

Abstract
Immunodeficiency typically appears many years after initial HIV infection. This long, essentially asymptomatic period contributes to the transmission of HIV in human populations. In rare instances, clearance of HIV-1 infection has been observed, particularly in infants. There are also reports of individuals who have been frequently exposed to HIV-1 but remain seronegative for the virus, and it has been hypothesized that these individuals are resistant to infection by HIV-1. However, little is known about the mechanism of immune clearance or protection against HIV-1 in these high-risk individuals because it is difficult to directly demonstrate in vivo protective immunity. Although most of these high-risk individuals show an HIV-1-specific cell-mediated immune response using in vitro assays, their peripheral blood lymphocytes (PBLs) are still susceptible to HIV infection in tissue culture. To study this further in vivo, we have established a humanized SCID mouse infection model whereby T-, B-, and natural killer-cell defective SCID/beige mice that have been reconstituted with normal human PBLs can be infected with HIV-1. When the SCID/beige mice were reconstituted with PBLs from two different multiply exposed HIV-1 seronegative individuals, the mice showed resistance to infection by two strains of HIV-1 (macrophage tropic and T cell tropic), although the same PBLs were easily infected in vitro. Mice reconstituted with PBLs from non-HIV-exposed controls were readily infected. When the same reconstituted mice were depleted of human CD8 T cells, however, they became susceptible to HIV-1 infection, indicating that the in vivo protection required CD8 T cells. This provides clear experimental evidence that some multiply exposed, HIV-1-negative individuals have in vivo protective immunity that is CD8 T cell-dependent. Understanding the mechanism of such protective immunity is critical to the design and testing of effective prophylactic vaccines and immunotherapeutic regimens.
The time course of disease progression after HIV infection varies depending on a number of factors including viral strains and host immune responses. While the majority of HIV-infected subjects develop AIDS at a median of ≈10 years, up to 5–10% of infected individuals do not appear to have clinical symptoms even after 10 years (1, 2). Indeed, epidemiological evidence suggests that some individuals may be resistant to infection by HIV; despite multiple high-risk exposures, these individuals remain HIV negative (3–5).
Both host genetic factors and the infection-immunization process following HIV exposure may play roles in the development of protective immunity (6–9). The mechanism of resistance to HIV infection in the HIV-exposed but uninfected individuals may be associated with HIV-specific cytotoxic T cell activity (4, 10–13), a switch from Th2 to Th1 cytokine response (6, 14), genetic loci linked to HLA, transporter associated with antigen processing (TAP), and HIV coreceptors (9, 15, 16), or development of anti-HLA antibodies (17). While strong indirect evidence supports the hypothesis that these exposed but uninfected individuals have protective immunity against HIV infection, direct in vivo experiments attesting to this hypothesis are still lacking. In an attempt to test this hypothesis directly, we selected two highly exposed but HIV-1 seronegative individuals, and conducted in vivo challenge experiments in human-peripheral blood lymphocyte SCID/beige (hu-PBL-SCID/bg) mice using both T cell and macrophage tropic laboratory strains of HIV-1. This study provides direct evidence demonstrating in vivo protective immunity in these multiply exposed, seronegative individuals.
MATERIALS AND METHODS
Study Subjects.
Two individuals were identified who have engaged in regular unprotected heterosexual or homosexual intercourse with HIV-positive partners over a period of up to 10 years, but have remained HIV negative as determined by repeated serological assay, polymerase chain reaction (PCR) and PBL cocultivation. High-risk (HR) donor 1 is the heterosexual partner of a bisexual individual who had a CD4 count of 400–500/μl at the time of positive HIV diagnosis in July 1992. An estimated 144 episodes of vaginal intercourse between the HR donor 1 and the partner took place before his diagnosis, and the HIV-positive partner’s CD4 count has decreased to 30/μl at the time of study. HR donor 2 is the homosexual partner of an individual known to be HIV-1 positive since 1986 and whose CD4 count remained in the 300–400/μl range. We estimate that unprotected anal sex took place ≈225 times before this study, and unprotected sexual contact has “continued” since diagnosis. Infectious virus had been isolated from phytohemagglutinin (PHA)-blasted PBLs of both HIV-positive partners. Additional multiply exposed, uninfected individuals who fit into the above criteria were also included in the in vitro studies. Blood provided by Canadian Red Cross and volunteers in the laboratory with no prior HR exposures to HIV were the source of control PBLs.
Virus Preparation.
HIV-1 strains used in this study include laboratory-established T cell tropic (HIVNL4-3) and macrophage tropic HIV-1 (HIVNLAD8) (18). Virus stocks were prepared from infected peripheral blood mononuclear cells or macrophages as described (19). The same virus stocks were used for both in vitro and in vivo experiments. The multiplicity of infection (moi) or tissue culture infectious dose (TCID50) was determined by serial dilution of virus stock on HIV-1 negative, PHA-stimulated PBLs as described previously (19, 20).
