

Abstract
Ocular cicatricial pemphigoid (OCP) is an autoimmune disease that affects mainly conjunctiva and other squamous epithelia. OCP is histologically characterized by a separation of the epithelium from underlying tissues within the basement membrane zone. Immunopathological studies demonstrate the deposition of anti-basement membrane zone autoantibodies in vivo. Purified IgG from sera of patients with active OCP identified a cDNA clone from a human keratinocyte cDNA library that had complete homology with the cytoplasmic domain of β4-integrin. The sera recognized a 205-kDa protein in human epidermal, human conjunctiva, and tumor cell lysates that was identified as β4-integrin by its reaction with polyclonal and monoclonal antibodies to human β4-integrin. Sera from patients with bullous pemphigoid, pemphigus vulgaris, and cicatricial pemphigoid-like diseases did not recognize the 205-kDa protein, indicating the specificity of the binding. These data strongly implicate a role for human β4-integrin in the pathogenesis of OCP. It should be emphasized that multiple antigens in the basement membrane zone of squamous epithelia may serve as targets for a wide spectrum of autoantibodies observed in vesiculobullous diseases. Molecular definition of these autoantigens will facilitate the classification and characterization of subsets of cicatricial pemphigoid and help distinguishing them from bullous pemphigoid. This study highlights the function and importance of β4-integrin in maintaining the attachment of epithelial cells to the basement membrane.
Ocular cicatricial pemphigoid (OCP) is an uncommon potentially blinding systemic vesiculobullous autoimmune disease that affects mainly conjunctiva and other squamous epithelia (1, 2). OCP has some common pathophysiological features with other bullous diseases such as linear IgA bullous disease (LABD) and cicatricial pemphigoid (CP). In addition, the sera of patients with bullous pemphigoid (BP), epidermolysis bullosa aquisita, LABD, and CP (3, 4) frequently have demonstrable circulating autoantibodies that bind to different antigens of the basement membrane zone (BMZ) of skin and mucous membranes (5). Indirect immunofluorescence techniques are often inadequate to detect the circulating antibodies in sera of patients with OCP, because the autoantibody titer is very low. Immunoblot is now being used to study and characterize the OCP antigens and antibodies. Recent studies by immunoblot analysis indicated that OCP patients’ sera recognize 230-, 205-, 180-, and 85-kDa proteins when normal human skin, human conjunctiva, and tumor cell lines were used as substrates (6). Some investigators have reported that like BP, CP patients’ sera recognize the 230- and 180-kDa proteins when human epidermal lysate (HEL) and BPAg1 fusion proteins were used as substrates. They did not find binding of their sera to a 205-kDa protein (7). In contrast, we have shown that when the lysates containing the OCP antigens were first repeatedly preabsorbed with BP sera and then were tested with OCP patients’ sera, only binding to the 205-kDa protein was observed. The additional step of preabsorbing test lysates with BP serum explains the discrepancy between our work (6, 8) and those of other investigators (7). These observations suggest that the putative target antigen for OCP is probably the 205-kDa protein (8). It is possible that the autoantibodies against the 230-kDa protein (BPAg1) and 180-kDa proteins (BPAg2) are produced in response to their availability to the immune system, once the integrity of the basement membrane and that of the basal epidermal cell are compromised. These autoantibodies are probably entirely nonpathogenic in OCP patients. A subset of CP patients has been reported to be characterized by the presence of autoantibodies against epiligrin, which is now identified as the α3 subunit of laminin 5 (9, 10). Interestingly, many of the OCP sera used in this and our earlier studies (6, 8) did not bind to epiligrin (Kim Yancey, personal communication). CP is a heterogeneous disease with a wide spectrum of clinical presentations and clinical course. In addition it appears that these clinical subsets correlate with a variety of anti-BMZ autoantibodies with different specificities that recognize different target molecules within the complex of cell surface proteins and extracellular matrix proteins in the BMZ. At the present time the only available method to define these subsets accurately is by long-term close clinical follow-up.
The purpose of this report is to describe studies that characterize the 205-kDa protein recognized by OCP sera and to partially sequence this antigen. Our results indicate that the putative target antigen for OCP is human β4-integrin (CD104).
MATERIALS AND METHODS
Serum Samples and Antibodies.
Sera used in this study were obtained from 12 patients with OCP in the acute and active phase of the disease prior to the institution of therapy. The diagnosis of OCP was confirmed in each patient by immunohistochemical analysis of biopsied conjunctiva. When tested on salt split skin, the anti-BMZ autoantibody was bound to the epidermal side of the split. All the sera studied demonstrated binding to a 205-kDa protein on an immunoblot using normal human epidermis or conjunctiva as substrate (8). Control sera were obtained from 10 healthy individuals, from 4 patients with established BP (without oral involvement), from 4 patients with pemphigus vulgaris (PV), and from 2 with pemphigus foliaceus. The study has been reviewed by Institutional Review Board of Center for Blood Research. Sera were obtained after appropriate informed consent procedures.
