calf serum.
K562 is an erythroleukemia cell line that is sensitive to natural killer cell cytotoxicity.
Immunocytochemistry.
Immunocytological evaluation was performed as described (20). In short, SCCHN cells were grown on chamber slides (Nunc), fixed in 10% formaldehyde for 2 h, and incubated with mAb against p53 (D0-7; Dako). The antibody recognizes an epitope in the N terminus between amino acids 35–45 and reacts with wild-type and most mutant forms of p53 protein. The avidin–biotin–peroxidase complex method was used to detect the primary antibody according to the instructions from the manufacturer (Dako). Reactivity was obtained after heating the slides for 10 min in a microwave oven at 600 W. The chromogen used was diaminobenzidine (Dako). The sections were counterstained with hematoxylin and mounted for light microscopic examination.
p53 Mutational Analysis.
This procedure was carried out on genomic DNA extracted from SCCHN cells [with the exception of the SCC-9 line, which already has been analyzed (24)] by a combination of denaturating gradient gel electrophoresis and direct sequence analysis as described (25). In brief, 12 genomic DNA fragments covering the entire coding region and all splice sites of the p53 gene were amplified by PCR and scanned for mutations by denaturating gradient gel electrophoresis. Fragments showing an altered electrophoretic band pattern were subjected to sequence analysis. The results are shown in Table 1.
Table 1.
Phenotype of tumor cells and recognition specificity of the anti-HLA*0201/p53 L9V peptide-specific CTL clone (3C5)
| Cell line | HLA
|
3C5 CTL response | ||||||
|---|---|---|---|---|---|---|---|---|
| Type | FACS
(MFI)
|
p53
|
TNF release, pg/ml | Cytotoxicity, % ± SD | % blocking by α-MHC-I | |||
| HLA-A2 | MHC-I | Exp. | Mutation | |||||
| SCC-4 | A*0201/ND | 260 | 304 | 57% | pmP151 to S | <1 | 5 ± 4 | |
| SCC-9 | A*0201/*0101 | 190 | 160 | 0% | dc274-85 (fs) | 125 | 25 ± 5 | 40 |
| SC-31 | A*0201/*3201 | 290 | 327 | 0% | dY236 (fs) | <1 | 1 ± 4 | |
| SC-46 | A*0301/*0301 | 0 | 160 | 0% | G/A sm int4 | <1 | −1 ± 2 | |
| SC-49 | A*0201/*0201 | 80 | 315 | 63% | dc250 (if) | 325 | 63 ± 15 | 50 |
| K562 | <1 | 2 ± 2 | ||||||
SCC cell lines were purchased from the American Type Culture Collection, whereas the SC-31, SC-46, and SC-49 lines were established from SCCHN patients in our laboratory. The HLA-type was verified by a DNA sequencing-based typing method (26) and expression further confirmed by flow cytometry (mean fluorescence intensity, MFI) (ND, not determined). HLA-A2 and MHC-I expression was analyzed by using saturating amounts of the mAbs BB7.2 and W6/32, respectively. p53 protein overexpression (Exp.) was estimated by immunocytochemistry and expressed as the percentage of p53 protein stained cells. p53 mutation was confirmed by a combination of denaturating gradient gel electrophoresis and direct sequence analysis (25). pm, Point mutation; dc, deletion of codon; d, deletion; fs, frameshift; sm, splicing mutation; int, intron; if, in-frame. TNF secretion and percent specific 51chromium release mediated by 3C5 CTL cells exposed to each of the five SCCHN cell lines and the natural killer cell-sensitive K562 cells are shown. Blocking of cytotoxicity was performed by an ascites form of the mAb W6/32 at a final concentration of 1:1000.
HLA-A Subtyping.
SCCHN cells were incubated with an anti-HLA-A2 mAb (HB82, American Type Culture Collection) and their HLA-A2 phenotype confirmed by flow cytometry (Becton Dickinson). The verification of the A*0201 subtype was established with a DNA sequencing-based typing method (26). In brief, tumor cell mRNA was reverse transcribed into cDNA using standard techniques and PCR amplified with an HLA-A locus-specific primer pair. The typing was performed by direct cycle-sequencing of the PCR product using an automated sequencer (ABI 373; Applied Biosystems). The actual assignment of the alleles was done by comparing the sequence obtained with a data base containing all known alleles within the HLA-A locus.
