

Abstract
Stimulation of naive T cells by antigen-presenting cells (APC) is thought to involve two qualitatively different signals: signal one results from T-cell receptor (TCR) recognition of antigenic peptides bound to major histocompatibility complex (MHC) molecules, whereas signal two reflects contact with one or more costimulatory molecules. The requirements for stimulating naive T cells were studied with MHC class I-restricted CD8+ T cells from a T-cell receptor transgenic line, with defined peptides as antigen and transfected Drosophila cells as APC. Three main findings are reported. First, stimulation of naive T cells via signal one alone (MHC plus peptide) was essentially nonimmunogenic; thus T cells cultured with peptides presented by MHC class I-transfected Drosophila APC lacking costimulatory molecules showed little or no change in their surface phenotype. Second, cotransfection of two costimulatory molecules, B7-1 and intercellular adhesion molecule 1 (ICAM-1), converted class I+ Drosophila cells to potent APC capable of inducing strong T-proliferative responses and cytokine (interleukin 2) production. Third, B7-1 and ICAM-1 acted synergistically, indicating that signal two is complex; synergy between B7-1 and ICAM-1 varied from moderate to extreme and was influenced by both the dose and affinity of the peptide used and the parameter of T-cell activation studied. Transfected Drosophila cells are thus a useful tool for examining the minimal APC requirements for naive T cells.
Via antigen-specific αβ T-cell receptors (TCR), T cells respond to peptide fragments of antigen bound to cell-surface major histocompatibility complex (MHC) molecules on antigen-presenting cells (APC) (1, 2). TCR binding to peptide/MHC complexes is aided by CD4 or CD8 coreceptors and leads to T-cell proliferation and differentiation. TCR-mediated triggering is generally ineffective, however, unless accompanied by “costimulatory” signals (3, 4). These signals control T-cell production of growth-promoting cytokines such as interleukin 2 (IL-2) and result from interactions between multiple sets of complimentary molecules on T cells and APC—e.g., between CD28 on T cells and B7 on APC. A wide variety of molecules on APC, including B7-1 (5–7), B7-2 (8, 9), intercellular adhesion molecule 1 (ICAM-1) (10–12), and heat stable antigen (13, 14), are known to express costimulatory function under defined conditions in vitro. Which of these molecules are crucial for costimulation, however, is unclear. This question can be addressed by transfecting cells with genes for known costimulatory molecules and then testing the cells for APC function (15, 16). A drawback of this approach is that nearly all cell types express at least a low level of certain costimulatory molecules. Such background expression of costimulatory molecules is less of a problem if nonmammalian cells (e.g., Drosophila cells) are used for transfection.
Although Drosophila cells can be stably transfected with mammalian class I molecules (17), the fact that Drosophila cells die rapidly at 37°C makes these cells an unlikely source of APC. In their favor, however, Drosophila cells lack TAP-1,2 peptide transporters, with the result that the class I molecules reaching the cell surface fall apart rapidly unless stabilized with exogenous peptide (18). This means that class I-transfected Drosophila cells can be induced to express very high levels of specific peptide/class I complexes—i.e., the ligands for CD8+ T cells.
Using defined peptides as antigen and TCR transgenic T cells as responders, we show here that class I-transfected Drosophila cells are nonimmunogenic for naive CD8+ T cells. When cotransfected with B7-1 and ICAM-1, however, class I-transfected Drosophila cells exhibit potent APC function in the absence of added lymphokines and induce strong proliferative responses, cytokine (IL-2) production, and cytotoxic activity to peptide antigens.
MATERIALS AND METHODS
Mice.
2C TCR transgenic mice were obtained (19) and were bred and maintained in the rodent breeding colony at the Scripps Research Institute.
Cell Lines, Cytokines, and mAbs.
The following mAbs were used (20): 3.168 (anti-CD8), RL172 (anti-CD4), J11d (anti-heat stable antigen), 28–16-8s (anti-I-Ab), S4B6 (anti-IL-2) (PharMingen), and BVD4-1D11 (anti-IL-4) (PharMingen). The RMA-S.Ld cell line and the hybridoma producing the anticlonotypic 1B2 mAb were kindly provided by H. Eisen (Massachusetts Institute of Technology) (21). Recombinant IL-2 was purchased from Genzyme.
Peptides.
Peptides used in this study were synthesized on an Applied Biosystems model 431 A synthesizer. All peptides were purified with C18 reverse-phase HPLC. The concentrations of peptides was determined by quantitative amino acid analysis (22).
Media.
RPMI 1640 medium was supplemented with 10% fetal calf serum (Irvine Scientific), 5% NCTC 109, 2 mM glutamine, 5 × 10−5 M 2-mercaptoethanol, and antibiotics (20).