In Vitro HIV-1 Infection.
Heparinized peripheral blood was collected from HR and low-risk (LR) HIV-negative donors. PBLs were separated by gradient centrifugation with Histopaque (Sigma). Infection was performed as described previously (19). Briefly, PBLs were treated with PHA (5 μg/ml, Sigma) and infected with T cell or macrophage tropic HIV-1 at 0.2 moi. The culture was split at a 1:3 ratio every 3–4 days. The supernatant was harvested for reverse transcriptase (RT) assay, and infections were also confirmed by immunohistochemical staining as described (19).
Establishment of hu-PBL-SCID/bg Mice and in Vivo HIV-1 Infection.
The human PBL reconstitution was established using a new strain of SCID mouse, the SCID/bg, which lacks mature T, B, and natural killer cells (21). C.B.17 SCID/beige (SCID/bg) mice, 6–8 weeks old, were purchased from Taconic Farms or Charles River Breeding Laboratories and reconstituted with human PBLs as described for SCID mice (22–25). Briefly, each mouse was intraperitoneally (i.p.) injected with 2–3 × 107 PBLs resuspended in 0.5 ml of Hanks’ balanced salt solution. A near 100% success rate had been obtained when fresh PBLs were used (ref. 26, and C.Z. and L.-J.C., unpublished results). Two weeks after reconstitution, mice were bled from the tail and the human Ig level was assessed by enzyme-linked immunosorbent assay (ELISA). Reconstituted SCID/bg mice (hu-PBL-SCID/bg) were challenged with 100 TCID50 of T cell or macrophage tropic HIV-1 by i.p. injection under metofane-induced anesthesia. Two weeks after HIV-1 challenge, mice were killed, and single cell suspensions were prepared from peritoneal lavage, spleen, and peripheral blood and processed for immunostaining, flow cytometry analysis, and PCR. All experiments were performed in a biosafety level 3 facility and protocols were approved by the Biosafety Committee, Research Ethics Board, and Health Sciences Animal Welfare Committee at the University of Alberta.
Immunohistochemical Staining and Fluorescence-Activated Cell Sorter (FACS) Analysis.
Immunohistochemical staining was performed as described previously (19). Briefly, the cells were washed with phosphate-buffered saline (PBS) and attached to a 24-well plate that was pretreated with poly-d-lysine (1 mg/ml, Sigma). Nonspecific peroxidase activities were eliminated by treating PBLs with 0.01% H2O2 at room temperature for 5 min. Serum from an HIV-1-positive patient was used as the first antibody and a biotinylated sheep anti-human antibody (Amersham) was used as the secondary antibody. After washing, the cells were incubated with the ABC staining solution (Pierce) and stained with 3,3′-diaminobenzidine tetrahydrochloride (DAB, Sigma) containing 0.3% NiCl2 for 1 min. HIV-positive cells were counted under an inverted microscope, and the percentage of HIV-1 positive cells was determined by taking the average of more than three representative counts of 1,000–10,000 cells. For FACS analysis, red blood cells were lysed with ammonium chloride solution [containing 9 ml of 0.16 M NH4Cl and 1 ml of 0.17 M Tris (pH 7.62 to final pH 7.2)]. The cells were stained with PE-anti-mouse-H-2Kd (Pharmingen), fluorescein isothiocyanate (FITC)-anti-human-CD45 (anti-HLe-1, Becton Dickinson), phycoerythrin-anti-human-CD4 or FITC-anti-human CD8 (Becton Dickinson) for 30 min on ice followed by three washes with immunostaining buffer (PBS containing 0.1% FBS, 0.02% NaN3). Isotype-matched PE- and FITC-mouse Igs were used for control staining. The samples were analyzed using the lysis ii program on a FACScan (Becton Dickinson).
ELISPOT Analysis of Interferon γ (IFN-γ) Production and in Vitro Lymphocyte Depletion.
ELISPOT was performed as described (27) with the following modifications (Y.C. and L.-J.C., unpublished work). To quantify the IFN-γ producing cells, a 96-well nitrocellulose-bottomed plate (MultiScreen-HA, Millipore) was coated with 75 μl per well mouse anti-human IFN-γ (10 μg/ml, Pharmingen) at room temperature overnight. HIV-1-infected PBLs were used as stimulators in the ELISPOT assay. Infection was confirmed by RT assay and by immunohistochemical staining (ranged 3–5% of HIV-1 positive). No difference was observed between HR and LR individuals in either RT or HIV antigen expression in the infected PBLs. HIV-1-infected PBLs were treated with mitomycin C (5 μg/ml) for 2.5 h, washed, and resuspended in RPMI medium 1640 containing 10% FBS and 20 units/ml interleukin 2 (Boehringer Mannheim), and seeded in the anti-IFN-γ-coated 96-well plate at 1 × 105 per well as target cells (T). Frozen unstimulated autologous PBLs, which were thawed and used as effector cells (E), were added to the wells at E/T ratio of 0.4:1, 2:1, and 10:1 in triplicates, and incubated in RPMI medium 1640 containing 10% FBS and 20 units/ml IL-2 at 37°C, 95% O2/5% CO2 for 24 h.
