

Abstract
β2-Microglobulin-deficient (β2m−) mice generate a CD4+ major histocompatibility complex class II-restricted cytotoxic T-lymphocyte (CTL) response following infection with lymphocytic choriomeningitis (LCM) virus (LCMV). We have determined the cytotoxic mechanism used by these CD4+ CTLs and have examined the role of this cytotoxic activity in pathogenesis of LCM disease in β2m− mice. Lysis of LCMV-infected target cells by CTLs from β2m− mice is inhibited by addition of soluble Fas-Ig fusion proteins or by pretreatment of the CTLs with the protein synthesis inhibitor emetine. In addition, LCMV-infected cell lines that are resistant to anti-Fas-induced apoptosis are refractory to lysis by these virus-specific CD4+ CTLs. These data indicate that LCMV-specific CD4+ CTLs from β2m− mice use a Fas-dependent lytic mechanism. Intracranial (i.c.) infection of β2m− mice with LCMV results in loss of body weight. Fas-deficient β2m−.lpr mice develop a similar wasting disease following i.c. infection. This suggests that Fas-dependent cytotoxicity is not required for LCMV-induced weight loss. A potential mediator of this chronic wasting disease is tumor necrosis factor (TNF)-α, which is produced by LCMV-specific CD4+ CTLs. In contrast to LCMV-induced weight loss, lethal LCM disease in β2m− mice is dependent on Fas-mediated cytotoxicity. Transfer of immune splenocytes from LCMV-infected β2m− mice into irradiated infected β2m− mice results in death of recipient animals. In contrast, transfer of these splenocytes into irradiated infected β2m−.lpr mice does not cause death. Thus a role for CD4+ T-cell-mediated cytotoxicity in virus-induced immunopathology has now been demonstrated.
In recent years considerable advances have been made in our understanding of the mechanisms of cytotoxic T-lymphocyte (CTL)-mediated cytotoxicity. Elegant studies using cultured cell lines and mutant mouse strains have demonstrated that at least two independent cytotoxic mechanisms can be deployed by CTLs (1–3). Many CTL have cytoplasmic granules which contain perforin and granzyme proteases (4). When such CTLs encounter target cells that display appropriate major histocompatibility complex (MHC)–peptide complexes, the granule contents are secreted toward the target cell. The target cell membrane is permeabilized by perforin, and the subsequent uptake of granzymes initiates a cascade of events that ultimately results in cell death. In addition to this perforin-dependent pathway, certain T cells can lyse target cells by a Fas-dependent mechanism (1–3). The interaction between the Fas molecule (CD95) on target cells and its ligand, FasL, on activated T cells results in receptor aggregation, which transduces signals leading to apoptosis of the target cell. Thus, unlike perforin-dependent CTLs, Fas-dependent T cells can lyse only target cells that express Fas. Although perforin-dependent and Fas-dependent killing represent the major pathways of cell-mediated cytotoxicity, other lytic mechanisms exist. For example, production of either membrane-associated or secreted tumor necrosis factor (TNF)-α by T cells can induce apoptosis in certain cell types (5).
CTLs play an important role in the control of intracellular pathogens and in the elimination of tumorigenic cells (3, 6). In the case of lymphocytic choriomeningitis (LCM) virus (LCMV) infection of mice, virus-specific CD8+ CTLs are required for clearance of virus following peripheral infection and for the development of lethal meningitis following intracranial (i.c.) inoculation (7). Unlike normal mice, mice that lack perforin fail to control LCMV infection and do not develop lethal meningitis following i.c. infection, despite having high levels of Fas-dependent CD8+ CTLs (8, 9). Similar to CD8+ CTLs, both perforin-dependent and Fas-dependent CD4+ CTLs have been described (2, 3, 10). In contrast to CD8+ CTLs, however, the biological role of CD4+ T-cell-mediated cytotoxicity is poorly understood.
To further investigate the role of CD4+ CTLs in vivo, we have studied LCMV infection of β2-microglobulin-deficient (β2m−) mice. β2m− mice are unable to express MHC class I complexes efficiently and, therefore, are deficient in CD8+ T cells (11, 12). We have previously shown that these mice do not elaborate CD8+, MHC class I-restricted, CTLs following LCMV infection, but instead generate virus-specific, CD4+, MHC class II-restricted CTLs that are detectable directly ex vivo (13–15). Despite the induction of this alternative subset of CTLs, β2m− mice are unable to clear LCMV infection and, following i.c. inoculation, succumb to a chronic wasting disease that is dependent on CD4+ T cells (13–16). In addition, we demonstrated, using adoptive transfer experiments, that these LCMV-induced CD4+ T cells can cause lethal disease in irradiated infected β2m− recipient mice (13).
In this report, we have defined the cytolytic mechanism used by LCMV-specific CTLs in β2m− mice. Furthermore, we have determined the role of this CD4+ T-cell-mediated cytotoxicity in causing lethal LCM disease in these animals.
MATERIALS AND METHODS
Mice and Virus.
The 129×B6.β2m− mice used in these investigations have been previously described (13). In certain experiments C57BL/6J-B2 mtm1 UNC or STOCK B2 mtm1 UNC were used (The Jackson Laboratory). Fas-deficient β2m− C57BL6.β2m−.lpr (β2m−.lpr) mice (17) and C57BL/6 (B6) mice were bred and maintained at the University of North Carolina. All of these strains of mice are H2b. Male and female mice between 8 and 15 weeks of age were used.