Generation of Drosophila Cell APC Expressing Murine Class I Molecules and Costimulation Molecules.
cDNA encoding B7-1 (23) or ICAM-1 (24) were generated by reverse transcription–PCR of template mRNA from concanavalin A-stimulated spleen cells from BALB/c mice using oligonucleotides based on the published sequences. These cDNA were fully sequenced and thereafter inserted into the Drosophila expression vector pHMRa-3 containing the metallothionein promoter. The generation of pHMRa-3-based expression constructs for Ld and murine β2-microglobulin has been described (17). Generation of stable cell lines expressing the various combinations of MHC class I and costimulatory molecules was achieved as follows: 24 μg of the appropriate recombinant plasmid DNA (equal amounts of each construct) was transfected together with 1 μg of phshneo DNA into Schneider SC2 cells by the calcium phosphate method. Stably transfected cells were selected by culture in Schneiders Drosophila medium containing 8% fetal calf serum (GIBCO/BRL) and 500 μg/ml geneticin (GIBCO/BRL). Twenty-four hours before use of the stable cell line, expression of the transfected genes was induced by addition of CuSO4 to a final concentration of 1 mM.
Purification of CD8+ and CD8− T Cells.
As described previously (25), cell suspensions prepared from pooled lymph nodes of young adult 2C mice were first treated with a mixture of mAbs (anti-CD4, anti-HSA, anti-I-Ab) plus C (complement) for 45 min at 37°C. The surviving cells were further separated into CD8+ cells by panning at 4°C for 60–90 min on Petri dishes coated with anti-CD8 mAb.
Proliferation Assay.
Purified populations of CD8+ 2C cells were cultured with transfected Drosophila cells in 200-μl wells in the presence or absence of peptides. Unless stated otherwise, responder cells were used at 5 × 104 per well; the dose of stimulator cells varied but was generally 2 × 105 per well. To measure T-cell proliferation, cultures were pulsed with 1 μCi [3H]thymidine 8 hr before harvest (1 Ci = 37 GBq). All of the data shown in the table and figures refer to the mean of triplicate cultures; SD were generally within 5–15% of the mean.
IL-2 Production.
The biological activity of IL-2 produced by 2C cells was measured by using an IL-2-dependent cell line, CTLL (25). At the time indicated, 50 μl of supernatants were collected from each culture well and added to 5000 CTLL cells for 24 hr; 1 μCi [3H]thymidine was added and the cultures were harvested 16 hr later.
Generation of Cytotoxic T Lymphocytes (CTLs).
Using established procedures (25), 5 × 105 CD8+ 2C cells were cultured with 2 × 106 transfected Drosophila cells plus 10 μM peptides in a volume of 2 ml in a 24-well culture plate. After 4 days, the cells were pooled and adjusted to the required number. To prepare targets, RMA-S.Ld cells were labeled with 51Cr (100 μCi/1-2 × 106 cells) at 37°C for 90 min in the presence or absence of peptides. After labeling, the cells were thoroughly washed and resuspended in medium with or without peptides. Specific 51Cr release was calculated as described (25).
Flow Cytometric Analysis.
For analysis of surface expression of Ld on transfected Drosophila cells, 1 × 106 cells were incubated with anti-Ld mAb (PharMingen) for 30 min on ice, washed, and then stained with fluorescein isothiocyanate (FITC)-conjugated goat F(ab′)2 anti-mouse Fcγ antibody (Jackson ImmunoResearch). FITC-conjugated anti-B7-1 and anti-ICAM-1 mAbs were used to analyze the expression of B7-1 and ICAM-1 on the transfected Drosophila cells. For analysis of surface expression of CD25 and CD69 on CD8+ 2C cells, resting and activated 2C cells were stained with biotinylated anti-CD25 or biotinylated anti-CD69 mAb (PharMingen) and then stained with FITC-conjugated 1B2 mAb (21), Red 613-conjugated streptavidin and phycoerythrin-conjugated anti-CD8 mAb (GIBCO/BRL). Propidium iodide (PI) was included in the last step of staining at 0.5 μg/ml. Live cells (PI negative) were acquired and analyzed on a FACScan using lysys software (Becton Dickinson).
RESULTS
Experimental Approach.
To test for APC function, Drosophila cells were transfected with a murine class I gene, Ld, linked to the metallothionein promoter and then tested for their capacity to present antigen to T cells from the 2C line of transgenic mice (19). With mouse cells as APC, this line displays strong alloreactivity for Ld molecules complexed with an endogenous 8-mer peptide, p2Ca (LSPFPFDL) derived from a Krebs cycle enzyme, 2-oxoglutarate dehydrogenase (OGDH) (26, 27). The p2Ca peptide is exposed naturally on the surface of H-2d (e.g., B10.D2) cells bound to Ld; p2Ca has intermediate binding affinity for soluble Ld molecules (4 × 106 M−1) and high affinity for 2C TCR molecules (2 × 106 M−1 to 1 × 107 M−1) (22, 28). A closely related 9-mer peptide, QL9, has even higher affinity for these molecules (2 × 108 M−1 for Ld and 2 × 107 M−1 for 2C TCR) (29). Except for one additional amino acid (glutamine) at the N terminus, QL9 has an identical sequence to p2Ca and, like p2Ca, forms part of the native sequence of OGDH.