Immunoaffinity-purified OCP autoantibodies were eluted from the nitrocellulose blots as described (11). This 205-kDa-specific autoantibody was used for detecting and characterizing the putative cDNA clones containing OCP antigen by screening the cDNA libraries from human keratinocytes. Polyclonal and monoclonal β4-integrin antibodies were obtained from Martin Hemler (Dana–Farber Cancer Institute) and from Ancell (Bayport, MN), respectively.
Preparation of Lysate from Human Skin and Conjunctiva.
The epidermis of the human skin was separated from underlying dermis and clear lysates of antigens were prepared as reported (12). Briefly, the human epidermis and conjunctiva were homogenized on ice with 1.5% SDS/62.5 mM Tris·HCl, pH 6.8, supplemented with 5% 2-mercaptoethanol, 2 mM phenylmethylsulfonyl fluoride, and pepstatin A (10 μg/ml), antipain (10 μg/ml), leupeptin (10 μg/ml), and chymostatin (10 μg/ml; Sigma). The lysates were then boiled for 5 min, vortex mixed, and centrifuged at 15,000 × g for 30 min. The clear supernatant of HEL and human conjunctival lysate (HCL) was centrifuged again at high speed in microfuge centrifuge for 30 min and supernatant was concentrated. Protein content was measured and aliquots were stored at −70°C until used.
Preparation of MDA-435 and UM-SCC-22 Cells Extract.
MDA-435 cells provided by Martin Hemler and UM-SCC-22 cells obtained from Thomas Carey (University of Michigan) were grown to confluence in T-25/T-75 flasks in cell culture medium (RPMI 1640 and DMEM). Cells were harvested by vigorous scraping and pipetting in minimum volume. Typically, 1–5 × 107 cells per ml were lysed with buffer containing 20 mM Tris·HCl (pH 6.8), 75 mM NaCl, 5% glycerol, 1% Nonidet P-40, 1 μM pepstatin A, 1 μM leupeptin, 100 μM idoacetimide, aprotinin (50 μg/ml), and 100 μM phenylmethylsulfonyl fluoride; vortex mixed vigorously; incubated for 45–60 min on ice; and centrifuged at 1500 × g for 30 min to remove cell debris, nuclei, etc. Supernatant was removed. Protein was estimated by Bio-Rad protein assay and aliquots were frozen at −70°C.
Immunoblot Analysis.
SDS/PAGE was performed as described by Laemmli (13). A sensitive Western blot analysis assay was used in this study as described by Hashimoto et al. (14) and modified by this laboratory (12). In brief, blotted nitrocellulose membrane were blocked with 3% skimmed milk. Blotted proteins (nitrocellulose strips) after extensive washing with TBS-containing 0.05% Tween 20 were incubated with diluted test sera or antibodies. After four washes, nitrocellulose strips were incubated with horseradish peroxidase-conjugated secondary antibodies (anti-human, anti-rabbit, and anti-mouse) then developed by ECL detection kit (Amersham).
Indirect Immunofluorescence Studies.
Normal human bulbar conjunctiva was used as a substrate for binding monoclonal and polyclonal anti-β4-integrin antibody to the conjunctival BMZ as described (1, 2). Four-micrometer sections of the conjunctiva were incubated for 30 min with mouse monoclonal anti-CD104 (human β4-integrin from Ancell), rabbit polyclonal anti-human β4-integrin (diluted 1:40), goat anti-human collagen IV (diluted 1:100; Biodesign International, Kennebunkport, ME), or goat anti-human albumin; then washed three times in PBS; and incubated for an additional 30 min with the appropriate fluorescein isothiocyanate-conjugated secondary antibody. In control experiments, incubation with the primary antibody was omitted. Anti-human albumin antibody served as a negative control, and anti-collagen IV antibody served as a positive control.
Immunoprecipitation of OCP Antigen/β4-Integrin.
Immunoprecipitation was performed as described (15, 16). Briefly, 100–200 μl (0.5–1.0 mg of protein) of ammonium sulfate-precipitated IgG from OCP patients’ sera was added to 200 μl of tumor cell lysates. After incubating at 4°C for an hour, 100 μl of protein A (from Sigma) was added and incubated for one more hour. Finally, samples were centrifuged/washed and resuspended in 1× sample buffer and examined by immunoblot analysis.
Absorption Studies.