The Armstrong-3 strain of LCMV was kindly provided by J. L. Whitton (Scripps Research Institute, La Jolla, CA). Virus was propagated in BHK-21 cells and was titered by plaque assay on Vero cell monolayers. Mice were infected by intraperitoneal (i.p.) inoculation with 2 × 104 plaque-forming units (pfu) of LCMV or were anesthetized with Metofane (Pittmann-Moore, Mundelein, IL) and inoculated i.c. with 2 × 103 pfu of virus in a volume of 20 μl.
Cell Lines.
EL 4 (MHC class I+, class II−; H2b) cells were cultured in RPMI-1640 medium supplemented with 10% (vol/vol) fetal calf serum (FCS), 2 mM l-glutamine, 10 mM Hepes, and 50 μM 2-mercaptoethanol. P815 (MHC class I+, class II−; H2d) and LB 27.4 (MHC class I+, class II+; H2b/d) cells were cultured in Dulbecco’s modified Eagle’s medium containing glucose at 4.5 g/liter and supplemented with 10% FCS, 2 mM l-glutamine, and 50 μM 2-mercaptoethanol. All cell lines were obtained from the American Type Culture Collection.
Selection of Anti-Fas-Resistant Cell Lines.
LB27.4 cells (1 × 108) were γ-irradiated [450 rad (1 rad = 0.01 Gy)] in a GammaCell-40 irradiator and allowed to recover in tissue culture for 5 days. The apoptosis-inducing anti-Fas antibody Jo2 (PharMingen) was then added to the culture to give a final concentration of 2 μg/ml. Twelve days later, surviving cells were cloned by limiting dilution on a feeder layer of γ-irradiated (3000 rad) mouse embryonic fibroblasts. The anti-Fas-resistant phenotype of these clones was maintained by the periodic addition of Jo2 to the cultures at 1 μg/ml. One of these anti-Fas-resistant clones, 3B10, was selected for further study.
Cytotoxicity Assays.
LCMV-specific CTL activity was determined by using 5-hr 51Cr release assays as described previously (14). Effector cells were prepared from the spleens of i.p. inoculated B6 or β2m− mice at 7–8 or 10–11 days after infection, respectively. These time points correspond to the peak of the LCMV-specific CTL response in the respective mouse strains (14). For “cold target” inhibition assays, various numbers of nonradiolabeled competitor cells (“cold targets”) were mixed with 2.5 × 105 effector cells. 51Cr-labeled LCMV-infected LB27.4 cells (5 × 103) were then added to give an effector-to-labeled-target ratio of 50:1. Following incubation, supernatants were harvested by using a Skatron harvesting system (Skatron Instruments, Sterling, VA). The amount of 51Cr released was quantified by γ counting, and the percent specific lysis was calculated as previously described (14). All assays were performed in triplicate.
To determine if effector cell protein synthesis is required for CTL activity, effector cells were pretreated with the irreversible protein synthesis inhibitor emetine (Sigma), for 1 hr at 37°C. Treated effector cells were washed three times prior to use in CTL assays. In certain experiments nylon wool-purified splenic effector cells were preincubated with 7 μg of Fas-Ig fusion protein (ref. 18; kindly provided by P. Leder and B. Stanger, Harvard Medical School, Boston) or with an equivalent concentration of human IgG1 (Sigma) for 30 min at 37°C. These effector cells were then used in CTL assays as described above.
Standard 51Cr-release assays were also used to measure anti-Fas-induced cytotoxicity. 51Cr-labeled target cells (5 × 103) were added into wells of U-bottom 96-well plates, which contained either no antibody or the anti-Fas mAb Jo2, to give a final volume of 200 μl. After incubation for 5 hr at 37°C in a 6% CO2/94% air atmosphere, the supernatants were harvested and radioactivity was measured as described above.
TNF-α production by LCMV-specific class II-restricted CTLs was determined in bystander lysis assays exactly as described by Hou et al. (19), using the TNF-α-sensitive EL 4 cell line and the TNF-α-resistant P815 cell line (19, 20). The assays were allowed to proceed for 6 hr at 37°C in 6% CO2, and the supernatants were harvested and radioactivity was measured as described above. For neutralization of TNF, rabbit antiserum to murine TNF-α (Genzyme) was added to the wells to give a final dilution of 1:200.
Flow Cytometry.
Staining and analysis of spleen cells were performed essentially as previously described (14). Briefly, spleen cells were prepared from untreated β2m− mice or from β2m− mice that had been infected i.p with LCMV 10 days earlier. Erythrocytes were removed by osmotic lysis and Fc receptors were blocked with mAb 2.4G2 (0.4 μg per 106 cells). Cells were stained with phycoerythrin (PE)-conjugated anti-CD4 (RM4-5; 0.5 μg per 106 cells) together with fluorescein isothiocyanate (FITC)-conjugated anti-CD45RB (16A; 1 μg per 106 cells) or FITC-conjugated anti-CD49d (9C10; 1 μg per 106 cells). For analysis of CD44 expression, cells were stained with FITC-conjugated anti-CD4 (0.5 μg per 106 cells) together with PE-conjugated anti-CD44 (IM7; 1 μg per 106 cells). All antibodies were obtained from PharMingen. After washing, stained cells were analyzed with a FACScan flow cytometer (Becton Dickinson), and collected data were examined using the computer program cicero (Cytomation, Fort Collins, CO). Dead cells were excluded on the basis of forward and side scatter.