For the depletion of CD4, CD8, and CD56 cells, thawed PBLs were incubated with mouse anti-human CD4-, CD8-, CD56-, or mouse IgG1-labeled BioMeg magnetic beads (PerSeptive Diagnostics, Cambridge, MA) at 50 beads per cell on ice for 30 min. The depletion was carried out by sorting the cells on a magnet for 5 min two times using mouse IgG1 sorting as control. After depletion, cells were washed with culture medium and seeded in triplicates with the HIV-1-infected autologous PBLs as target cells in an anti-IFN-γ-coated 96-well plate for 24 h as described above. To evaluate the contribution of individual subpopulation of lymphocytes in the IFN-γ production, comparison was based on the initial number of PBL used. Thus, the E/T ratio (10:1) was based on the starting PBL number before depletion. After overnight incubation, the wells were washed four times using PBS-Tween 20 (PBS-T, 0.05%), blocked with 20% FBS in PBS-T at room temperature for 15 min, and incubated with 100 μl of biotinylated-mouse-anti-human IFN-γ (Pharmingen, 2.5 μg/ml in PBS-T) at 4°C overnight. Each well was then washed four times using PBS-T and incubated with peroxidase-labeled streptoavidin (Caltag, South San Francisco, CA) in PBS/T at room temperature for 1 h. IFN-γ-producing cells were detected as purple brown spots after DAB (Sigma) and 0.3% NiCl2 staining. The wells were washed four times with double distilled H2O and air-dried, and the number of IFN-γ producing cells was counted using a video-imaging (Appligene, Strasbourg, France) and computer analysis system (Y.C. and L.-J.C., unpublished work). HIV-1-specific ELISPOT was shown as the mean ± SE in triplicate wells after subtracting the background from controls. The control samples were prepared using the same effector cells plus autologous PBLs that were not infected with HIV-1.
Depletion of CD8 T Cells in hu-PBL-SCID/bg Mice.
CD8 T cells were depleted as described (29) with the following modifications. Anti-human CD8 antibody was purified from ascites of OKT8 (American Type Culture Collection) hybridoma using protein A affinity column (ImmunoPure Plus, Pierce). Mice were i.p. injected with 20 μg of the purified antibody five times in total: 2 days before PBL reconstitution, at the time of PBL injection, and at 1, 2, and 3 weeks after PBL injection. To confirm the depletion of CD8 T cells, hu-PBL-SCID/bg mice were killed, and the lymphocytes were analyzed by FACS analysis as described above. The cells were stained with PE-anti-mouse-H-2Kd (Pharmingen) and FITC-anti-human-CD45 (Anti-HLe-1, Becton Dickinson) or with PE-anti-human-CD4 and FITC-anti-human CD8 (Becton Dickinson) for 30 min on ice followed by three washes with immunostaining buffer (PBS containing 0.1% FBS and 0.02% NaN3). Isotype-matched PE-mouse-Ig and FITC-mouse Ig were used as controls.
RESULTS
Susceptibility of HIV-1-Exposed, HR Individuals’ PBLs to HIV-1 Infection.
To determine if lymphocytes from the HR individuals were susceptible to HIV-1 infection, their PBLs were collected, activated with PHA in culture and then incubated with T cell or macrophage tropic HIV-1. Infection was scored on the basis of HIV-1 RT activity and immunohistochemical staining with AIDS patients’ sera. Both the RT assay and the immunostaining results demonstrated that PBLs from these two HR, HIV-1-seronegative individuals were as susceptible as PBLs from normal LR donors to HIV-1 infection in tissue culture (Fig. 1a). To further examine whether the HR individuals’ PBLs were somewhat resistant to HIV-1 infection, the tissue culture infection was performed using a series of diluted virus preparations (from moi of 10−1 to 10−4). The result showed that even with moi of 10−4, the HR donors’ PBLs were still infected (Fig. 1b). A comparable dose-dependent kinetics of infection with different concentrations of virus was observed between LR and HR individuals (not shown). Similar results have been reported for HR, HIV-1-seronegative hemophiliacs (30). However, little is known about the possible nature of the protective immunity in these individuals.
Figure 1.