Peptide Pulsing and Cytotoxicity Assay.
For peptide pulsing of T2 cells, L9V (p53, 264–272) or G11V (p53, 187–197) synthetic peptides and human β2 microglobulin (β2m) were added to 51chromium-labeled T2 cells (see below) at 50 μg/ml and 3 μg/ml, respectively, for 1 h at 37°C. The L9V and the G11V peptides show comparable efficiency in upregulation of HLA-A2 expression on T2 cells following peptide pulsing (21). Target cells were labeled with sodium–51chromate and specific lysis was measured in replicates of four cytotoxicity assay cultures at titered effector:target cell ratios after 4 h incubation (20). Spontaneous release never exceeded 20% of the total 51chromium release. Cold target cell blocking experiments were performed with titered numbers of “cold” to “hot” targets. Cold T2 target cells were pulsed for 1 h with L9V or G11V peptides.
An ascites form of the anti-monomorphic MHC-I antibody w6/32, used at a final concentration of 1:1000, was tested for inhibition of killing. An ascites preparation containing mAb of an irrelevant specificity was used for comparison.
TNF Release Assay.
Stimulator cells, (3 × 104) were added to 3 × 103 CTL in 100 μl of RPMI 1640 containing 10% human serum supplemented with 10 units/ml of recombinant (r) interleukin 2 (Eurocetus, Amsterdam). After 24 h, 50 μl of the supernatant was harvested and mixed with 3 × 104 tumor necrosis factor (TNF)-sensitive WEHI-172-2 cells (27) in 50 μl of medium. The amount of released TNF was estimated by comparing the effect of known concentrations of rTNF-β (Pepro Tech, Rocky Hill, NJ) on WEHI-172-2 survival. TNF release from CTL cultured without tumor cells (20–30 pg/ml) was subtracted from TNF values shown in Table 1.
RESULTS
SCCHN Carcinoma Lines and 3C5 CTL Recognition.
The HLA*0201-restricted p53 L9V peptide-specific CTL clone (21, 28), 3C5, was tested for recognition and cytotoxic activity on five different SCCHN cell lines depicted in Table 1. Except for the SC-46 line, all the SCCHN cell lines were HLA-A*0201-positive as revealed by flow cytometry and HLA-A subtyping. The carcinoma cell lines established in our own laboratory (SC-31, SC-46, SC-49) were studied from their second in vitro passage onwards. Using an immunocytological technique, more than 50% of the SCC-4 and SC-49 cells stained positive for the p53 gene product, whereas the remaining three cell lines were negative. Except for the SCC-9 line, the p53 mutations shown in Table 1 were analyzed by denaturating gradient gel electrophoresis and direct sequence analysis. As shown in Table 1, exposure of 3C5 to SCC-9 or SC-49 cells resulted in high levels of CTL-derived TNF production, whereas no TNF could be shown by 3C5 CTL cocultured with the three remaining SCCHN cell lines. In agreement with the TNF data, Table 1 further demonstrates that the 3C5 clone killed only the SCC-9 and SC-49 carcinoma cells and that this killing could be blocked 40–50% in the presence of anti-MHC-I antibody (w6/32 mAb) in the cytotoxicity culture. It should be noted that the natural killer cell-sensitive target, K562, was not killed by 3C5 CTLs.
HLA-A2/p53 L9V Peptide-Specific Killing Is Blocked by SC-49 and SCC-9 Carcinoma Cells.
Data in Fig. 1A show the HLA-A2 restriction and p53 peptide specificity of the 3C5 CTL clone. As documented previously (22), the 3C5 CTL clone specifically kills HLA-A2+ peptide transporter-deficient (26) L9V-pulsed T2 cells, but not T2 cells pulsed with an irrelevant p53-derived, HLA-A2 binding peptide (G11V). The cytotoxic activity was blocked more than 85% in the presence of cold, i.e., 51chromium-unlabeled, T2 target cells pulsed with the L9V peptide, and more than 95% by cold SC-49 or SCC-9 (data not shown) carcinoma cells. The cytotoxic activity was not blocked by cold T2 target cells pulsed with G11V peptide or by K562 cells. Specific cytotoxicity was inhibited more than 95% in the presence of anti-CD8 mAb, suggesting CD8 dependence of killing.