Cell surface TNF-α expression on purified CD4+ cells was also determined by flow cytometry (5). Spleen cells were prepared as described above and CD4+ cells were isolated from these cell suspensions by using anti-CD4-coated magnetic beads (L3T4 microbeads, Miltenyi Biotec, Auburn, CA) in accordance with the manufacturer’s instructions. The purified cells were greater than 95% CD4+ as assessed by flow cytometry. Cell surface TNF expression was determined by staining the cells with anti-TNF (MP6-XT3; 1 μg per 106 cells) for 30 min at 4°C. Following washing, FITC-conjugated anti-rat IgG1 (RG11/39.4; 1 μg per 106 cells) was added and the cells were incubated for a further 30 min at 4°C. Control samples were stained with the secondary antibody only. FACScan analysis was performed as described above.
Adoptive Transfer of Immune Splenocytes.
Single-cell suspensions were prepared from the spleens of donor β2m− mice that had been infected i.p with LCMV 10–11 days previously. Erythrocytes were removed by osmotic lysis and the remaining cells were resuspended in PBS prior to transfer. Aliquots of these cells (1.7 × 108 or 1.8 × 108) were injected i.p. into recipient mice that had been irradiated (600 rad) and infected i.c. with LCMV 5 days previously. Control recipient mice were irradiated and infected but did not receive donor cells. Mice were monitored daily for morbidity and mortality.
RESULTS
Anti-Fas-Resistant Cells Are Refractory to Lysis by CD4+ CTLs.
Following infection with LCMV, β2m− mice generate virus-specific CD4+ CTLs which are detectable directly ex vivo (12). Cell lines resistant to anti-Fas-induced cell death were used to determine if these CTLs use a Fas-dependent lytic mechanism. Addition of the anti-Fas mAb Jo2 to 51Cr-labeled LB27.4 cells results in cell death as assessed by 51Cr release (Fig. 1A) and microscopic examination (data not shown). In contrast, the 3B10 cell line, derived from LB27.4 by irradiation mutagenesis and selection using Jo2, is resistant to anti-Fas-induced cell death (Fig. 1A). Standard 51Cr release assays were performed to determine if the Jo2-resistant 3B10 cells are susceptible to lysis by CTLs. Virus-specific CTLs from either LCMV-infected B6 mice, which generate CD8+ perforin-dependent CTLs (8, 9), or LCMV-infected β2m− mice, which elaborate CD4+ CTLs, were used as effectors. As expected, LCMV-infected LB27.4 cells are lysed by effector cells from both B6 and β2m− mice (Fig. 1B). In contrast, LCMV-infected, anti-Fas-resistant, 3B10 cells are lysed by effector cells from B6 mice but are not lysed above background levels by LCMV-specific CD4+ CTLs from β2m− mice (Fig. 1C).
Figure 1.
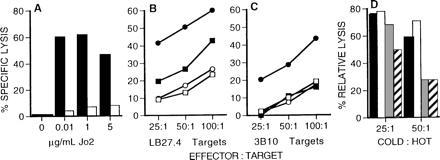
Anti-Fas-resistant cells are refractory to lysis by LCMV-specific CD4+ CTLs. (A) LB27.4 cells (solid bars) or anti-Fas-selected 3B10 cells (open bars) were tested in 51Cr release assays for their ability to be killed by Jo2 mAb. (B and C) LB27.4 (B) and 3B10 cells (C) were tested for their ability to be lysed by splenic effector cells prepared from B6 mice (○, •) or β2m− mice (□, ▪) at 7 or 10 days after i.p. infection with LCMV, respectively. Target cells were either noninfected (○, □) or infected with LCMV 48 hr previously (•, ▪). (D) Unlabeled target inhibition assays were performed using effector cells from LCMV-infected β2m− mice assayed at an effector to 51Cr-labeled target cell ratio of 50:1. The 51Cr-labeled targets (“hot targets”) were LCMV-infected LB27.4 cells. Unlabeled competitor cells (“cold targets”) were added to give cold:hot target ratio of either 25:1 or 50:1. Cold targets were noninfected LB27.4 cells (solid bars), noninfected 3B10 cells (open bars), LCMV-infected LB27.4 cells (shaded bars) and noninfected 3B10 cells (hatched bars). Percent relative lysis was calculated as follows: 100 × (% specific lysis in the presence of cold targets/% specific lysis in the absence of cold targets).