Ammonium sulfate-precipitated IgG fraction of OCP patients’ sera was coupled to cyanogen bromide-activated Sepharose 4B by the method of Porath and Axen (17). Briefly, 100–200 μl (1–2.0 mg) of lysates from MDA-435/UM SCC-22 cells were incubated with 0.5 ml of Sepharose 4B coupled with IgG fraction of OCP patients sera on a shaker at 4°C. After centrifugation the supernatant was incubated with a second aliquot of 0.5 ml of OCP antibodies coupled with Sepharose 4B for an hour. Supernatant was collected, and equal amounts of protein were loaded on 8% SDS/PAGE gel and subjected to immunoblot analysis as described earlier by using sera from OCP patients and polyclonal and monoclonal antibodies to β4-integrin.
Screening of cDNA Libraries.
A phage λgt11 cDNA library was constructed from mRNA of human keratinocyte and screened using cDNA insert screening amplimers (CLONTECH) and as described by Young and Davis (18). The cDNA library and putative plaques were screened by immunoaffinity-purified OCP patients’ antibodies that recognize a 205-kDa protein. Initially two positive clones were selected and sequentially screened until the progeny plaques were identified by OCP antibodies. Finally 10 clones were picked and stored at 4°C.
Polymerase Chain Reaction (PCR).
Two putative clones (clones 8 and 10) were PCR-amplified (Fig. 1) by using 5′ and 3′ amplimers from CLONTECH, plaque lysates, and Taq polymerase in a Perkin–Elmer PCR machine (GeneAmp model 9600). Briefly, after mixing all the components of PCR, initial denaturation was for 1.0 min at 94°C. The total number of cycles was 30 and the PCR machine was programmed as follows: denaturation for 30 sec (94°C), then annealing for 10 sec (60°C), then amplification for 60 sec (72°C), followed by a 5-min amplification (72°C) that held at 4°C until further processing.
Figure 1.
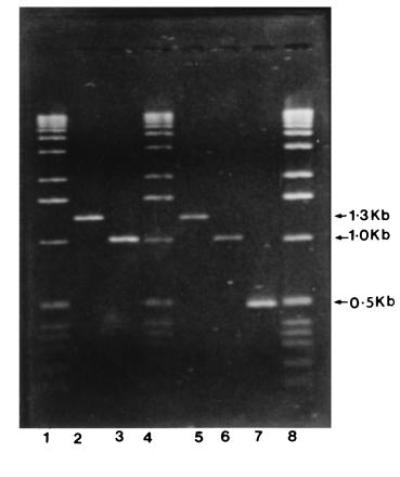
PCR amplification of clone 10 of OCP antigen. Lanes: 1, 4, 0.5 μg of DNA ladder (GIBCO/BRL); 8, 1.0 μg of DNA ladder; 2 and 5, clone 8 (1.3 kb) as internal standard; 3 and 6, replicates of clone 10 (1.0 kb, OCP antigen clone); 7, internal standard for PCR amplification kit (Perkin–Elmer and CLONTECH).
DNA Sequencing.
A cDNA clone (clone 10) was PCR-amplified (≈1.0 kb, Fig. 1) and purified by a Qiagen kit (Qiaquick spin PCR purification kit) according to the manufacturer’s protocol. The purified product was checked on 1.2% agarose gel and used in the sequencing. Nucleotide sequences were determined by using the dideoxynucleotide chain-termination method (19) in an automatic sequencer (Applied Biosystems, model 373A, version 1.2.1). Both the strands of cDNA clone were fully sequenced by using insert screening 5′-GACTCCTGGAGCCCG and 3′-GGTAGCGACCGGCGC amplimers (CLONTECH) as well as new primers (5′-ATAGAGTCCCAGGATGGA, and AGGAGTTTGTGAGCCGGA, and GGAAGGGTCCTCCATCCT-3′) synthesized on an Applied Biosystems model 3948 DNA synthesizer using solid-phase cyanoethyl phosphoramidite chemistry at Molecular Biology Core facility at Dana–Farber Cancer Institute. Nucleotide and amino acid sequences were analyzed by using the sequence analysis software package of the National Center for Biotechnology Information under the blast network services (Cruncher) as described by Altschul et al. (20).
RESULTS
Isolation of OCP Antigen cDNA.
A λgt11 cDNA library was prepared from poly(A)+ RNA isolated from human keratinocytes and screened using immunoaffinity-purified OCP patients’ sera autoantibodies that bind to a 205-kDa protein in epidermal lysates. After screening more than ≈300,000 clones/plaques, we identified two positive clones: clone 10 contained an insert of ≈1.0 kb and clone 8 contained an insert of ≈1.3 kb (Fig. 1).
Partial cDNA (Nucleotide) Sequence of OCP Antigen and Deduced Amino Acid Sequence.
The partial cDNA sequence and the deduced amino acid sequence of the OCP antigen are shown in Fig. 2. The partial nucleotide sequence of the OCP antigen is 964 bp encoding a peptide of 321 amino acids. The 3′ region contains the polyadenylylation signal TGCAAA, followed by 26 bases followed by a poly(A) stretch. The deduced nucleotide sequence and amino acid sequence of clone 10 (nt 42–964) showed complete homology with cytoplasmic domain of human β4-integrin.