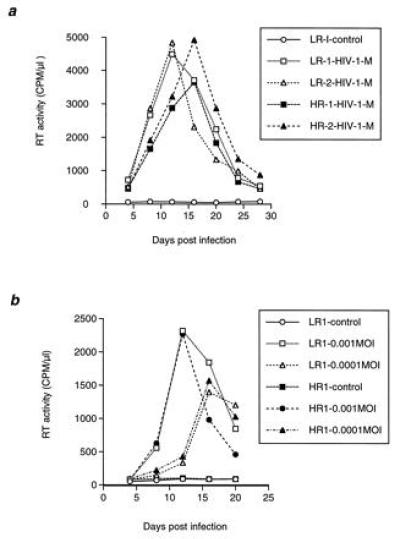
Infection of PBLs from LR and HR seronegative individuals with macrophage tropic HIV-1. Heparinized PBLs were collected from HR and LR HIV-negative donors and infected with different cytotropic strains of HIV-1NL4-3 as described previously (19). (a) Infection of PBLs from two LR (LR-1 and LR-2) and two HR (HR-1 and HR-2) seronegative individuals with HIVNLAD8 (macrophage tropic, M) at 0.2 moi. Similar result was obtained with the T-cell tropic strain HIV-1NL4-3 (not shown). (b) Infection of PBLs from LR-1 and HR-1 with HIVNL4-3 at moi 0.001–0.0001. The moi of 0.005 is equivalent to a TCID50 of 100.
HIV-1 Exposed-but-Uninfected Individuals Are Resistant to HIV-1 Infection in Vivo.
To overcome the problem of the lack of an in vivo HIV-1 infection model, we used the human PBL-reconstituted severe combined immunodeficiency mouse model (hu-PBL-SCID) to investigate the nature of the protective immunity in the HIV-exposed but uninfected individuals. We have demonstrated that these mice can be efficiently (near 100%) reconstituted with human PBLs by direct intraperitoneal injection (i.p.), and the reconstituted mice can be subsequently infected with HIV-1 (26). We used this in vivo model to test the susceptibility of the two HR, HIV-negative individuals to HIV-1 infection. PBLs were isolated and cells from each donor were used to reconstitute 6–7 SCID/bg mice each time as described in the Materials and Methods. Two weeks after reconstitution, the hu-PBL-SCID/bg mice were challenged with T cell or macrophage tropic HIV-1 using a TCID50 of 100. Mice were killed 2 weeks after HIV-1 challenge, and peritoneal lavage, splenocytes, and peripheral blood mononuclear cells were collected and analyzed by flow cytometry using anti-mouse major histocompatibility complex class I antibody (H-2Kd) and anti-human CD45, CD3, CD4, and CD8 antibodies. At the same time, HIV-1 infection was examined by a sensitive single-cell immunohistochemical staining method using either HIV-positive patients’ sera or a monoclonal anti-p24 antibody as described previously (19). The results of immunostaining demonstrated that all of the 10 hu-PBL-SCID/bg mice reconstituted with PBLs from control individuals were infected by either T cell or macrophage tropic HIV-1, at frequencies of about 1–4% of cells from the peritoneal lavage (Fig. 2a), 0.8–5% of splenocytes (Fig. 2b), and 0.1–0.5% of peripheral blood mononuclear cells (not shown). In contrast, all nine mice reconstituted with PBLs from the two HR, HIV-uninfected individuals were negative for the HIV immunostaining (Fig. 2 c and d, peritoneal lavage and spleen, respectively), but one of the negative mice was positive in the spleen for HIV-1 DNA sequences by PCR analysis (not shown), suggesting that infection was established at the time of challenge. These results are summarized in Fig. 2e. The in vivo challenge experiment was repeated once with eight mice reconstituted for each donor’s PBLs and similar conclusion was obtained (data not shown).
Figure 2.
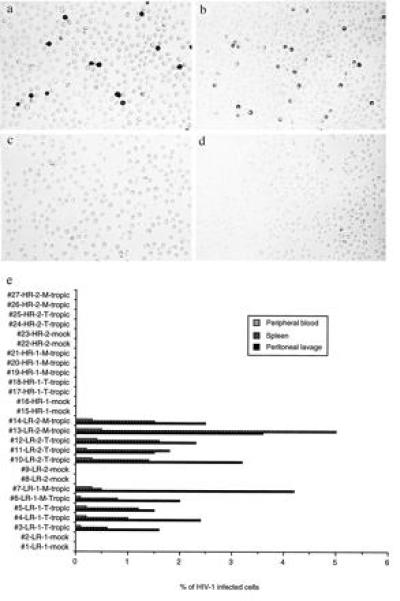
In vivo infection of hu-PBL-SCID/bg mice. (a and b) Representative HIV-1-positive immunostaining of peritoneal lavage and splenocytes, respectively, of mice reconstituted from a LR individual. (c and d) Representative HIV-1-negative immunostaining of peritoneal lavage and splenocytes, respectively, of mice reconstituted from a HR individual. (e) T cell and macrophage tropic HIV-1 challenge of hu-PBL-SCID/bg mice reconstituted with LR and HR donors’ PBLs. Reconstitution of mice with PBLs from the LR-1 donor (#1–#7), the LR-2 donor (#8–#14), the HR-1 donor (#15–#21), and the HR-2 donor (#22–#27)) were confirmed by ELISA for human Ig and FACS analysis for the human CD45 leukocyte marker. Reconstituted hu-PBL-SCID/bg mice were challenged with 100 TCID50 of T cell (T) or macrophage tropic (M) HIV-1 by i.p. injection. All the mice were killed 4 weeks after reconstitution, and single cell suspensions were prepared from the three different organ compartments for immunostaining. HIV-1-positive cells were counted under an inverted microscope and the percentage of infected cells was determined by taking the average of more than three representative counts of 1000–10,000 cells as previously described (19).