With p2Ca and QL9 peptides (prepared in synthetic form) we studied the APC requirements for mature unprimed 2C CD8+ cells in vitro. The responder CD8+ cells were purified from lymph nodes of 2C mice on a C57BL/6 (B6, H-2b) background by mAb plus C treatment followed by positive panning (Materials and Methods); >95% of the CD8+ cells obtained were clonotype-positive (IB2+) and 98% of these cells displayed a naive (CD44lo) phenotype. To study APC function, purified CD8+ 2C cells were cultured in the presence of peptides with a panel of Ld-transfected Drosophila cells expressing Ld alone (Ld), Ld plus B7-1 (Ld.B7), Ld plus ICAM-1 (Ld.ICAM), or Ld plus B7-1 plus ICAM-1 (Ld.B7.ICAM). By fluorescence-activated cell sorter (FACS) analysis, cell-surface expression of these molecules on Drosophila cells was moderate (Fig. 1) and no higher than on normal Ld (B10.D2) spleen cells (not shown). When cultured at 37°C, the viability of Drosophila cells declined to <1% by 12 hr as measured by PI exclusion. However, most of the cells failed to disintegrate and remained intact for several days.
Figure 1.
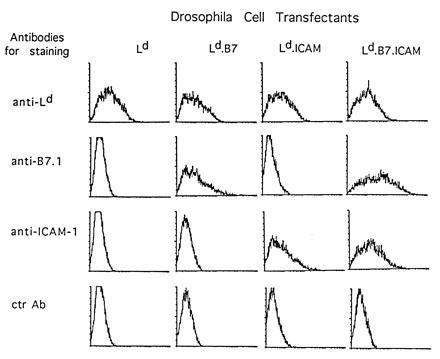
Expression of Ld, B7-1, and ICAM-1 by transfected Drosophila cells. Drosophila cells were transfected with cDNAs encoding Ld, β2-microglobulin, B7-1, and ICAM-1 under the control of the metallothionein promoter as described. Transfected cells were separated with a FACS to obtain cells expressing Ld molecules and were maintained in vitro. The data show Ld, B7-1, and ICAM-1 expression on the cell lines 24 hr after induction with 1 mM CuSO4. The higher staining for B7-1 on Ld.B7.ICAM than Ld.B7 cells was not seen in other experiments.
Early Events.
TCR stimulation elicits a complex pattern of intracellular events which lead to early up-regulation of CD25 [IL-2 receptor (IL-2R)] and CD69 on the cell surface (1, 2, 30, 31). These changes are apparent within a few hours of stimulation and are followed by cytokine synthesis and cell proliferation. Fig. 2a shows CD25 and CD69 expression on purified naive CD8+ 2C cells stimulated for 12 hr with transfected Drosophila cells and a high concentration (10 μM, 10−5 M) of QL9 or p2Ca peptide. The effects of reducing the concentrations of these peptides are illustrated in Fig. 2b for CD25 expression; near-identical results applied to CD69 expression (not shown).
Figure 2.
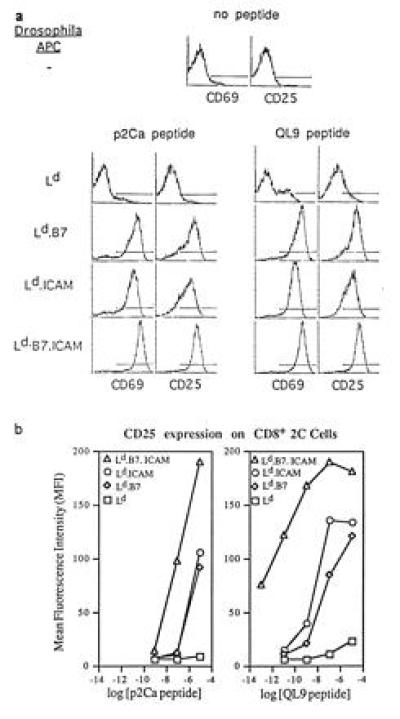
Expression of activation markers on CD8+ 2C cells is influenced by peptide and costimulation. (a) Expression of CD69 and CD25 on CD8+ 2C cells stimulated with 10 μM peptides presented by transfected Drosophila cells. Purified CD8+ 2C cells were incubated with transfected Drosophila cells plus p2Ca or QL9 peptides (10 μM) in bulk (2 ml) culture for 12 hr and then stained for the markers shown. The data show staining of gated CD8+ cells. (Top) Staining of noncultured 2C cells is shown as a control. (b) Influence of peptide dose on expression of CD25 on CD8+ 2C cells. Purified CD8+ 2C cells were cultured with the indicated concentration of peptides presented by Drosophila cell APC for 12 hr and then stained for CD25 expression on gated CD8+ cells.
With Ld APC, the stronger QL9 peptide caused a slight increase in CD25 and CD69 expression on 2C T cells, but only at a high concentration of peptide; no increase in the expression of these markers was seen with p2Ca peptide. With Ld.B7 and Ld.ICAM APC, both peptides at high concentration caused moderately strong up-regulation of CD25 and CD69; with lower concentrations, QL9 peptide was much more effective at inducing these markers than p2Ca peptide. With Ld.B7.ICAM APC, up-regulation of CD25 and CD69 was even stronger than for Ld.B7 and Ld.ICAM APC and was apparent with very low concentrations of peptide—e.g., 100 fM (10−13 M) for QL9 peptide. With Ld.B7 or Ld.ICAM APC, by contrast, a 100-fold higher concentration of QL9 peptide (10−11 M) caused no up-regulation of CD25 or CD69. Thus, at low concentrations of peptide, the up-regulation of CD25 and CD69 involved a marked synergy between B7 and ICAM.
Proliferation and IL-2 Production.