The results shown in Fig. 1C demonstrate that LCMV-infected 3B10 cells are resistant to lysis by virus-specific class II-restricted CTLs. To determine if 3B10 cells are capable of presenting LCMV antigen to these CD4+ CTLs, unlabeled target competition assays were performed. Because LCMV-infected LB27.4 cells are recognized by CTLs, the addition of excess nonradiolabeled infected LB27.4 cells competitively inhibits lysis of infected 51Cr-labeled LB27.4 cells (Fig. 1D, shaded bars). Similarly, the addition of nonradiolabeled LCMV-infected 3B10 cells also blocks lysis of infected 51Cr-labeled LB27.4 cells (Fig. 1D, hatched bars). In contrast, the addition of noninfected LB27.4 or noninfected 3B10 cells does not markedly inhibit CTL-mediated lysis (Fig. 1D, solid and open bars, respectively). Since both infected LB27.4 cells and infected 3B10 cells can block lysis by virus-specific, class II-restricted CTLs, these data show that both cell types are capable of being recognized by these CTLs. Taken together, these data demonstrate that 3B10 cells are not defective in their ability to be recognized by class II-restricted CTLs, but they are impaired in their ability to be killed by these CTLs.
Inhibitors of Fas-Dependent Cytotoxicity Block CTL-Mediated Lysis.
We next investigated whether inhibitors of Fas-dependent CTL activity affected the ability of LCMV-specific class II-restricted CTLs to lyse target cells. Inhibition of T-cell protein synthesis prevents the induction of FasL expression that occurs when a Fas-dependent CTL engages an appropriate target cell. Consequently, such treatment can inhibit the killing by Fas-dependent CTLs but not by perforin-dependent CTLs (5). Treatment of β2m− class II-restricted CTLs with the irreversible protein synthesis inhibitor emetine prior to assay markedly inhibited the ability of these CTLs to kill virus-infected target cells (Fig. 2A). Treatment with emetine had no effect on effector cell viability over the course of the assay as assessed by trypan blue dye exclusion (data not shown). These data further suggest that LCMV-specific class II-restricted CTLs employ a Fas-dependent lytic mechanism.
Figure 2.
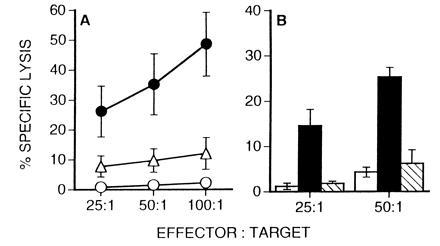
Inhibitors of Fas-dependent CTL activity prevent cytolysis by LCMV-specific CD4+ CTLs. Effector cells were prepared from β2m− mice at 10 days after i.p. infection and tested for their ability to lyse 51Cr-labeled LB27.4 cells. (A) Effector cells were preincubated in medium only (•, ○) or with 5 μM emetine (▵) and tested for their ability to lyse either infected target cells (▵, •) or noninfected target cells (○). (B) In separate experiments effector cells were assayed for their ability to lyse LCMV-infected target cells in the presence (hatched bars) or absence (solid bars) of 7 μg of Fas-Ig. Lysis of noninfected targets in the absence of Fas-Ig is also shown (open bars). Addition of 7 μg of human IgG1 does not inhibit CTL activity (data not shown).
To confirm that the LCMV-specific CTLs from β2m− mice use a Fas-dependent lytic mechanism, we attempted to inhibit CTL-mediated killing in 51Cr-release assays by the addition of soluble Fas-Ig fusion proteins (18). As illustrated in Fig. 2B, the addition of Fas-Ig prevents CTL-mediated target cell death. In contrast, the addition of control human IgG1 has no effect on CTL-mediated target cell death (data not shown). Taken together with the results shown in Fig. 1, these data demonstrate that LCMV-specific class II-restricted CTLs from β2m− mice primarily employ a Fas-dependent lytic mechanism.
β2m− and β2m−.lpr Mice Lose Weight Following i.c. Infection with LCMV.
CD4+ cells are required for the development of LCMV-induced weight loss in β2m− mice (12–15). Since LCMV-specific CD4+ CTL exhibit Fas-dependent lytic activity in vitro, we investigated whether Fas expression is required for the development of the wasting disease. We previously reported that Fas-expressing β2m− mice lose a marked amount of body weight following i.c. infection (12–14). Similarly, i.c infected β2m−.lpr mice also develop a wasting disease which closely resembles that observed in Fas-expressing β2m− mice (Fig. 3). In addition to weight loss, both strains showed other signs of illness, including ruffled fur and lethargy, which were most pronounced 10–15 days after infection. The finding that β2m−.lpr mice lose weight after i.c. infection with LCMV indicates that Fas expression, and therefore Fas-dependent cytotoxicity, is not required for the development of LCMV-induced wasting disease.
Figure 3.
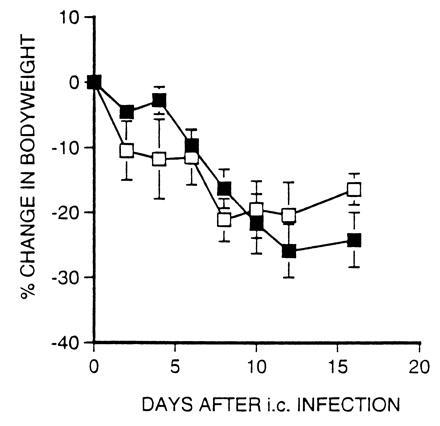
β2m− and β2m−.lpr mice lose weight after i.c. inoculation with LCMV. β2m− (▪) and β2m−.lpr (□) mice were inoculated i.c. with 2 × 103 pfu of LCMV. Mice were weighed at regular intervals and their weights were expressed as percent change from their initial (day 0) body weights (mean percent change ± SEM; n = 4).