Cell-Mediated Immune Responses in the HR Individuals.
To investigate if the in vivo protection is due at least in part to HIV-1-specific, cell-mediated immune responses in the HR, seronegative individuals, we adapted a convenient and very sensitive ELISPOT assay to determine the fraction of IFN-γ producing cells after HIV antigen presentation. IFN-γ is produced by T cells and natural killer cells activated by antigens and is the key mediator of CD4+ Th1 cell development (31). Four LR and three HR donors’ PBLs, unstimulated, were incubated with mitomycin C-treated, HIV-infected autologous PBLs in a 96-well nitrocellulose-bottomed plate that had been coated with an anti-IFN-γ antibody. Production of IFN-γ was detected 24 h later using a secondary horseradish peroxidase-conjugated anti-IFN-γ antibody. As shown in Fig. 3, no HIV-specific IFN-γ producing responders were observed in PBLs of four LR donors except for donor 3, who showed a frequency of IFN-γ producing cells at one in 1 × 106 PBLs at the highest E/T ratio. In contrast, the existence of HIV-specific responders in the PBLs of HR seronegative donors were easily detected; at the E/T ratio of 10:1, the frequencies of HIV-specific IFN-γ producing cells were 14, 21, and 28 per 106 cells for the three HR donors, which was significantly different (P < 0.05, t test) from that of the LR donors. Even at an E/T ratio of 0.4:1, HIV-specific IFN-γ-producing cells were still detectable in all three HR donors. The contribution of the individual subset of immune effector cells in this HIV-specific reaction was further studied by depleting CD4, CD8, or CD56 cells from PBLs of two of these HR donors prior to the in vitro ELISPOT assay (not shown). Results of this quantitative analysis indicated that CD8 T cells contributed significantly to the production of IFN-γ (48–71%).
Figure 3.
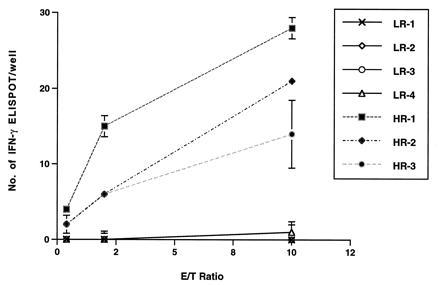
ELISPOT analysis of HIV-1-specific IFN-γ production in PBLs of HR and LR uninfected individuals. ELISPOT assays were performed as described. The number of IFN-γ producing cells was determined using a computerized video-imaging system as described previously (Y.C. and L.-J.C., unpublished work). HIV-1-specific ELISPOT was shown as the mean ± SE in triplicate wells after subtracting the background from controls that were prepared using the same effector cells incubated with autologous HIV-1-negative PBLs and went through the same treatment as described.
In Vivo Resistance to HIV-1 Infection Is CD8 T Cell-Dependent.
The above information was used to further delineate the mechanism of in vivo protection from HIV-1 in the reconstituted SCID/bg mice. We used HR donor 1’s PBLs and depleted CD8 T cells from the reconstituted mice using a monoclonal antibody (OKT8) i.p. injected before, during, and after PBL reconstitution (CD4 cells are the targets of HIV-1 infection and thus cannot be depleted in this study). Reconstitution and CD8 depletion were confirmed by FACS analysis 4 weeks later. In mice receiving no antibody, we observed 14% human CD45 cells (Fig. 4a), 7% human CD4 cells, and 8% human CD8 cells (Fig. 4c) in spleens 4 weeks after reconstitution. In mice treated with anti-CD8 antibody, there were similar numbers of CD45 cells in the spleen (12%, Fig. 4b), but the CD8 cells were virtually absent (0.1%, Fig. 4d). These mice were inoculated with the macrophage-tropic HIV-1NLAD8 on day 14 after reconstitution and killed 14 days later for analysis as before. Results of this study are summarized in Fig. 4e. Mice reconstituted with PBLs from HR donor 1 continued to be resistant to HIV-1 infection as long as CD8 cells were present. However, after CD8 cell depletion, similarly reconstituted mice became susceptible to HIV-1 infection (Fig. 4e, hu-PBL-SCID/bg mice #14, #15, and #16). These results strongly suggest that in vivo protection from HIV-1 infection in the reconstituted mice is mediated by human CD8 lymphocytes.
Figure 4.