Figure 2.

Partial nucleotide sequence and deduced amino acid sequence of clone 10 of OCP antigen. Nucleotide sequence was analyzed on an Applied Biosystems model 373A automated DNA sequencer and deduced amino acid were performed at National Center for Biotechnology Information using blast network service. Nucleotide sequences from positions 42 to 964 have complete homology with published sequence of human β4-integrin.
Indirect Immunofluorescence Studies.
The monoclonal antibody against human β4-integrin (CD104) bound in a smooth uniform linear pattern along the BMZ of human conjunctiva. The staining pattern was very similar to that observed with binding of OCP sera and collagen IV. The monoclonal anti-β4-integrin antibody exhibited more intense staining than polyclonal β4-integrin antibody and OCP sera. No binding was seen with antibodies to human albumin.
Immunoblot Studies of OCP Antigen.
Sera from 12 active OCP patients and antibodies against human β4-integrin (polyclonal and/or monoclonal) recognize an ≈205-kDa protein in human epidermal, human conjunctival, and MDA 435 and UM-SCC-22 tumor cell line lysates. However, MDA 435 cell lysates showed a slightly higher molecular weight (≈210 kDa) protein band compared with UM-SCC-22 cells. This size difference may be due to the increased glycosylation of the 205-kDa protein in MDA 435 cells. Besides ≈205-kDa protein bands, we have also noticed three additional protein bands (180, 160, and 85 kDa), particularly in UM-SCC-22 cell lysates (Fig. 3). Normal human serum and sera from patients with other related bullous diseases did not show any binding to the ≈205-kDa protein. BP sera bound to 230-kDa and 180-kDa proteins, PV sera bound to 130-kDa protein, and pemphigus foliaceus sera bound to 160-kDa protein on HELs, indicating the validity of the assay system.
Figure 3.
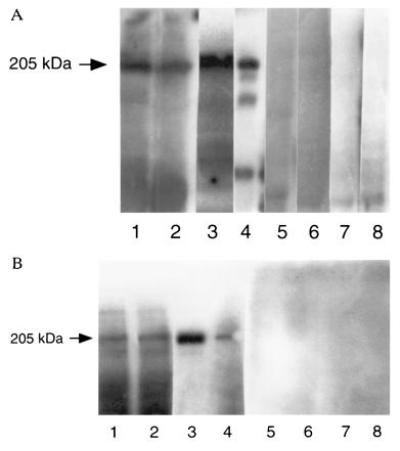
(A) Immunoblot of OCP antigen/β4-integrin (205/210 kDa) using human epidermal, human conjunctival, and tumor cell lines (MDA 435 and UM-SCC-22) and detected by polyclonal antibody to β4-integrin. Lanes: 1, HEL; 2, HCL; 3, MDA 435 cell lysates; 4, UM-SCC-22 cell lysate reacted with polyclonal antibody to β4-integrin; 5–8, same lysates reacted with normal human serum. In all four groups (lanes 1–4), a 205/210-kDa protein band was observed when β4-integrin antibody was used. No similar protein band was detected when normal human serum was used (lanes 5–8). (B) The identical setup of experiment was used except lanes 3 and 4 were UM-SCC-22 and MDA 435, respectively, and monoclonal anti-β4-integrin was used instead of polyclonal.
Absorption Studies.
In these studies we used tumor cell lysates for two reasons. (i) Tumor cell lysates demonstrating a binding pattern of 205-kDa protein were identical in both HEL and HCL. (ii) The binding pattern of a 205-kDa protein was clearer and stronger in tumor cell lysates compared with HEL and HCL. The tumor cell lysates (MDA 435 and UM-SCC-22) were first absorbed with OCP patients’ sera and analyzed by immunoblot and then binding was detected either by β4-integrin antibodies or with another high-titer OCP patient’s serum. The 205-kDa protein band was completely depleted, whereas other lower molecular weight proteins were unabsorbed (Fig. 4). In control samples where tumor cell lysates were preabsorbed with normal human serum, we did not see any loss of 205-kDa protein (Fig. 4). This indicates that our 205-kDa protein is specifically recognized by sera of OCP patients.
Figure 4.