Figure 1.
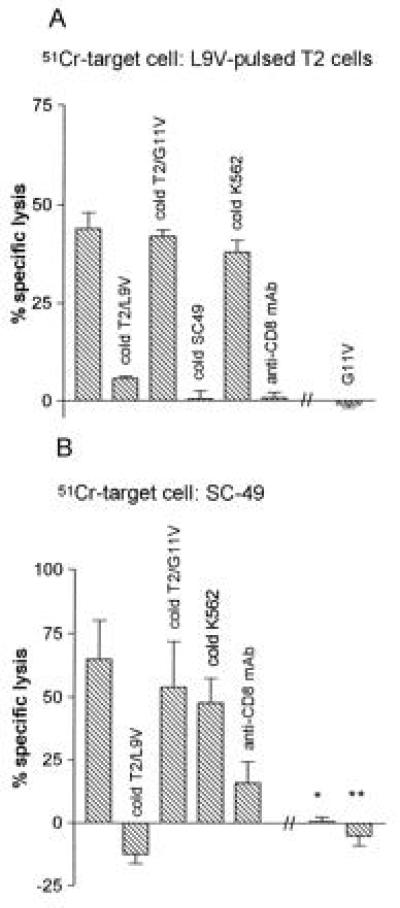
HLA-A2 restricted, peptide-specific killing by 3C5 CTLs of T2 cells pulsed with the L9V peptide (p53264–272) (A) and of SC-49 SCCHN cells (B). Columns represent mean specific lysis in replicates of four assay cultures at an effector:target ratio of 25:1. Bars represent one SD. Notice the absence of killing of two autologous cell lines. ∗, An Epstein–Barr virus-transformed B-cell line; ∗∗, fibroblasts. Blocking data represent a cold:hot ratio of 20:1.
SC-49-Specific Killing Is Blocked by HLA-A2/p53 L9V and Is CD8 Dependent and MHC-I Restricted.
Data in Fig. 1B show that 3C5 CTL-mediated killing of SC-49 carcinoma cells was specifically blocked by cold T2 target cells pulsed with the L9V peptide, whereas cold T2 cells pulsed with the irrelevant G11V peptide or K562 cells did not block lysis of SC-49. Similar data were obtained for killing of the SCC-9 carcinoma cell line (data not shown). As with L9V-pulsed T2 cells, the presence of anti-CD8 mAb nearly completely blocked killing of SC-49 carcinoma cells. Fig. 1B also shows that an Epstein–Barr virus transformed autologous B cell line (∗) and an autologous fibroblast cell line (∗∗) were not killed by the 3C5 CTL clone.
DISCUSSION
This study demonstrates for the first time, to the best of our knowledge, the existence of CTL-mediated, HLA/p53-peptide-specific and MHC-restricted killing of human carcinoma cells overexpressing the tumor suppressor protein p53. The HLA-A2 restricted, peptide-specific killing of SC-49 and SCC-9 carcinoma cells was documented by its sensitivity to cold target cell inhibition by L9V- (but not G11V) pulsed T2 cells and by the ability of cold SC-49 and SCC-9 cells to block killing of L9V-pulsed T2 cells. In addition, the killing was blocked by anti-CD8 and anti-MHC-I mAbs. The absence of blocking with cold K562 cells and the lack of killing of autologous normal cells further argue against the possibility that 3C5 CTL-mediated killing is due to promiscuous natural killer cell-like activity or alloreactivity, respectively.
CTL clones recognizing target cells exogenously pulsed with peptide have been generated against a number of wild-type and mutant p53 peptides (21, 28). However, p53 peptide-specific killing by human CTL clones of tumor cells expressing endogenous p53 protein has not been reported previously. However, a few murine studies have demonstrated MHC-I/mutant p53 peptide-restricted, CTL-mediated killing (12, 13). As suggested from the data in Table 1, overexpression of p53 protein is not an absolute requirement for killing, since SCC-9 carcinoma cells, in which p53 protein was not detectable, could also induce both TNF secretion and killing by 3C5 CTLs, although at lower levels than the p53 overexpressing SC-49 carcinoma cells. Also, overexpression of the p53 protein by SCCHN cells does not necessarily lead to killing of the cells. Thus, HLA-A2-positive, p53 protein overexpressing SCC-4 cells were not killed by the 3C5 CTL clone. Most probably, differences exist in the endogenous p53 protein processing machinery of SCCHN cell lines generated from different patients.