Consistent with their minimal capacity to induce CD25 and CD69 expression, Drosophila cells transfected with Ld alone failed to cause proliferation of 2C CD8+ cells to either p2Ca or QL9 peptide in the absence of exogenous lymphokines (Fig. 3). When supplemented with exogenous IL-2 (the ligand for CD25), QL9 peptide induced a significant proliferative response, but only at a high dose of peptide (10 μM) (Fig. 3). Responses elicited by p2Ca peptide, however, were virtually undetectable.
Figure 3.
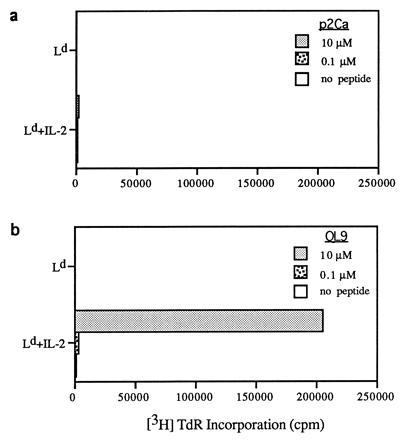
IL-2-dependent proliferative responses of CD8+ 2C cells to peptides presented by Drosophila cells transfected with Ld only. Responses to p2Ca (a) and QL9 (b) peptides were measured by culturing 5 × 104 purified CD8+ 2C cells with 2 × 105 Drosophila cells in the presence or absence of the indicated concentrations of peptides for 3 days. [3H]thymidine was added during the last 8 hr of culture; recombinant IL-2 was added at a final concentration of 20 units/ml. The data refer to the mean of triplicate cultures.
These findings on the effects of adding exogenous IL-2 are thus in close accord with the data on CD25 expression (Fig. 2). With Drosophila cells as APC, the capacity of signal one alone to cause signs of T-cell activation (up-regulation of CD25 leading to responsiveness to IL-2) was therefore apparent only with a very high affinity peptide (QL9), and then only with a high concentration of this peptide.
The data in Table 1 show day 3 proliferative responses elicited by Drosophila cells in the absence of added lymphokines; peptides were added at a high concentration, 10 μM. With the weaker p2Ca peptide, proliferative responses of CD8+ 2C cells to Ld, Ld.B7, or Ld.ICAM Drosophila APC were undetectable. The lack of proliferation with these APC correlated with a failure to synthesize IL-2 (Table 1). In marked contrast, Ld.B7.ICAM APC induced quite strong proliferative responses and high IL-2 production in response to p2Ca peptide. This finding implies that, as for CD25 and CD69 up-regulation, B7 and ICAM acted synergistically, both for proliferation and for IL-2 production. Interestingly, responses to p2Ca were very low with a mixture of Ld.B7 and Ld.ICAM APC, indicating that both molecules had to be expressed on the same cell. Similar synergy between B7 and ICAM applied to the response to the stronger QL9 peptide. With this peptide, however, synergy was more stringent for IL-2 production than for proliferation (but see below for low doses of peptide). Thus, in contrast to p2Ca, QL9 peptide elicited significant proliferative responses with APC expressing either B7 or ICAM.
Table 1.
Capacity of transfected Drosophila cells to stimulate primary proliferative responses and IL-2 production by CD8+ 2C lymph node cells
| Assay | Peptides added | [3H]Thymidine incorporation (cpm ×
103) with transfected Drosophila cells
expressing
|
||||
|---|---|---|---|---|---|---|
| Ld | Ld.B7 | Ld.ICAM | Ld.B7.ICAM | Ld.B7 + Ld.ICAM* | ||
| Proliferation | — | 0.2* | 0.1 | 0.3 | 0.2 | — |
| (day 3) | p2Ca | 0.2 | 0.3 | 1.5 | 142.0 | 1.5 |
| QL9 | 0.3 | 60.9 | 73.9 | 263.7 | 132.9 | |
| IL-2 production | — | 0.3 | 0.2 | 0.1 | 1.2 | — |
| (day 2) | p2Ca | 0.2 | 0.2 | 0.1 | 64.6 | 0.3 |
| QL9 | 0.1 | 0.4 | 0.2 | 158.6 | 0.5 | |
Doses of 5 × 104 highly-purified naive-phenotype CD8+ 2C lymph node cells were cultured at 37°C with 3 × 105 transfected Drosophila cells ± peptides (10 μM final concentration) in 0.2 ml in 96-well plates (25). Proliferative responses were measured by adding [3H]thymidine (3HTdR) (1 μCi/well) 8 hr before harvest. IL-2 production was measured by removing supernatants from the cultures at 48 hr and adding 50 μl supernatant to the IL-2-responsive indicator line, CTLL (25). Proliferation of the indicator line was measured by addition of [3H]thymidine. It should be noted that Drosophila cells die rapidly at 37°C and fail to incorporate [3H]thymidine at this temperature. Data are given as the mean of triplicate cultures; SD were generally within 5–15% of the mean.
Values are 1.5 × 105 of each population.
For p2Ca peptide, optimal proliferative responses elicited by Ld.B7.ICAM APC on day 3 required high concentrations of peptide—e.g., 10 μM (10−5 M) (Fig. 4a). With QL9 peptide, by contrast, proliferative responses with Ld.B7.ICAM APC on day 3 were maximal with 100 nM (10−7 M) and were clearly apparent with doses as low as 10 pM (10−11 M) (Fig. 4a).