LCMV-Specific CD4+ CTLs Produce TNF-α.
The finding that LCMV-induced wasting in β2m− mice is dependent on CD4+ cells but does not require the Fas-dependent cytotoxic activity prompted us to examine TNF-α production by these class II-restricted T cells. TNF-α is a potent cachectic cytokine (21) and, therefore, is a potential mediator of LCMV-induced weight loss. We stained CD4+ cells from LCMV-infected β2m− mice with a mAb to detect cell-associated TNF-α, an indicator of TNF-α production (5). CD4+ cells from LCMV-infected β2m− mice express increased levels of surface TNF-α compared with CD4+ cells from noninfected β2m− mice (Fig. 4A). Production of TNF-α was confirmed in bystander lysis experiments. LCMV-specific CTLs from β2m− mice were cultured with either noninfected or LCMV-infected nonradiolabeled LB27.4 cells. Addition of 51Cr-labeled TNF-α-sensitive EL 4 cells to cultures containing infected LB27.4 cells resulted in lysis of the EL 4 cells (Fig. 4B). This bystander lysis of EL 4 cells was inhibited by addition of neutralizing antiserum against TNF-α. In contrast, TNF-α-resistant P815 cells were not killed in these bystander lysis experiments. Overall, these data demonstrate that LCMV-specific class II-restricted T cells from β2m− mice produce the cachectic cytokine TNF-α.
Figure 4.
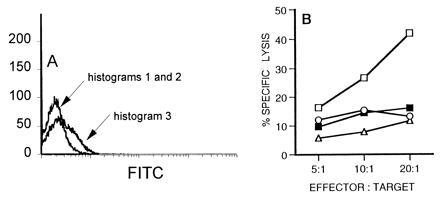
CD4+ cells from LCMV-infected β2m− mice produce TNF-α. (A) CD4+ cells were isolated from the spleens of noninfected β2m− mice (histogram 1) or from β2m− mice that had been infected 10 days previously (i.p.) with LCMV (histograms 2 and 3). Fc receptors were blocked with mAb 2.4G2, and surface expression of TNF-α was detected by using rat anti-TNF-α followed by FITC-conjugated anti-rat IgG1 (histograms 1 and 3). Control cells from infected mice were stained with FITC-conjugated anti-rat IgG1 only (histogram 1). (B) Production of biologically active TNF-α by LCMV-specific class II-restricted cells was assessed in bystander lysis assays. Spleen cells from LCMV-infected β2m− mice were cultured with infected LB27.4 cells and either 51Cr-labeled TNF-α-sensitive EL 4 cells (▪) or 51Cr-labeled TNF-α-resistant P815 cells (○) were added. Bystander lysis of EL 4 cells was blocked by addition of 0.5% (final concentration) of rabbit anti-mouse TNF-α (□). Spleen cells from infected mice were also cultured with noninfected LB27.4 cells, and bystander lysis of 51Cr-labeled EL 4 cells was tested (▵).
Fas Expression Is Required for Adoptive Transfer of Lethal LCM Disease.
The results shown in Fig. 3 demonstrate that Fas-dependent cytotoxicity is not required for LCMV-induced weight loss in β2m− mice. We have previously demonstrated that adoptively transferred LCMV-specific CD4+ cells can cause lethal disease in irradiated, i.c. infected, recipient β2m− mice (13). We therefore examined if Fas-dependent cytotoxicity is necessary for adoptive transfer of lethal LCM disease. Donor cells were prepared from the spleens of i.p. infected β2m− mice 10 days after infection. As shown in Fig. 5, compared with spleen cells from noninfected mice, the spleens of infected β2m− mice contain an increased percentage of CD4+ cells which are CD45RBlow (47.3% compared with 37%), CD44high (52.9% compared with 31.3%), and CD49dhigh (45.4% compared with 17.6%). Thus, 10 days after LCMV infection of β2m− mice, approximately 50% of splenic CD4+ cells have a phenotype characteristic of activated T cells (22, 23).
Figure 5.
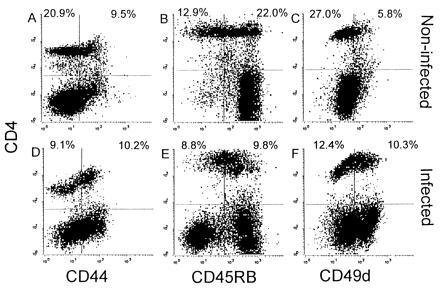
Activation of splenic CD4+ cells after LCMV infection. Two-color flow cytometric analysis of splenocytes from noninfected β2m− mice (A–C) or from β2m− mice that had been infected i.p. with LCMV 10 days previously (D–F) was performed. Cells were stained for CD4 and CD44 (A and D), CD45RB (B and D), or CD49d (α4-integrin) (E and F). The percentages of CD4+ cells are indicated in the upper two quadrants of each plot.