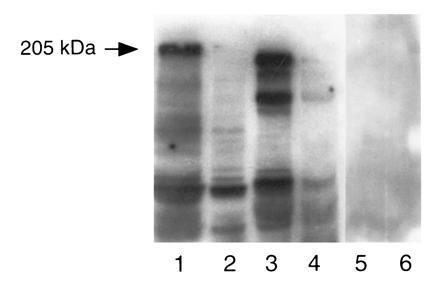
Preabsorption/blocking studies by OCP patients’ sera using MDA 435 and UM-SCC-22 cell lysates. MDA 435 and UM-SCC-22 cell were preabsorbed with OCP patients’ sera and normal human serum that was previously bound to cyanogen bromide-activated Sepharose 4B. Samples were prepared for SDS/PAGE, transferred to nitrocellulose, examined by immunoblot analysis with β4-integrin polyclonal antibody and normal human serum. Lanes 1 and 2 contain MDA 435 cell lysate. In lane 1 lysate was absorbed with normal human serum (NHS) and then blotted with anti-β4-antibody. Note the 205-kDa band. In lane 2, cell lysate was first absorbed with OCP sera then blotted with anti-β4 antibody. Note the absence of the 205-kDa band. Lanes 3 and 4 represent UM-SCC-22 cell lysate treated similarly. Lanes 5 and 6 tumor cell lysates preabsorbed with NHS then reacted with NHS.
Specificity Experiments.
To characterize OCP antigen specificity, we studied OCP patients’ sera, anti-β4-integrin antibodies, anti-laminin-5 antibodies, and anti-epiligrin antibodies in an immunoblot assay. The results are shown in the Fig. 5. OCP patients’ sera and anti-β4-integrin antibodies showed reactivity with a 205-kDa protein, whereas anti-epiligrin human sera and antibodies against laminin 5 did not show any reactivity in this region. This specific binding of OCP patients’ sera to a 205-kDa protein indicates that the OCP antigen is different from BP antigens, laminin 5, and epiligrin proteins. Additionally, BP patients’ sera bound to 230-kDa and 180-kDa proteins, PV sera bound to a 130-kDa protein, and pemphigus foliaceus sera bound to a 160-kDa protein in HEL, confirming the sensitivity and validity of the assay system.
Figure 5.
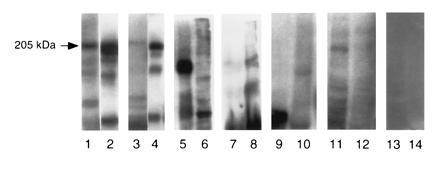
Specificity experiment using MDA 435 and UM-SCC-22 cell lysates and detected by various patients sera and anti-β4-integrin antibody. Lanes: 1, β4-integrin antibody; 3, OCP patients’ sera; 5, polyclonal anti-laminin 5; 7, anti-epiligrin human sera; 9, PV patients’ sera; 11, BP patients’ sera; 13, normal human sera. All these sera reacted with MDA cell lysates. Lanes: 2, β4-integrin antibody; 4, OCP patients’ sera; 6, anti-laminin 5; 8, anti-epiligrin human sera; 10, PV patients’ sera; 12, BP patients’ sera; 14, normal human sera. All these sera reacted with UM-SCC-22 cell lysates.
DISCUSSION
There has been considerable interest in identifying various proteins that make up the cell-membrane–hemidesmosome/BMZ attachment complex, which could be important in the pathogenesis of various bullous diseases. Our aim was to characterize the OCP antigen. We have identified and characterized a cDNA clone of the OCP antigen that has complete homology with the cytoplasmic domain of human β4-integrin. Immunoblot analysis using preabsorption and blocking experiments demonstrated that this 205-kDa protein specifically binds to sera of OCP patients. Both OCP patients’ sera and antibodies against β4-integrin recognized the same 205-kDa protein, whereas sera from patients with related autoimmune bullous diseases did not. Sera of patients with minimal disease and those in clinical remission do not demonstrate binding with the 205-kDa protein. These studies strongly suggest that the OCP antigen has complete homology with cytoplasmic domain of β4-integrin. These observations further our efforts to better understand the pathophysiology of OCP and characterize some of the biological functions of this cell adhesion molecule. Indeed it is known that integrins play an important role in ocular biology, influencing ocular development, cell migration, and wound healing as well as other processes (21). Besides the observations reported in this manuscript on the possible role of β4-integrin in the pathogenesis of OCP, the only other known hemidesmosomal component with a transmembrane domain implicated in a human autoimmune disease is BPAg2, which has been shown to be a major target of BP and herpes gestationis autoantibodies (7, 22–24).
Integrins are heterodimeric molecules, each composed of an α and β subunit; at least 15 α and 8 β subunits have been reported (21). Several combinations of α and β integrins mediate, perform, facilitate, or enhance cell adhesion. Epidermal keratinocytes express two types of integrins. The heterodimer α6β4 is associated with binding of keratin filaments to hemidesmosomes; α3β1 facilitates binding of the actin filaments to the cytoskeleton (25, 26). The most frequently observed integrin heterodimer in association with hemidesmosomes in epidermal cells is α6β4 (27, 28). It probably plays a vital role in cell–cell adhesion and in cell and extracellular matrix adhesion and interactions within the BMZ (21). Ultrastructural studies have shown that in skin (22, 29) and cornea (30), the α6β4 integrin is located in hemidesmosomes. Molecular cloning studies of β4-integrin indicate that it is unique since it has a long cytoplasmic domain (31, 32). Immunohistochemical analysis has shown that it is mainly expressed in epithelial cells (27). The laminins are ligand for β4-integrin (21, 33).