Figure 4.
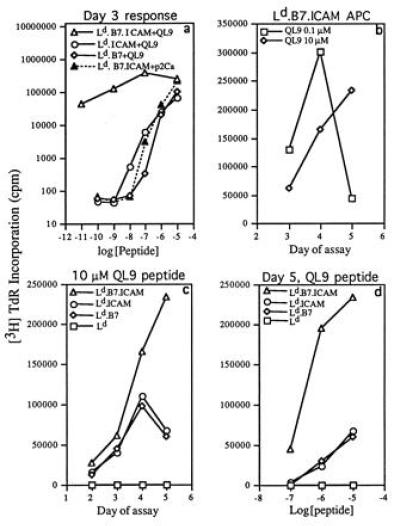
Features of proliferative responses of CD8+ 2C cells to peptides presented by transfected Drosophila cells. (a) Influence of peptide concentration on the day 3 response of CD8+ 2C cells to different Drosophila APC. (b) Influence of the dose of QL9 peptide on the kinetics of the response of CD8+ 2C cells with Ld.B7.ICAM APC. (c) Kinetics of the response of CD8+ 2C cells to 10 μM QL9 peptide presented by different Drosophila APC. (d) Influence of peptide dose on the day 5 response of CD8+ 2C cells with different Drosophila APC. CD8+ cells (5 × 104) were cultured with 2 × 105 Drosophila APC. The data show the mean of triplicate cultures.
The kinetics of the proliferative response of CD8+ 2C cells to QL9 peptide are shown in Fig. 4 b and c. With an intermediate dose of 0.1 μM (10−7 M) QL9, proliferative responses to Ld.B7.ICAM APC were high on day 3, reached a peak on day 4, and then declined to low levels on day 5 (Fig. 4b). With a high dose of 10 μM (10−5 M) QL9, however, the response was low on day 3 but then increased markedly to reach a high peak on day 5 (Fig. 4 b–d). This prolonged proliferative response was associated with high and sustained production of IL-2 in the cultures (Table 1 and data not shown).
These data applied with the highly immunogenic Ld.B7.ICAM APC. With the less immunogenic Ld.B7 or Ld.ICAM APC, proliferative responses to QL9 peptide reached a peak on day 3–4 (rather than day 5) (Fig. 4c) and required a high dose of peptide (Fig. 4 a and d). With low doses of QL9 [e.g., 1 nM (10−9 M)], proliferative responses with Ld.B7 and Ld.ICAM APC were completely undetectable (<100 cpm) (Fig. 4a). This was in striking contrast to Ld.B7.ICAM APC where 1 nM QL9 led to high responses (>100,000 cpm). Thus, in contrast to the results with high doses of QL9 peptide (Table 1), the synergistic interaction between B7 and ICAM for proliferative responses became extreme at low peptide doses.
CTL Generation.
The capacity of Ld.B7, Ld.B7.ICAM, and Ld.ICAM APC to induce CTL activity to 10 μM QL9 peptide in bulk cultures is shown in Fig. 5a; CTL activity was tested on 51Cr-labeled RMA.S-Ld targets sensitized with p2Ca peptide or an irrelevant peptide (P1A). As expected, strongly immunogenic Ld.B7.ICAM APC were highly efficient in generating CTL (Fig. 5a Right). Significantly, Ld.B7 APC were also effective in generating CTL (Fig. 5a Left). The surprising finding, however, was that Ld.ICAM APC were totally unable to stimulate CTL generation (Fig. 5a Center) unless stimulated with exogenous IL-2 (Fig. 5b Right). This finding was unexpected because Ld.ICAM APC were no less efficient than Ld.B7 APC at inducing proliferative responses (Fig. 4 a, c, and d). As mentioned earlier, neither Ld.B7 nor Ld.ICAM APC elicited detectable IL-2 production in culture supernatants. Nevertheless, IL-2 mRNA was clearly apparent for Ld.B7 APC, though very low for Ld.ICAM APC (data not shown). Hence the capacity of Ld.B7 but not Ld.ICAM APC to elicit CTL generation could reflect low but significant IL-2 production by Ld.B7 APC. In support of this possibility, adding anti-IL-2 mAb to Ld.B7 APC substantially reduced (Fig. 5b Left) or abolished (Fig. 5c) CTL generation, depending on the concentration used; anti-IL-4 mAb was ineffective (Fig. 5 b Right and c).
Figure 5.
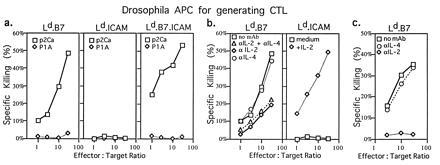
Generation of primary CTL from naive CD8+ 2C cells cultured with QL9 peptide presented by Drosophila cell APC. (a) CTL activity of CD8+ 2C cells stimulated with Ld.B7, Ld.B7.ICAM, or Ld.ICAM APC plus QL9 peptide (10 μM) in the absence of exogenous cytokines. (b Left) CTL activity of CD8+ 2C cells stimulated with Ld.B7 APC plus QL9 peptide (10 μM) in the absence or presence of anti-IL-2 and/or anti-IL-4 mAb (2 μg/ml). (b Right) CTL activity of CD8+ 2C cells stimulated with Ld.ICAM APC plus QL9 peptide (10 μM) in the absence or presence of recombinant IL-2 (20 unit/ml). (c) CTL activity of CD8+ 2C cells stimulated with Ld.B7 APC plus QL9 peptide in the absence or presence of either anti-IL-2 or anti-IL-4 mAb (20 μg/ml). Purified CD8+ 2C cells (5 × 105) were cultured with 2 × 106 Drosophila APC. After 4 days, the cells were harvested and CTL activity was tested against [51Cr]-labeled RMA-S.Ld target cells loaded with p2Ca or control P1A.35-43 peptide (32); P1A peptide binds strongly to Ld but has no detectable affinity for the 2C TCR (32). The data show the mean level of specific 51Cr release from duplicate cultures.