These activated spleen cells were injected i.p. into either irradiated i.c. infected β2m− mice or irradiated i.c. infected β2m−.lpr mice. As expected, adoptive transfer of immune spleen cells into the β2m− recipients caused lethal LCM disease (Table 1). In contrast, adoptive transfer of these cells into β2m−.lpr recipient mice did not result in death of the recipient animals. Irradiated infected control mice of either strain that did not receive donor cells did not die. In addition, we have previously shown that transfer of splenocytes from noninfected β2m− mice does not cause any disease in recipient mice (13). These data show that expression of Fas in recipient β2m− mice is required for adoptive transfer of lethal LCM disease.
Table 1.
Fas expression is required for lethal disease following cell transfer
| Exp. | Transferred*
|
Not
transferred†
|
||
|---|---|---|---|---|
| β2m− | β2m−.lpr | β2m− | β2m−.lpr | |
| 1 | 3/3 | ND | 0/3 | ND |
| 2 | 2/2 | 0/4 | 0/3 | 0/3 |
| 3 | 2/2 | 0/2 | 0/2 | 0/2 |
| Total | 7/7‡ | 0/6‡ | 0/8 | 0/5 |
Numbers are deaths/total. ND, not done.
Recipient mice received 1.7 × 108 (Exp. 1) or 1.8 × 108 (Exps. 2 and 3) donor splenocytes.
Control recipient mice did not receive donor cells.
These two groups are statistically different (P < 0.001, Fisher’s exact test).
DISCUSSION
In this report, we have determined the mechanism of cytotoxicity of the LCMV-specific CD4+ CTLs in β2m− mice and have examined the requirement for cytotoxic effector function in LCM disease. We show that the LCMV-specific, MHC class II-restricted CTLs in β2m− mice predominantly use a Fas-dependent lytic mechanism. Furthermore, we demonstrate that this Fas-dependent CTL activity is required for causing lethal LCM disease but is not necessary for causing chronic wasting following i.c. infection of β2m− mice with LCMV.
Normal mice elaborate virus-specific, class I-restricted, CD8+ CTLs following infection with LCMV. These antiviral CTL predominantly exhibit perforin-dependent cytotoxic activity (8, 9). β2m− (H2b) mice are unable to mount a detectable CD8+ antiviral CTL response but instead generate CD4+ class II-restricted CTLs (12–15). CD4+ CTLs have also been detected in normal mice after infection with certain viruses or after immunization with certain antigens. In these instances the cytotoxic mechanisms used by CD4+ CTLs have frequently been found to be Fas-dependent (24–26). Recent studies, however, have demonstrated that the acquisition of perforin-dependent or Fas-dependent lytic mechanisms by either CD4+ or CD8+ CTLs can be determined by the stimulation conditions used (10, 27, 28). The LCMV-specific CD4+ CTL activity in β2m− mice is detectable directly ex vivo, therefore, the cytotoxic mechanism used by these CTLs can be studied without the need for restimulation in vitro. The inability of the LCMV-specific β2m− CTLs to kill anti-Fas-resistant target cells (Fig. 1) and the inhibition of this CTL activity by using Fas-Ig (Fig. 2) demonstrates that these CTLs use a Fas-dependent cytotoxic mechanism. This conclusion is supported by the observation that CTL activity is inhibited following treatment of the effector cells with emetine (Fig. 2). Expression of FasL on the surface of T cells requires de novo protein synthesis (5), therefore, Fas-dependent cytotoxicity is sensitive to protein synthesis inhibitors such as emetine. Although β2m− mice elaborate Fas-dependent LCMV-specific CTLs, these CTLs are unable to clear the infection (12, 14, 15). LCMV can infect a wide range of cells in vivo; however, Fas-dependent CTLs can kill only target cells that express the Fas molecule. Therefore, infected cells that do not express Fas in vivo will not be directly eliminated by these CTLs. Consequently, such cells may serve as a reservoir of virus and lead to persistent infection. The limited tissue distribution of MHC class II molecules in vivo also restricts the target cell range of these class II-restricted CTLs; however, even class I-restricted, Fas-dependent, CD8+ CTLs do not clear LCMV infection (8, 9). These observations emphasize the limited efficacy of Fas-dependent class II-restricted CTLs in controlling systemic viral infections.
The elucidation of the cytotoxic mechanism used by the β2m− CTLs has enabled us to investigate the role of this cytotoxic effector function in LCM disease in β2m− mice. Following i.c. infection with LCMV, β2m− mice succumb to a CD4+ T-cell-dependent, chronic wasting disease (12–16). Like β2m− mice, β2m−.lpr mice also develop similar symptoms after i.c. infection (Fig. 3). Interestingly, i.c infection of perforin-deficient mice with LCMV also results in a marked loss of body weight. Since these strains of mice all succumb to wasting disease which is similar in both severity and kinetics, this suggests that neither Fas-dependent nor perforin-dependent cytotoxicity is required for LCMV-induced weight loss. Therefore, non-cytolytic lymphocyte effector functions are likely to mediate this disease. The potent cachectic cytokine TNF-α is a potential mediator of this wasting disease. We have found that LCMV-specific class II-restricted T cells from β2m− mice produce TNF-α in vitro (Fig. 4). Production of TNF-α by such cells in vivo could result in weight loss. LCMV infection of β2m− mice leads to activation of CD4+ T cells (Fig. 5), resulting in increased expression of CD49d on splenic CD4+ cells. CD49d plays an important role in directing specific T cells to sites of viral infection (29). Mild to moderate infiltrates of mononuclear cells are apparent in the brains of i.c. infected β2m− mice (12, 16). In the brain, infiltrating LCMV-specific CD4+ T cells may react against viral antigen presented on class II molecules, leading to production of TNF-α within the central nervous system, a situation that results in dramatic weight loss (21).