Do p53-derived wild-type peptides presented by MHC-I molecules on tumor cells play any role as target molecules for CD8+ T-cell-mediated immune surveillance? The feasibility to generate anti-p53 peptide-specific CD8+ CTL clones (21, 28) and to measure CTL precursor frequencies (29) from normal peripheral T cells of healthy individuals may support this idea. In addition, according to a recent report, CTL lines were generated in HLA-A2 transgenic mice immunized with p53 wild-type peptides, including the L9V peptide (30). Such murine CTL lines killed HLA-A2-positive, p53 overexpressing breast and colon carcinomas in a CD8-independent fashion, strongly indicating that p53 derived peptides can serve as high-affinity target molecules for T-cell receptors in the process of immune surveillance.
In conclusion, mutations of the p53 gene (6–8) or other conditions (31) leading to an overexpression of p53 protein, are present in more than 50% of all human malignancies. This demonstration of an HLA-A2/p53 wild-type peptide, recognizable in an MHC-I-restricted fashion by an in vitro generated CTL clone, suggests that natural tolerance against endogenous processed p53 protein-derived self-epitopes can be broken by peptide-specific priming in vitro. p53 protein-derived wild-type peptides might thus represent tumor associated target molecules for the immune surveillance and, in addition, for immunotherapeutical approaches.
Acknowledgments
This work was supported by Grant 78-6000 from the Danish Cancer Society.
Footnotes
Abbreviations: MHC, major histocompatibility complex; CTL, cytotoxic T lymphocyte; SCCHN, squamous cell carcinomas of head and neck; TNF, tumor necrosis factor.
References
- 1.Cheever M A, Disis M L, Bernhard H, Gralow J R, Hand S L, Huseby E S, Qin H L, Takhashi M, Chen W. Immunol Rev. 1995;145:33–59. doi: 10.1111/j.1600-065x.1995.tb00076.x. [DOI] [PubMed] [Google Scholar]
- 2.Fearon E R. Adv Intern Med. 1994;39:123–147. [PubMed] [Google Scholar]
- 3.Devilee P, Cornelisse C J. Biochim Biophys Acta. 1994;1198:113–130. doi: 10.1016/0304-419x(94)90009-4. [DOI] [PubMed] [Google Scholar]
- 4.Melief C J. Adv Cancer Res. 1992;58:143–175. doi: 10.1016/s0065-230x(08)60294-8. [DOI] [PubMed] [Google Scholar]
- 5.Germain R N. Cell. 1994;76:287–299. doi: 10.1016/0092-8674(94)90336-0. [DOI] [PubMed] [Google Scholar]
- 6.de Fromentel C C, Soussi T. Genes Chromosome Cancer. 1992;4:1–15. doi: 10.1002/gcc.2870040102. [DOI] [PubMed] [Google Scholar]
- 7.Lane D P, Benchimol S. Genes Dev. 1990;4:1–8. doi: 10.1101/gad.4.1.1. [DOI] [PubMed] [Google Scholar]
- 8.Dursun G, Sak S D, Akyol G, Dursun A, Erekul S, Demireller A, Akiner M. Ear Nose Throat J. 1995;74:645–648. [PubMed] [Google Scholar]
- 9.Möller G. Immunol Rev. 1995;145:1–250. [Google Scholar]
- 10.Boon T, Cerottini J C, van den Eynde B, van der Bruggen P, van der Pel A V. Annu Rev Immunol. 1994;12:337–365. doi: 10.1146/annurev.iy.12.040194.002005. [DOI] [PubMed] [Google Scholar]
- 11.Melief C J M, Kast W M. Curr Opin Immunol. 1993;5:709–713. doi: 10.1016/0952-7915(93)90125-c. [DOI] [PubMed] [Google Scholar]
- 12.Yanuck M, Carbone D P, Pendleton D, Tsukui T, Winter S F, Berzofsky J A. Cancer Res. 1993;53:3257–3261. [PubMed] [Google Scholar]