DISCUSSION
Under physiological conditions, stimulation of naive T cells is controlled by professional APC such as dendritic cells (33). To define the features of professional APC, we constructed artificial APC from Drosophila cells by gene transfection. Using a TCR transgenic model where the binding affinities involved in TCR/peptide/MHC interaction are clearly defined, we show here that class I-transfected Drosophila cells expressing two costimulatory molecules, B7 (B7-1) and ICAM (ICAM-1), act as highly potent APC for naive CD8+ cells. These artificial APC elicit strong IL-2 production, a sustained T-cell-proliferative response and differentiation into effector T cells. In addition, we show that the qualitative features of the primary response of CD8+ cells are influenced by a number of factors, including the concentration of peptide used, the affinity of TCR/peptide/MHC interaction, and the particular costimulatory molecules expressed on APC.
The finding that Ld-transfected Drosophila cells were nonimmunogenic unless these cells coexpressed costimulatory molecules confirms the crucial importance of costimulation for activating naive T cells (7). However, the data are difficult to reconcile with a simple two-signal model for T-cell activation. Here, the prevailing view is that signal one (reflecting TCR recognition of MHC/peptide) leads to partial T-cell activation (e.g., IL-2R expression), whereas signal two (costimulation) elicits cytokine production. In the case of signal one, exposing 2C CD8+ cells to a high concentration of the strong QL9 peptide presented by Drosophila cells expressing Ld alone led to significant proliferative responses in the presence of exogenous cytokines, indicative of IL-2R up-regulation. Despite this finding, FACS analysis showed that IL-2R (and CD69) expression elicited by Ld APC plus QL9 was very low relative to the high levels induced by Ld.B7 or Ld.ICAM APC. Moreover, with the weaker p2Ca peptide, even high doses of peptide elicited no detectable evidence of T-cell activation with Ld APC; yet this peptide was strongly immunogenic when presented by Ld.B7.ICAM APC. Thus, in terms of surface phenotype, the capacity of Signal one to elicit signs of T-cell activation was minimal.
Another problem with the two-signal model is that, at least for the weaker p2Ca peptide, IL-2 synthesis was virtually undetectable at the protein level unless the APC expressed three different ligands—i.e., Ld, B7, and ICAM. The data are thus more compatible with a “three-signal” model than a two-signal model (but see below). The marked synergy observed between B7 and ICAM has been reported previously for CD4+ cells and transfected fibroblasts as APC (16) and is shown here to apply to CD8+ cells and to affect three different parameters for T-cell activation, namely IL-2R (and CD69) expression, IL-2 synthesis and proliferation. Although the data are consistent with a three-signal model, it should be stressed that the nature of the synergy between B7 and ICAM is still unclear. In particular, it has yet to be proved that T-cell interaction with B7 and ICAM activates two discrete signaling pathways. In the case of B7, it is well accepted that B7 interaction with CD28 on T cells initiates a distinct signal transduction pathway that synergizes with TCR/CD3 signals (5–9). The situation with ICAM is more complex. Through binding to lymphocyte function-associated antigen 1 (LFA-1) and CD43 counterreceptors on T cells, ICAM is known to facilitate cell adhesion (34, 35). Hence the “costimulation” induced by ICAM may not reflect the induction of a unique signaling pathway but simply enhanced T/APC interaction leading to more effective TCR/CD3 crosslinking. Alternatively, ICAM may be bifunctional and induce both signal transduction and enhanced cell adhesion (10, 35–37). The data in this paper do not discriminate between these two possibilities.
In the case of Ld.ICAM cells, it is of interest that these APC were able to elicit proliferative responses to QL9 peptide but failed to generate CTL unless supplemented with exogenous IL-2. These findings correlated with a failure of Ld.ICAM APC to elicit IL-2 production and are in agreement with the evidence from IL-2−/− mice that exposure to IL-2 is not essential for proliferation of CD8+ cells but is obligatory for CTL generation (38). It is notable that Ld.B7 APC generated strong CTL responses even though IL-2 production was apparent only at the level of mRNA. Yet, adding anti-IL-2 mAb ablated CTL generation. The concentration of IL-2 required for CTL production thus appears to be extremely low.