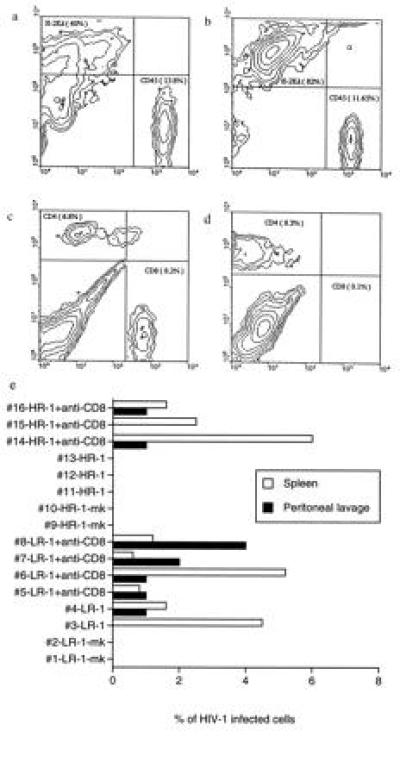
HIV-1 infection of human lymphocytes in hu-PBL-SCID/bg mice treated or untreated with anti-CD8 antibody. (a and c) FACS analysis of splenocytes from the reconstituted hu-PBL-SCID/bg mouse #11 immunostained with anti-mouse H-2Kd, anti-human CD45, anti-human CD4, or anti-human CD8 as depicted; (b and d) FACS analysis of splenocytes derived from the reconstituted hu-PBL-SCID/bg mouse #14, after anti-CD8 antibody treatment. (e) Infection of SCID/bg mice reconstituted with HR donor 1’s PBLs after CD8 T cell depletion. CD8 T cells were depleted by i.p. injection with 20 μg of purified OKT8 monoclonal antibody five times in total. The reconstituted mice, untreated or anti-CD8 treated as indicated, were infected with HIV-1NLAD8, and the percentage of infected cells was determined in the spleen and in the peritoneal lavage 2 weeks after virus challenge as described. The mock-infected (mk) mice were not treated with anti-CD8 antibody.
DISCUSSION
The difference observed between the in vivo and the in vitro studies indicates that anti-HIV-1 protective immunity cannot be properly assessed by tissue culture infection study or by the in vitro assays for cell-mediated immunity alone. The culture condition used in vitro is clearly different from body fluids encountered in vivo. An in vivo model is thus urgently needed. Here we demonstrate that multiply exposed, HIV-1-seronegative individuals have in vivo protective immunity against HIV-1 even though their PBLs are susceptible to HIV-1 infection in tissue culture. To obtain statistically valid results, a large number of reconstituted SCID/bg mice for each study subject is necessary. Due to the difficulty in acquiring large volumes of blood from HR, HIV-negative donors and the laborious nature of handling and analyzing a large number of reconstituted SCID/beige mice, we chose two representative HR, HIV-1-negative subjects for in vivo infection study in depth. The reconstitution efficiency and the percentage of infection of human lymphocytes in three different organ compartments were analyzed for each reconstituted mouse. Results of the in vivo HIV-1 challenge suggest that the cell-mediated immunity developed in these HR subjects also protects against alternative strains of HIV-1 since the challenge strains used in this study, T cell and macrophage tropic HIV-1, are unrelated to the endogeneous HIV-1 strains in the corresponding partners of these HR individuals under investigation.
Other studies have demonstrated the development of cell-mediated immune responses in HIV-1-exposed humans (4, 7, 10–12, 32, 33). Studies of the simian AIDS models have also suggested that cell-mediated immune responses play an important role in protection against simian immunodeficiency virus infection (34). It is possible that some genetic factors including HLA haplotype may also contribute to the development of resistance to HIV-1 infection (1, 8, 9). However, the two HR subjects who participated in the hu-PBL-SCID mouse challenge study do not share common HLA haplotypes (not shown).
In vivo protective immunity against HIV-1 has also been hypothesized to involve the functions of CD8 T cells that secret anti-HIV chemokines (35). In addition, CD4 T cells from 2 of 25 frequently exposed but uninfected individuals have recently been shown to have relative resistance to HIV-1 infection (28); subsequent genotype analysis of these two individuals by PCR and sequencing indicates that they have homozygous defects in the CCR-5 loci that encode the macrophage-tropic HIV-1 coreceptor (16). Results of PCR analysis of six multiply-exposed, uninfected individuals including the two HR subjects in our in vivo study showed that one is heterozygous and five are wild type (not shown). Both of the HR subjects participating the in vivo studies have homozygous wild-type CCR-5 loci. This is consistent with the result that PBLs of these two HR subjects were susceptible to macrophage-tropic HIV-1 infection in vitro.
While the in vitro ELISPOT analysis for the HIV-1-reactive effector functions showed that when exposed to autologous HIV-1-infected cells, the Th1-associated, HIV-1 antigen-specific IFN-γ production was higher from CD8 cells than from CD4 or natural killer cells in the HR donors, the possible contribution of CD4 cells to resistance due to the loss of double positive cells (CD4+ CD8+) in the depletion assay cannot be excluded. Thus, it is possible that these two HR, HIV-seronegative individuals participating the in vivo SCID/beige mouse challenge study may also have protective immunity developed in their CD4 T cell population. Further studies are necessary to characterize the possible mechanisms of CD4 or natural killer cell-mediated resistance.