In OCP the pathognomonic feature is the presence of a subepithelial vesicle or bullae at the BMZ. The fact that autoantibodies in OCP patients are directed against the β4 component of α6β4 integrin emphasizes the importance of the β4 molecules to the integrity of the conjunctival BMZ and that anti-β4 autoantibodies may be pathogenic. Indeed, studies by two groups of investigators indicate that monoclonal antibody BM-165 targeted against the α3 subunit of laminin can cause deepithelization of human skin in vitro and can also inhibit in vitro attachment of epidermal cells (33, 34). Interestingly, in one study Venning et al. (28) found no abnormality of α6β4 integrin expression in uninvolved normal perilesional skin of BP patients but did find absence of BPA and α6β4 integrin in fully developed BP blisters (29). Our studies demonstrate that β4 is present in the conjunctival BMZ; but its absence or derangement in OCP lesions has yet to be determined.
Several studies have suggested a role for epiligrin/laminin 5 in the pathogenesis of a group of patients with a cicatricial pemphigoid-like disease (29, 35–37). Kirtschig et al. (35) have demonstrated that the α3 subunit of laminin 5 is the target antigen for the anti-BMZ antibodies present in the sera of these patients (29, 35–37). Recently, Ghohestani et al. (38) have demonstrated that in patients with CP limited to the oral cavity have an antibody directed against a 168-kDa oral mucosa protein. Anti-BMZ antibodies in these patients do not cross-react with BP antibodies (38). These observation clearly support our view that CP is a heterogeneous disease and that different subsets of the disease may be associated with different target molecules involved in the cell–extracellular matrix and BMZ interaction.
There is increasing evidence to suggest that membrane-bound α6β4 heterodimeric molecules, while providing the functions of cell adhesion, also are critical in transmitting extracellular signals to cytoplasmic processes (39). In recent studies involving a knockout mice in which the BPAg1 gene was deleted, the skin epidermal cell hemidesmosomes were normal but the cells lacked the inner plate and cytoskeleton attachment, with resultant compromised mechanical integrity and cell migration (40). Interestingly, these mice did not develop clinical BP but did develop severe dystonia and sensory nerve degeneration similar to that observed in dystonia musculorum (dt/dt) mice (40). Two recent reports on β4-integrin-deficient mice demonstrate that the absence of β4 in these mice produces a phenotype similar to the human analogue of junctional epidermolysis bullosa (41, 42). The mice exhibit extensive separation of epidermis from dermis and die shortly after birth.
In earlier studies (43), our group demonstrated a highly statistically significant association between OCP and the HLA-DQB1∗0301 allele. Now that the target antigen is known and the sequence of β4-integrin is available, both the antibody binding epitope and T-cell response can be identified and characterized. When such epitope mapping is available, it will enable us to study the regulation of anti-BMZ autoantibody production in patients with OCP.
We believe that the observations reported herein will help advance our understanding of how the conjunctival cicatrization process is initiated at the molecular, cellular, and tissue level. We also now believe that OCP is a heterogeneous disease with polymorphic phenotypic presentations, with other molecules as possible targets in the pathogenesis of the various subsets of the disease. Indeed, more than one molecule may be involved for the pathogenic process to be completed.
Acknowledgments
We are grateful to Dr. Martin Hemler for providing MDA 435 cells and polyclonal antibody against β4-integrin, Dr. Robert Burgeson for anti-laminin-5 antibody, Dr. Kim Yancey for anti-epiligrin and anti-laminin antibodies, Dr. Thomas Carey and Ancell Corporation for providing UM-SCC-22 cell line and lysate and UMA-9 antibody, Dr. Arthur Mercurio for his constructive comments on the manuscript, and Dr. Chris Boles from Mosaic Technologies for his suggestions. This study was supported by National Institutes of Health Grants EYO8378 and DEO9978 and in part by grants from the Rubenstein Foundation and the Pemphigus Foundation.
Footnotes
Abbreviations: OCP, ocular cicatricial pemphigoid; CP, cicatricial pemphigoid; BP, bullous pemphigoid; BMZ, basement membrane zone; PV, pemphigus vulgaris; HEL, human epidermal lysate; HCL, human conjunctival lysate.