Bearing in mind that Drosophila cells die rapidly at 37°C, the capacity of transfected Drosophila cells to act as an extremely potent source of APC for naive CD8+ cells might seem surprising. One has to consider the possibility that Drosophila cells are not a completely neutral vector. Thus one might argue that some of the constitutive surface molecules on Drosophila cells (e.g., carbohydrate moieties) can be recognized by T cells and provide some form of costimulation. Although it is difficult to exclude this possibility, it is striking that peptide-induced up-regulation of IL-2R and CD69 elicited by Drosophila cells expressing Ld alone was very limited. This finding implies that the “background” level of costimulatory molecules on Drosophila cells must be extremely low.
Despite their rapid loss of viability at 37°C, Drosophila cells remained intact for several days and thus presumably continued to display high levels of peptide/MHC complexes on the cell surface. We attribute the strong APC function of Drosophila cells to the lack of TAP-1,2 peptide transporters in these cells. Because of this deficit, the density of antigen (peptide/MHC) on the cell surface is very high. TCR stimulation via Signal one is thus intense and, when combined with appropriate costimulation, induces the responding T cells to mount a prolonged proliferative response and strong and sustained production of IL-2. Similar findings apply with professional APC—e.g., with B10.D2 (Ld) dendritic cells (DC). When CD8+ 2C cells respond to B10.D2 DC without exogenous peptide, the low level of endogenous p2Ca and QL9 peptides on these cells stimulates only low IL-2 production and a brief proliferative response (39). However, augmenting the avidity of T/APC interaction by adding exogenous QL9 peptide stimulates high IL-2 production and a prolonged proliferative response (39).
The capacity of Drosophila cells to present a high density of peptide/MHC complexes suggests that Drosophila cells could be used as APC for normal (nontransgenic) CD8+ cells. In fact, we have found that Ld.B7.ICAM APC plus QL9 peptide elicit significant primary responses by normal allogeneic B6 (H-2b) CD8+ cells (unpublished data). In addition, transfected Drosophila cells are also capable of inducing normal syngeneic T cells to mount primary proliferative and CTL responses to several different peptides, including tumor-specific peptides (32). Drosophila cells could thus be a useful tool for tumor immunotherapy.
Acknowledgments
We wish to express our thanks to Dr. Herman Eisen for sending us RMAS-Ld and 1B2 hybridoma lines and Ms. Barbara Marchand for typing the manuscript. This work was supported by Grants CA38355, CA25803, AI21487, and AI32068 from the U.S. Public Health Service. This is Publication no. 9546-IMM from the Scripps Research Institute.
Footnotes
Abbreviations: TCR, T-cell receptor(s); MHC, major histocompatibility complex; APC, antigen-presenting cell(s); IL-2, interleukin 2; IL-2R, IL-2 receptor; ICAM-1, intercellular adhesion molecule 1; CTL, cytotoxic T lymphocyte; PI, propidium iodide; FACS, fluorescence-activated cell sorter.
References
- 1.Germain R N. In: Fundamental Immunology. 3rd Ed. Paul W E, editor. New York: Raven; 1993. pp. 629–676. [Google Scholar]
- 2.Janeway C A, Bottomly K. Cell. 1994;76:275–285. doi: 10.1016/0092-8674(94)90335-2. [DOI] [PubMed] [Google Scholar]
- 3.Shimizu Y, van Seventer G A, Horgan K J, Shaw S. Immunol Rev. 1990;114:109–143. doi: 10.1111/j.1600-065x.1990.tb00563.x. [DOI] [PubMed] [Google Scholar]
- 4.Schwartz R H. Cell. 1992;71:1065–1068. doi: 10.1016/s0092-8674(05)80055-8. [DOI] [PubMed] [Google Scholar]
- 5.Linsley P S, Brady W, Grosmaire L, Aruffo A, Damle N K, Ledbetter J A. J Exp Med. 1991;173:721–730. doi: 10.1084/jem.173.3.721. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Azuma M, Cayabyab M, Buck D, Phillips J H, Lanier L L. J Exp Med. 1992;175:353–360. doi: 10.1084/jem.175.2.353. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Harding F A, Allison J P. J Exp Med. 1993;177:1791–1796. doi: 10.1084/jem.177.6.1791. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Azuma M, Ito D, Yagita H, Okumura K, Phillips J H, Lanier L L, Somoza C. Nature (London) 1993;366:76–79. doi: 10.1038/366076a0. [DOI] [PubMed] [Google Scholar]
- 9.Freeman G J, Gribben J G, Boussiotis V A, Ng J W, Restivo V A, Jr, Lombard L A, Gray G S, Nadler L M. Science. 1993;262:909–911. doi: 10.1126/science.7694363. [DOI] [PubMed] [Google Scholar]