Our study demonstrates the usefulness of the in vivo hu-PBL-SCID/bg mouse model for evaluating the protective immunity of individuals exposed to HIV-1. The SCID/bg mice can be reconstituted with human PBLs and infected with HIV-1 at a near 100% success rate. Further studies including different doses and different strains of HIV challenge and a large number of HR cohort will be necessary. In addition, this model should also be very useful for the assessment of the immune status of HIV-1 vaccinees.
Acknowledgments
We thank the cooperation of the multiply exposed individuals who participated in this study; the Canadian Red Cross, the individuals who donated blood; T. Birse and G. McCarthy for collecting blood and coordinating patients’ visit; J. F. Elliott, P. Dickie, and T. Mosmann for discussions; and Anne Hudson for secretarial assistance. L.-J.C. is a Research Scholar of Alberta Heritage Foundation for Medical Research and is supported by grants from National Health Research and Development Program (6609-1936-AIDS), the Medical Research Council of Canada (MT-12314) and the AIDS Network Research Trust Fund of Alberta.
Footnotes
Abbreviations: PBL, peripheral blood lymphocyte; HR, high risk; LR, low risk; moi, multiplicity of infection; PHA, phytohemagglutinin; RT, reverse transcriptase; FITC, fluorescein isothiocyanate; FACS, fluorescence-activated cell sorter; IFN-γ, interferon γ; E/T ratio, effector/target ratio.
References
- 1.Haynes B F, Pantaleo G, Fauci A S. Science. 1996;271:324–328. doi: 10.1126/science.271.5247.324. [DOI] [PubMed] [Google Scholar]
- 2.Baltimore D. N Engl J Med. 1995;332:259–260. doi: 10.1056/NEJM199501263320410. [DOI] [PubMed] [Google Scholar]
- 3.Detels R, Liu Z, Kan J, Visscher B R, Taylor J M, et al. J Acquired Immune Defic Syndr. 1994;7:1263–1269. [PubMed] [Google Scholar]
- 4.Rowland-Jones S, Sutton J, Ariyoshi K, Dong T, Gotch F, McAdam S, Whitby D, Sabally S, Gallimore A, Corrah T, Takiguchi M, Schultz T, McMichael A, Whittle H. Nat Med. 1995;1:59–64. doi: 10.1038/nm0195-59. [DOI] [PubMed] [Google Scholar]
- 5.Taylor R. J NIH Res. 1994;6:29–31. [Google Scholar]
- 6.Barcellini W, Paolo G, Velati C, Borghi M O, Fain C, Lazzarin A, Meroni P L. AIDS. 1995;9:691–694. doi: 10.1097/00002030-199507000-00004. [DOI] [PubMed] [Google Scholar]
- 7.Taylor R. J NIH Res. 1994;6:68–71. [Google Scholar]
- 8.Kaslow R A, Carrington M, Apple R, Park L, Munoz A, Saah A J, Goedert J J, Winkler C, O’Brien S J, Rinaldo C, Detels R, Blattner W, Phair J, Erlich H, Mann D L. Nat Med. 1996;2:405–411. doi: 10.1038/nm0496-405. [DOI] [PubMed] [Google Scholar]
- 9.Detels R, Mann D, Carrington M, Hennessey K, Wu Z, Hirji K F, Wiley D, Visscher B R, Giorgi J V. AIDS. 1996;10:102–104. doi: 10.1097/00002030-199601000-00016. [DOI] [PubMed] [Google Scholar]
- 10.Clerici M, Giorgi J V, Chou C C, Gudeman V K, Zack J A, Gupta P, Ho H N, Nishanian P G, Berzofsky J A, Shearer G M. J Infect Dis. 1992;165:1012–1019. doi: 10.1093/infdis/165.6.1012. [DOI] [PubMed] [Google Scholar]
- 11.Rowland-Jones S L, Nixon D F, Aldhous M C, Gotch F, Ariyoshi K, Hallam N, Kroll J S, Froebel K, McMichael A. Lancet. 1993;341:860–861. doi: 10.1016/0140-6736(93)93063-7. [DOI] [PubMed] [Google Scholar]
- 12.Pinto L A, Sullivan J, Berzofsky J A, Clerici M, Kessler H A, Landay A L, Shearer G M. J Clin Invest. 1995;96:867–876. doi: 10.1172/JCI118133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Langlade-Demoyen P, Ngo-Giang-Huong N, Ferchal F, Oksenhendler E. J Clin Invest. 1993;93:1293–1297. doi: 10.1172/JCI117085. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Clerici M, Shearer G M. Immunol Today. 1993;14:107–111. doi: 10.1016/0167-5699(93)90208-3. [DOI] [PubMed] [Google Scholar]