References
- 1.Foster C S. Trans Am Ophthalmol Soc. 1986;84:527–663. [PMC free article] [PubMed] [Google Scholar]
- 2.Ahmed A R, Hombal S M. Int J Dermatol. 1986;25:90–96. doi: 10.1111/j.1365-4362.1986.tb04544.x. [DOI] [PubMed] [Google Scholar]
- 3.Bernard P, Prost C, Aucoutuier P, Durepaire N, Denis F, Jean-Marie B. J Invest Dermatol. 1991;97:259–263. doi: 10.1111/1523-1747.ep12480369. [DOI] [PubMed] [Google Scholar]
- 4.Neiboer C, Boarsma D M, Woederma M J. Br J Dermatol. 1982;106:419–422. [Google Scholar]
- 5.Shimuzu H, Masunago T, Ishiko A, Matsumura K, Hashimoto T, Nishokawa T, Damloge-Hultsch N, Lazarova Z, Yancey K B. J Invest Dermatol. 1995;104:370–373. doi: 10.1111/1523-1747.ep12665840. [DOI] [PubMed] [Google Scholar]
- 6.Mohimen A, Neumann R, Foster C S, Ahmed A R. Curr Eye Res. 1993;12:741–752. doi: 10.3109/02713689308995770. [DOI] [PubMed] [Google Scholar]
- 7.Balding S D, Prost C, Diaz L A, Bernard P, Beadne C, Aberdam D, Giudice G J. Invest Dermatol. 1996;106:141–146. doi: 10.1111/1523-1747.ep12329728. [DOI] [PubMed] [Google Scholar]
- 8.Bhol K, Mohimen A, Neumann R, Yunis J, Foster C S, Yunis E J, Ahmed A R. Curr Eye Res. 1996;15:521–532. doi: 10.3109/02713689609000763. [DOI] [PubMed] [Google Scholar]
- 9.Domloge-Hultsch N, Anhalt G J, Gammon W R, Lazarova Z, Briggaman R A, Welch M, Jabs D A, Huff C, Yancey K B. Arch Dermatol. 1994;130:1521–1529. [PubMed] [Google Scholar]
- 10.Gudula Kirtschig M, Marinkovich P, Burgeson R E, Yancey K B. J Invest Dermatol. 1995;105:543–548. doi: 10.1111/1523-1747.ep12323431. [DOI] [PubMed] [Google Scholar]
- 11.Olmsted J B. J Biol Chem. 1981;256:11955–11957. [PubMed] [Google Scholar]
- 12.Mohimen A, Ahmed A R. Arch Dermatol Res. 1995;287:202–208. doi: 10.1007/BF01262333. [DOI] [PubMed] [Google Scholar]
- 13.Laemmli U K. Nature (London) 1970;277:680–685. doi: 10.1038/227680a0. [DOI] [PubMed] [Google Scholar]
- 14.Hashimoto T, Ogawa M M, Konohana A, Nishikawa T. J Invest Dermatol. 1990;94:327–331. doi: 10.1111/1523-1747.ep12874456. [DOI] [PubMed] [Google Scholar]
- 15.Qureshi T, Nagarwalla N, Sarela A, Ahmed A R. Clin Immunol Immunopathol. 1995;75:94–98. doi: 10.1006/clin.1995.1057. [DOI] [PubMed] [Google Scholar]
- 16.Pearson T W, Clark M W. In: Handbook of Experimental Immunology. Weir D M, editor. Oxford: Blackwell; 1986. pp. 65–67. [Google Scholar]
- 17.Porath J, Axen R. Methods Enzymol. 1976;44:19–45. doi: 10.1016/s0076-6879(76)44005-3. [DOI] [PubMed] [Google Scholar]
- 18.Young R A, Davis R W. Proc Natl Acad Sci USA. 1983;80:1194–1198. doi: 10.1073/pnas.80.5.1194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Sanger F, Nicklen S, Coulson A R. Proc Natl Acad Sci USA. 1977;74:5463–5467. doi: 10.1073/pnas.74.12.5463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Altschul S F, Gish W, Miller W, Myers E W, Lipman D J. J Mol Biol. 1990;215:403–410. doi: 10.1016/S0022-2836(05)80360-2. [DOI] [PubMed] [Google Scholar]
- 21.Elner S G, Elner V M. Invest Ophthalmol Visual Sci. 1996;37:696–701. [PubMed] [Google Scholar]
- 22.Bernard P, Prost C, Durepaire N, Basset-Seguin N, Didierjean L, Saurat J I I. J Invest Dermatol. 1992;99:174–179. doi: 10.1111/1523-1747.ep12616797. [DOI] [PubMed] [Google Scholar]
- 23.Chan L S, Hammerberg C, Cooper K D. Arch Dermatol. 1990;126:1466–1468. doi: 10.1001/archderm.126.11.1466. [DOI] [PubMed] [Google Scholar]