- 10.van Seventer G A, Shimizu Y, Horgan K J, Shaw S. J Immunol. 1990;144:4579–4586. [PubMed] [Google Scholar]
- 11.Kuhlman P, Moy V T, Lollo B A, Brian A A. J Immunol. 1991;146:1773–1782. [PubMed] [Google Scholar]
- 12.Jaraquemada D, Marti M, Martin R, Wagner A, MacFarland H F, Rosen-Bronson S. Eur J Immunol. 1994;24:947–951. doi: 10.1002/eji.1830240425. [DOI] [PubMed] [Google Scholar]
- 13.Liu Y, Jones B, Aruffo A, Sullivan K M, Linsley P S, Janeway C A., Jr J Exp Med. 1992;175:437–445. doi: 10.1084/jem.175.2.437. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Liu Y, Jones B, Brady W, Janeway C A, Jr, Linsley P S. Eur J Immunol. 1992;22:2855–2859. doi: 10.1002/eji.1830221115. [DOI] [PubMed] [Google Scholar]
- 15.Galvin F, Freeman G J, Razi-Wolf Z, Hall W, Jr, Benacerraf B, Nadler L, Reiser H. J Immunol. 1992;149:3802–3808. [PubMed] [Google Scholar]
- 16.Dubey C, Croft M, Swain S L. J Immunol. 1995;155:45–57. [PubMed] [Google Scholar]
- 17.Jackson M R, Song E S, Yang Y, Peterson P A. Proc Natl Acad Sci USA. 1992;89:12117–12121. doi: 10.1073/pnas.89.24.12117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Yang Y, Fruh K, Chambers J, Waters J B, Wu L, Spies T, Peterson P A. J Biol Chem. 1992;267:11669–11672. [PubMed] [Google Scholar]
- 19.Sha W C, Nelson C A, Newberry R D, Kranz D M, Russell J H, Loh D Y. Nature (London) 1988;335:271–274. doi: 10.1038/335271a0. [DOI] [PubMed] [Google Scholar]
- 20.Sprent J, Schaefer M. J Exp Med. 1985;162:2068–2088. doi: 10.1084/jem.162.6.2068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Kranz D M, Tonegawa S, Eisen H N. Proc Natl Acad Sci USA. 1984;81:7922–7926. doi: 10.1073/pnas.81.24.7922. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Sykulev Y, Brunmark A, Jackson M, Cohen R J, Peterson P A, Eisen H N. Immunity. 1994;1:15–22. doi: 10.1016/1074-7613(94)90005-1. [DOI] [PubMed] [Google Scholar]
- 23.Freeman G J, Gray G S, Gimmi C D, Lombard D B, Zhou L J, White M, Fingeroth J D, Gribben J G, Nadler L M. J Exp Med. 1991;174:625–631. doi: 10.1084/jem.174.3.625. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Siu G, Hedrick S M, Brian A A. J Immunol. 1989;143:3813–3820. [PubMed] [Google Scholar]
- 25.Cai Z, Sprent J. J Exp Med. 1994;179:2005–2015. doi: 10.1084/jem.179.6.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Udaka K, Tsomides T J, Eisen H N. Cell. 1992;69:989–998. doi: 10.1016/0092-8674(92)90617-l. [DOI] [PubMed] [Google Scholar]
- 27.Udaka K, Tsomides T J, Walden P, Fukusen N, Eisen H N. Proc Natl Acad Sci USA. 1993;90:11272–11276. doi: 10.1073/pnas.90.23.11272. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Corr M, Slanetz A E, Boyd L F, Jelonek M T, Khilko S, Al-Ramadi B K, Sang Kim Y, Maher S E, Bothwell A L M, Margulies D H. Science. 1994;265:946–949. doi: 10.1126/science.8052850. [DOI] [PubMed] [Google Scholar]
- 29.Sykulev Y, Brunmark A, Tsomides T J, Kageyama S, Jackson M, Peterson P A, Eisen H N. Proc Natl Acad Sci USA. 1994;91:11487–11491. doi: 10.1073/pnas.91.24.11487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Hommel-Berrey G A, Shenoy A M, Brahmi Z. J Immunol. 1991;147:3237–3243. [PubMed] [Google Scholar]
- 31.Graber M, Bockenstedt L K, Weiss A. J Immunol. 1991;146:2935–2943. [PubMed] [Google Scholar]
- 32.Sun S, Cai Z, Langlade-Demoyen P, Kosaka H, Brunmark A, Jackson M R, Peterson P A, Sprent J. Immunity. 1996;4:555–564. doi: 10.1016/s1074-7613(00)80482-3. [DOI] [PubMed] [Google Scholar]
- 33.Steinman R M. Annu Rev Immunol. 1991;9:271–296. doi: 10.1146/annurev.iy.09.040191.001415. [DOI] [PubMed] [Google Scholar]
- 34.Springer T A, Dustin M L, Kishimoto T K, Marlin S D. Annu Rev Immunol. 1987;5:223. doi: 10.1146/annurev.iy.05.040187.001255. [DOI] [PubMed] [Google Scholar]
- 35.Manjunath N, Correa M, Ardman M, Ardman B. Nature (London) 1995;377:535–538. doi: 10.1038/377535a0. [DOI] [PubMed] [Google Scholar]
- 36.van Seventer G A, Bouvini A, Yamada H, Conti A, Stringfellow S, June C H, Shaw S. J Immunol. 1992;149:3872–3880. [PubMed] [Google Scholar]
- 37.Kanner S B, Grosmaire L S, Ledbetter J A, Damle N K. Proc Natl Acad Sci USA. 1993;90:7099–7103. doi: 10.1073/pnas.90.15.7099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Kramer S, Mamalaki C, Horak I, Schimpl A, Kioussis D, Hunig T. Eur J Immunol. 1994;24:2317–2322. doi: 10.1002/eji.1830241009. [DOI] [PubMed] [Google Scholar]
- 39.Cai Z, Sprent J. J Exp Med. 1996;183:2247–2257. doi: 10.1084/jem.183.5.2247. [DOI] [PMC free article] [PubMed] [Google Scholar]