Adoptive transfer of spleen cells from LCMV-infected β2m− mice into irradiated infected β2m− mice results in death (13). Similar to the wasting disease described above, lethal disease is also dependent on CD4+ T cells; however, in contrast to LCMV-induced wasting, lethal LCM disease is Fas-dependent. Transfer of spleen cells from LCMV-infected β2m− mice into irradiated, infected, Fas-deficient β2m−.lpr mice does not result in death of the recipients (Table 1). These data are consistent with our observation that adoptive transfer of noncytolytic splenic CD4+ cells from LCMV-infected B6 mice into syngeneic β2m− recipient mice does not cause lethal disease (13). Furthermore, these data indicate that pathogenic mechanisms of LCMV-induced wasting disease and of lethal LCM disease are distinct. A recent study by Hilderman et al. (30) supports this hypothesis. Using recombinant vaccinia viruses expressing LCMV proteins, these authors were able to vaccinate β2m− mice against lethal disease, but not against wasting disease. Our results suggest that lethal immunopathology in LCMV-infected β2m− mice requires cytotoxic effector function but that the chronic wasting disease is independent of cell-mediated cytotoxicity.
Unlike the biological role of perforin-dependent cytotoxicity, that of Fas-dependent cytotoxicity is unclear. It has been proposed that the primary function of Fas-mediated cytolysis may be to maintain immune system homeostasis (23, 31). Thus, the induction of FasL on responding T cells may result in the elimination of other activated T cells (18, 31, 32) as well as B cells (32) and macrophages (33). In contrast, the Fas-dependent cytotoxic activity we observe in LCMV-infected β2m− mice is highly specific. Similar to perforin-dependent CD8+ CTLs in normal mice, these Fas-dependent CD4+ CTLs mediate antigen-specific MHC-restricted cytolysis in vitro and cause lethal immunopathology upon adoptive transfer. We believe, therefore that the induction of Fas-dependent CD4+ CTLs in β2m− mice is an attempt to compensate for the lack of perforin-dependent CD8+ CTLs.
In conclusion, we have demonstrated that LCMV-specific CD4+ CTLs in β2m− mice utilize a Fas-dependent lytic mechanism. This cytotoxic activity is not required for the development of virus-induced chronic wasting disease; however, adoptive transfer of lethal LCM disease is Fas-dependent. These data provide the first demonstration, to our knowledge, of a role for Fas-dependent CD4+ CTLs in virus-induced immunopathology.
Acknowledgments
We thank K. Lindley and A. Wolthusen for technical assistance and Drs. A. Peace-Brewer, M. Maldonado, J. Ye, and A. Whitmore for helpful advice and discussion. This work was supported by National Institutes of Health Grant A20288 to J.A.F and Training Grant AI07273 to A.J.Z. and D.G.Q.
Footnotes
Abbreviations: β2m−, β2-microglobulin-deficient; CTL, cytotoxic T lymphocyte; FITC, fluorescein isothiocyanate; i.c., intracranial; i.p., intraperitoneal; LCM, lymphocytic choriomeningitis; LCMV, LCM virus; MHC, major histocompatibility complex; pfu, plaque-forming units; TNF, tumor necrosis factor.
References
- 1.Kägi D, Vignaux F, Ledermann B, Bürki K, Depraetere V, Nagata S, Hengartner H, Golstein P. Science. 1994;265:528–530. doi: 10.1126/science.7518614. [DOI] [PubMed] [Google Scholar]
- 2.Henkart P A. Immunity. 1994;1:343–346. doi: 10.1016/1074-7613(94)90063-9. [DOI] [PubMed] [Google Scholar]
- 3.Kägi D, Ledermann B, Bürki K, Zinkernagel R M, Hengartner H. Annu Rev Immunol. 1996;14:207–232. doi: 10.1146/annurev.immunol.14.1.207. [DOI] [PubMed] [Google Scholar]
- 4.Podack E R, Kupfer A. Annu Rev Cell Biol. 1991;7:479–504. doi: 10.1146/annurev.cb.07.110191.002403. [DOI] [PubMed] [Google Scholar]
- 5.Walsh C M, Glass A A, Chiu V, Clark W R. J Immunol. 1994;153:2506–2514. [PubMed] [Google Scholar]
- 6.Berke G. In: Fundamental Immunology. Paul W E, editor. New York: Raven; 1993. pp. 965–1014. [Google Scholar]