- 24.Chan L S, Yancey K B, Hammerberg C, Soong H K, Regezi J A, Johnson K, Cooper K D. Arch Dermatol. 1993;129:448–455. doi: 10.1001/archderm.129.4.448. [DOI] [PubMed] [Google Scholar]
- 25.Carter W G, Kaur P, Gil S G, Ghar P J, Wayner E A. J Cell Biol. 1990;111:3141–3154. doi: 10.1083/jcb.111.6.3141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Hertle M D, Adams J C, Watt F M. Development (Cambridge, UK) 1991;112:193–206. doi: 10.1242/dev.112.1.193. [DOI] [PubMed] [Google Scholar]
- 27.Sonnenberg A, Calafat J, Janssen H, Daams H, van der Raaij-Helmer L M H, Falcioni R, Kennel S, Aplin J D, Baker J, Loizidou M, Garrod D. J Cell Biol. 1991;113:907–917. doi: 10.1083/jcb.113.4.907. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Hemler M E, Crouse C, Sonnenberg A. J Biol Chem. 1989;264:6529–6535. [PubMed] [Google Scholar]
- 29.Venning V A, Allen J, Alpin J D, Kirtschig G, Wojnarowska F. Br J Dermatol. 1992;127:103–111. doi: 10.1111/j.1365-2133.1992.tb08040.x. [DOI] [PubMed] [Google Scholar]
- 30.Stepp M A, Spurr-Michaud S, Tisdale A, Elwell J, Gipson I K. Proc Natl Acad Sci USA. 1990;87:8970–8974. doi: 10.1073/pnas.87.22.8970. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Hogervorst F, Kuikman I, von dem Borne A E G Kr, Sonnenberg A. EMBO J. 1990;9:765–770. doi: 10.1002/j.1460-2075.1990.tb08171.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Tamura R N, Razzo C, Starr L, Chambers J, Reichadt L F, Cooper H M, Quaranta V. J Cell Biol. 1990;111:1593–1604. doi: 10.1083/jcb.111.4.1593. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Niessen C M, Hogervorst F, Jaspars L H, De Melker A A, Delwel G O, Hulsman E H M, Kuikman I, Sonnenberg A. Exp Cell Res. 1994;211:360–367. doi: 10.1006/excr.1994.1099. [DOI] [PubMed] [Google Scholar]
- 34.Champliaud M-F, Lunstrum G P, Rousselle P, Nishiyama T, Keene D R, Burgeson R E. J Cell Biol. 1996;132:1189–1198. doi: 10.1083/jcb.132.6.1189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Kirtschig G, Marinkovich M P, Burgeson R E, Yancey K B. J Invest Dermatol. 1995;105:543–548. doi: 10.1111/1523-1747.ep12323431. [DOI] [PubMed] [Google Scholar]
- 36.Sonnenberg A, de Melker A A, Martinez de Velasco A M, Janssen H, Calafat J, Niessen C M. J Cell Sci. 1993;106:1083–1102. doi: 10.1242/jcs.106.4.1083. [DOI] [PubMed] [Google Scholar]
- 37.Burgeson R E, Chiquet M, Deutzmann R, Ekblom P, Engel J, Kleinman H, Martin G R, Meneguzzi G, Paulsson M, Sanes J, Timpl R, Tryggvason K, Yamada Y, Yurchenco P D. Matrix Biol. 1994;14:209–211. doi: 10.1016/0945-053x(94)90184-8. [DOI] [PubMed] [Google Scholar]
- 38.Ghohestani R F, Niclas J F, Rousselle P, Claudy A L. J Invest Dermatol. 1996;107:136–139. doi: 10.1111/1523-1747.ep12298424. [DOI] [PubMed] [Google Scholar]
- 39.Hynes R O. Cell. 1992;69:11–25. doi: 10.1016/0092-8674(92)90115-s. [DOI] [PubMed] [Google Scholar]
- 40.Guo L, Degenstein L, Dowling J, Yu Q-U, Wollman R, Perman B, Fuchs E. Cell. 1995;81:233–243. doi: 10.1016/0092-8674(95)90333-x. [DOI] [PubMed] [Google Scholar]
- 41.van der Neut, Kimpenfort P, Calafat J, Niessen C M, Sonnenberg A. Nat Genet. 1996;13:366–369. doi: 10.1038/ng0796-366. [DOI] [PubMed] [Google Scholar]
- 42.Dowling J, Yu Q-C, Fuchs E. J Cell Biol. 1996;134:559–572. doi: 10.1083/jcb.134.2.559. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Ahmed A R, Foster S, Zaltas M, Notani G, Awdeh Z, Alper C A, Yunis E. Proc Natl Acad Sci USA. 1991;88:11579–11582. doi: 10.1073/pnas.88.24.11579. [DOI] [PMC free article] [PubMed] [Google Scholar]