- 13.Noguchi Y, Chen Y T, Old L J. Proc Natl Acad Sci USA. 1994;91:3171–3175. doi: 10.1073/pnas.91.8.3171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Linehan D C, Goedegebuure P S, Peoples G E, Rogers S O, Eberlein T J. J Immunol. 1995;155:4486–4491. [PubMed] [Google Scholar]
- 15.Wolfel T, Van Pel A, Brichard V, Schneider J, Seliger B, Buschenfelde K H M, Boon T. Eur J Immunol. 1994;24:759–764. doi: 10.1002/eji.1830240340. [DOI] [PubMed] [Google Scholar]
- 16.Bakker A B, Schreurs M W, de Boer A J, Kawakami Y, Rosenberg S A, Adema G J, Figdor C G. J Exp Med. 1994;179:1005–1009. doi: 10.1084/jem.179.3.1005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Kawakami Y, Eliyahu S, Delgado C H, Robbins P F, Rivoltini L, Topalian S L, Miki T, Rosenberg S A. Proc Natl Acad Sci USA. 1994;91:3515–3519. doi: 10.1073/pnas.91.9.3515. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Boel P, Wildmann C, Sensi M L, Brasseur R, Renauld J C, Coulie P, Boon T, van der Bruggen P. Immunity. 1995;2:167–175. doi: 10.1016/s1074-7613(95)80053-0. [DOI] [PubMed] [Google Scholar]
- 19.Hald J, Rasmussen N, Claesson M H. Cancer Immunol Immunother. 1994;39:383–390. doi: 10.1007/BF01534425. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Hald J, Rasmussen N, Claesson M H. Cancer Immunol Immunother. 1995;41:243–250. doi: 10.1007/BF01516999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Houbiers J G A, Nijman H W, van der Burg S H, Drijfhout J W, Melief C J M. Eur J Immunol. 1993;23:2072–2077. doi: 10.1002/eji.1830230905. [DOI] [PubMed] [Google Scholar]
- 22.Sette A, Alexander J, Ruppert J, Snoke K, Franco A, Ishioka G, Grey H M. Annu Rev Immunol. 1994;12:413–431. doi: 10.1146/annurev.iy.12.040194.002213. [DOI] [PubMed] [Google Scholar]
- 23.Salter R D, Cresswell P. EMBO J. 1986;5:943–949. doi: 10.1002/j.1460-2075.1986.tb04307.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Jung M, Notario V, Dritschilo A. Cancer Res. 1992;52:6390–6393. [PubMed] [Google Scholar]
- 25.Guldberg, P., Nedergaard, T., Nielsen, H. J., Olsen, A. C., Ahrenkiel, V. & Zeuthen, J. (1996) Hum. Mutat. 8, in press. [DOI] [PubMed]
- 26.Santamaria P, Boyce-Jacino M T, Linström A L, Barbosa J J, Faras A J, Rich S S. Hum Immunol. 1992;33:69–81. doi: 10.1016/0198-8859(92)90056-s. [DOI] [PubMed] [Google Scholar]
- 27.Jaattela M, Wissing D, Bauer P A, Li G C. EMBO J. 1992;11:3507–3510. doi: 10.1002/j.1460-2075.1992.tb05433.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Nijman H W, van der Burg S H, Vierboom M P M, Houbiers J G A, Kast W M, Melief C J M. Immunol Lett. 1994;40:171–178. doi: 10.1016/0165-2478(94)90189-9. [DOI] [PubMed] [Google Scholar]
- 29.Röpke M, Regner M, Claesson M H. Scand J Immunol. 1995;42:98–103. doi: 10.1111/j.1365-3083.1995.tb03631.x. [DOI] [PubMed] [Google Scholar]
- 30.Theobald M, Biggs J, Dittmer D, Levine A J, Sherman L A. Proc Natl Acad Sci USA. 1995;92:11993–11997. doi: 10.1073/pnas.92.26.11993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Niedobitek G, Agathanggelou A, Barber P, Sallmann L A, Jones L E, Young L S. J Pathol. 1993;170:457–461. doi: 10.1002/path.1711700409. [DOI] [PubMed] [Google Scholar]