- 7.Doherty P C, Allan J E, Lynch F, Ceredig R. Immunol Today. 1990;11:55–59. doi: 10.1016/0167-5699(90)90019-6. [DOI] [PubMed] [Google Scholar]
- 8.Kägi D, Ledermann B, Bürki K, Seiler P, Odermatt B, Olsen K J, Podack E R, Zinkernagel R M, Hengartner H. Nature (London) 1994;369:31–37. doi: 10.1038/369031a0. [DOI] [PubMed] [Google Scholar]
- 9.Walsh, C. M., Matloubian, M., Liu, C.-C., Ueda, R., Kurahara, C. G., Christensen, J. L., Huang, M. T. F., Young, J. D.-E., Ahmed, R. & Clark, W. R. Proc. Natl. Acad. Sci. USA 91, 10854–10858. [DOI] [PMC free article] [PubMed]
- 10.Williams N S, Engelhard V H. J Immunol. 1996;156:153–159. [PubMed] [Google Scholar]
- 11.Raulet D H. Adv Immunol. 1994;55:381–421. doi: 10.1016/s0065-2776(08)60514-3. [DOI] [PubMed] [Google Scholar]
- 12.Quinn D G, Zajac A J, Frelinger J A. Immunol Rev. 1995;148:151–169. doi: 10.1111/j.1600-065x.1995.tb00097.x. [DOI] [PubMed] [Google Scholar]
- 13.Quinn D G, Zajac A J, Frelinger J A, Muller D. Int Immunol. 1993;5:1193–1198. doi: 10.1093/intimm/5.10.1193. [DOI] [PubMed] [Google Scholar]
- 14.Zajac A J, Muller D, Pederson K, Frelinger J A, Quinn D G. Int Immunol. 1995;7:1545–1556. doi: 10.1093/intimm/7.10.1545. [DOI] [PubMed] [Google Scholar]
- 15.Doherty P C, Hou S, Southern P J. J Neuroimmunol. 1993;46:11–17. doi: 10.1016/0165-5728(93)90228-q. [DOI] [PubMed] [Google Scholar]
- 16.Lehmann-Grube F, Löhler J, Utermöhlen O, Gegin C. J Virol. 1993;67:332–339. doi: 10.1128/jvi.67.1.332-339.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Maldonado M A, Eisenberg R A, Roper E, Cohen P L, Kotzin B L. J Exp Med. 1995;181:641–648. doi: 10.1084/jem.181.2.641. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Ju S-T, Panka D J, Cui H, Ettinger R, El-Khatib M, Sherr D H, Stanger B Z, Marshak-Rothstein A. Nature (London) 1995;373:444–448. doi: 10.1038/373444a0. [DOI] [PubMed] [Google Scholar]
- 19.Hou S, Fishman M, Murti K G, Doherty P C. J Virol. 1993;67:6299–6302. doi: 10.1128/jvi.67.10.6299-6302.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Fishman M. Cell Immunol. 1991;137:164–174. doi: 10.1016/0008-8749(91)90066-k. [DOI] [PubMed] [Google Scholar]
- 21.Tracey K J. In: Tumor Necrosis Factors: The Molecules and Their Emerging Role in Medicine. Beutler B, editor. New York: Raven; 1992. pp. 255–273. [Google Scholar]
- 22.Ewing C, Topham D J, Doherty P C. Virology. 1995;210:179–185. doi: 10.1006/viro.1995.1329. [DOI] [PubMed] [Google Scholar]
- 23.Butcher E C, Picker L J. Science. 1996;272:60–66. doi: 10.1126/science.272.5258.60. [DOI] [PubMed] [Google Scholar]
- 24.Stalder T, Hahn S, Erb P. J Immunol. 1993;152:1127–1133. [PubMed] [Google Scholar]
- 25.Ju S-T, Cui H, Panka D J, Ettinger E, Marshak-Rothstein A. Proc Natl Acad Sci USA. 1994;91:4185–4189. doi: 10.1073/pnas.91.10.4185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Hanabuchi S, Koyanagi M, Kawasaki A, Shiohara N, Matsuzawa A, Nishimura Y, Yonehara S, Yagita H, Okumura K. Proc Natl Acad Sci USA. 1994;91:4930–4934. doi: 10.1073/pnas.91.11.4930. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Cao W, Tykodi S S, Esser M T, Braciale V L, Braciale T J. Nature (London) 1995;378:295–298. doi: 10.1038/378295a0. [DOI] [PubMed] [Google Scholar]
- 28.Brossart P, Bevan M J. J Exp Med. 1996;183:2449–2458. doi: 10.1084/jem.183.6.2449. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Christensen J P, Andersson E C, Scheynius A, Marker O, Thomsen A R. J Immunol. 1995;154:5293–5301. [PubMed] [Google Scholar]
- 30.Hilderman D, Salvato M, Whitton L J, Muller D. Vaccine. 1996;14:1223–1229. doi: 10.1016/s0264-410x(96)00028-x. [DOI] [PubMed] [Google Scholar]
- 31.Hahn S, Gehri R, Erb P. Immunol Rev. 1995;146:57–79. doi: 10.1111/j.1600-065x.1995.tb00684.x. [DOI] [PubMed] [Google Scholar]
- 32.Scott D W, Grdina T, Shi Y. J Immunol. 1996;156:2352–2356. [PubMed] [Google Scholar]
- 33.Ashany D, Song X, Lacy E, Nikolic-Zugic J, Friedman S M. Proc Natl Acad Sci USA. 1995;92:11225–11259. doi: 10.1073/pnas.92.24.11225. [DOI] [PMC free article] [PubMed] [Google Scholar]