- 15.Hill A V S. Nat Med. 1996;2:395–396. doi: 10.1038/nm0496-395. [DOI] [PubMed] [Google Scholar]
- 16.Liu R, Paxton W A, Choe S, Ceradini D, Martin S R, Horuk R, MacDonald M E, Stuhlmann H, Koup R A, Landau N R. Cell. 1996;86:367–377. doi: 10.1016/s0092-8674(00)80110-5. [DOI] [PubMed] [Google Scholar]
- 17.Beretta A, Weiss S H, Rappocciolo G, Mayur R, De Santis C, Quirinale J, Cosma A, Robbioni P, Shearer G M, Berzofsky J A, Villa M L, Siccardi A G, Clerici M. J Infect Dis. 1996;173:472–476. doi: 10.1093/infdis/173.2.472. [DOI] [PubMed] [Google Scholar]
- 18.Adachi A, Gendelman H E, Koenig S, Folks T, Willey R, Rabson A, Martin M A. J Virol. 1986;59:284–291. doi: 10.1128/jvi.59.2.284-291.1986. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Chang L-J, Zhang C. Virology. 1995;211:157–169. doi: 10.1006/viro.1995.1388. [DOI] [PubMed] [Google Scholar]
- 20.Ho D D, Moudgil T, Alam M. N Engl J Med. 1989;321:1621–1625. doi: 10.1056/NEJM198912143212401. [DOI] [PubMed] [Google Scholar]
- 21.Froidevaux S, Loor F. J Immunol Methods. 1991;137:275–279. doi: 10.1016/0022-1759(91)90034-d. [DOI] [PubMed] [Google Scholar]
- 22.Mosier D E, Gulizia R J, Baird S M, Wilson D B. Nature (London) 1988;335:256–259. doi: 10.1038/335256a0. [DOI] [PubMed] [Google Scholar]
- 23.Torbett B E, Picchio G, Mosier D E. Immunol Rev. 1991;124:139–164. doi: 10.1111/j.1600-065x.1991.tb00620.x. [DOI] [PubMed] [Google Scholar]
- 24.Mosier D E, Gulizia R J, MacIsaac P D, Corey L, Greenberg P D. Proc Natl Acad Sci USA. 1993;90:2443–2447. doi: 10.1073/pnas.90.6.2443. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Safrit J T, Fung S C, Andrews C A, Braun D G, Sun N C, Chang T W, Koup R A. AIDS. 1993;7:15–21. doi: 10.1097/00002030-199301000-00002. [DOI] [PubMed] [Google Scholar]
- 26.Chang L-J, Zhang C, Robinson D, Dickie P. Transfusion Sci. 1996;17:89–98. [Google Scholar]
- 27.Miyahira Y, Murata K, Rodriguez D, Rodriguez J R, Esteban M, Rodrigues M M, Zavala F. J Immunol Methods. 1995;181:45–54. doi: 10.1016/0022-1759(94)00327-s. [DOI] [PubMed] [Google Scholar]
- 28.Paxton W A, Martin S R, Tse D, O’Brien T R, Skurnick J, VanDevanter N L, Padian N, Braun J F, Kotler D P, Wolinsky S M, Koup R A. Nat Med. 1996;2:412–417. doi: 10.1038/nm0496-412. [DOI] [PubMed] [Google Scholar]
- 29.Baskar S, Ostrand-Rosenberg S, Nabavi N, Nadler L M, Freeman G J, Glimcher L H. Proc Natl Acad Sci USA. 1993;90:5687–5690. doi: 10.1073/pnas.90.12.5687. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Lederman M M, Jackson J B, Kroner B L, White G C, III, Eyster M E, Aledort L M, Hilgartner M W, Kessler C M, Cohen A R, Kiger K P, Goedert J J. J Inf Dis. 1995;172:228–231. doi: 10.1093/infdis/172.1.228. [DOI] [PubMed] [Google Scholar]
- 31.Wenner C A, Guler M L, Macatonia S E, O’Garra A, Murphy K M. J Immunol. 1996;156:1442–1447. [PubMed] [Google Scholar]
- 32.Clerici M, Levin J M, Kessler H A, Harris A, Berzofsky J A, Landay A L, Shearer G M. J Am Med Assoc. 1994;271:42–46. [PubMed] [Google Scholar]
- 33.Shearer G M, Clerici M. Immunol Today. 1996;17:21–24. doi: 10.1016/0167-5699(96)80564-0. [DOI] [PubMed] [Google Scholar]
- 34.Salvato M S, Emau P, Malkovsky M, Schultz K T, Johnson E, Pauza C D. J Med Primatol. 1994;23:125–130. doi: 10.1111/j.1600-0684.1994.tb00112.x. [DOI] [PubMed] [Google Scholar]
- 35.Cocchi F, DeVico A, Garzino-Demo A, Arya S K, Gallo R C, Lusso P. Science. 1995;270:1811–1815. doi: 10.1126/science.270.5243.1811. [DOI] [PubMed] [Google Scholar]