

Abstract
Transcription factors and chromatin structural modifiers induce clinically relevant epigenetic modifications of blood leukocytes during severe systemic inflammation (SSI) in humans and animals. These changes affect genes with distinct functions, as exemplified by the silencing of a set of acute proinflammatory genes and the sustained expression of a group of antimicrobial and anti-inflammatory genes. This paradigm is closely mimicked in the THP-1 human promonocyte cell model of lipopolysaccharide (LPS) endotoxin tolerance. We previously reported that LPS-induced de novo expression of RelB is required for generating tolerance to interleukin-1β (IL-1β) and tumor necrosis factor alpha (TNF-α) expression. RelB represses transcription by binding with heterochromatic protein 1 α (HP1α) to the proximal promoters of IL-1β and TNF-α. In contrast, we report herein that RelB is required for sustained expression of anti-inflammatory IκBα in LPS-tolerant THP-1 cells. RelB transcription activation requires binding to the IκBα proximal promoter along with NF-κB p50 and is associated with an apparent dimer exchange with p65. We also observed that RelB induced during human SSI binds to the IκBα proximal promoter of circulating leukocytes. We conclude that RelB functions as a dual transcription regulator during LPS tolerance and human SSI by activating and repressing innate immunity genes.
Alterations in gene expression occur in circulating and tissue monocytes and neutrophils during severe systemic inflammation (SSI), such as sepsis, and in cell models of SSI, such as lipopolysaccharide (LPS)-mediated tolerance in THP-1 promonocytes (30). These genes have distinct functions, including persistent repression of a set of acute proinflammatory genes that initiate SSI and sustained expression of a group of anti-inflammatory and antimicrobial genes (7, 10, 17, 28).
The formation of the transcriptionally active NF-κB p65/p50 heterodimer on cognate DNA sequences plays a crucial role in initiating the expression of many innate immunity genes with acute proinflammatory functions that participate in SSI (24). We reported that disruption of the NF-κB p65/p50 heterodimer formation at the proximal interleukin-1β (IL-1β) and tumor necrosis factor alpha (TNF-α) promoters occurs during epigenetic transcription repression of these genes in LPS tolerance and in human SSI (6, 9). We also discovered that de novo LPS induction of NF-κB transcription factor RelB is required for sustaining repression of IL-1β and TNF-α transcription in the THP-1 cell model of LPS tolerance (31). The repressor effect of RelB is coupled to the heterochromatin silencing marks of dimethylation of histone H3 lysine 9 and heterochromatin protein 1 α (HP1α) binding (9). This epigenetic repressor profile also occurs in LPS-tolerant circulating leukocytes obtained from humans with SSI (31).
The mechanisms controlling the sustained expression of anti-inflammatory genes during endotoxin tolerance are unclear. One such critically important immune modulator is IκBα. IκBα has the most efficient inhibitory effect on NF-κB proteins (IκBα, IκBβ, IκBγ, and BCL-3) (2, 25, 27). Unlike NF-κB transcription factors, IκBα is synthesized and degraded very rapidly in the basal and stimulated states, where it dynamically exists in free and bound states, respectively (1, 13). IκBα acts as an anti-inflammatory mediator by binding to NF-κB factors p65 and p50 and sequestering them primarily in the cytosol (21). Other NF-κB dimers, such as RelB and p52, do not tightly interact with IκBα. Stimulation by LPS results in rapid IκBα phosphorylation and proteosome degradation, allowing the NF-κB p65/p50 heterodimer to translocate into the nucleus and activate the transcription of a large set of target genes, including a positive feedback signal from IκBα (14). The crucial role of IκBα in regulating inflammation is supported by the observation that either the genetic ablation of IκBα in mice generates SSI or the repression of IκBα degradation is immunosuppressive (11, 26). We previously reported that rapid and sustained resynthesis of IκBα contributes to the repressive signature associated with LPS tolerance in the THP-1 human promonocytic cell model of SSI (15).
The objective of this study was to determine whether RelB might differentially regulate gene transcription during LPS tolerance of THP-1 cells and in human SSI. We found that RelB functions in LPS tolerance of THP-1 promonocytes as an essential transcription activator at the core promoter of the IκBα gene, thus contributing to sustaining the expression of this important anti-inflammatory mediator. This process involves a dimer exchange at the IκBα core promoter, with the replacement of p65/p50 by RelB/p50 after RelB is induced by LPS. We also observed that RelB binds to the IκBα proximal promoter in circulating leukocytes obtained from humans with SSI. We conclude that RelB, following its induction by LPS or during SSI, acts on specific genes and is an indispensable transcription regulator with dual functions, acting both as a transcription activator and as a repressor in the same cell type, thereby influencing the epigenetic profile of innate immunity.
MATERIALS AND METHODS
Cell culture and stimulation.
THP-1 cells obtained from the American Type Culture Collection were maintained in RPMI 1640 medium (Invitrogen Life Technologies) supplemented with 10 U/ml penicillin G, 10 μg/ml streptomycin, 2 mM l-glutamine, and 10% fetal bovine serum (HyClone) at 37°C and 5% CO2 in a humidified incubator. LPS-mediated tolerance that mimics the SSI phenotype in THP-1 cells was previously described (6, 9). Briefly, LPS tolerance is generated by an initial stimulation with Escherichia coli O111:B4 LPS (1.0 μg/ml) for 16 h, followed by restimulation (1.0 μg/ml) for the times shown in Fig. 1 to 3. This LPS acts exclusively through TLR4 as determined with cells lacking TLR4. High concentrations of LPS are used to optimize the tolerance phenotype, although changes occur with doses as low as 10 to 100 ng/ml. Tolerance occurs within 3 h and is sustained for at least 96 h (unpublished data). LPS-induced tolerance is typified by repression of acute proinflammatory genes and sustained expression of anti-inflammatory and antimicrobial genes. Normal and LPS-tolerant THP-1 cells (1.0 × 106 cells/sample) were washed once with RPMI 1640 medium, resuspended in fetal bovine serum-supplemented RPMI 1640 medium at 1 × 106 cells/ml, and stimulated with LPS (1.0 μg/ml) for the times shown in Fig. 1 to 3. Low-passage-number and log-phase cells were used for all experiments.
FIG. 1.

IκBα mRNA and protein levels in normal and LPS-tolerant THP-1 cells. Normal and tolerant THP-1 cells were stimulated with 1 μg/ml LPS for the indicated times (0, 0.5, 1, 3, 6, 12 h), and IκBα levels were assessed by real-time PCR and Western blot analysis. (A) LPS stimulation of normal cells resulted in increases in steady-state IκBα mRNA that reached a peak by 6 h and were moderately diminished at 12 h. In contrast, IκBα mRNA was elevated prior to the stimulation of tolerant cells, increased slightly after secondary LPS stimulation, and remained elevated. (B) No significant differences were seen between the IκBα mRNA half-lives in normal and tolerant cells, supporting the observation that mRNA levels likely reflected transcription. (C) Western blots of whole-cell IκBα protein in normal cells and tolerant cells without and with LPS stimulation are shown. IκBα protein levels were prominent and stable in both normal and tolerant cells without LPS simulation (second lane, IκBα). IκBα protein was rapidly degraded and resynthesized following LPS stimulation of normal cells [lane IκBα (LPS)]. A reduction in IκBα protein also occurred in tolerant cells, although it was less prominent than that observed in normal cells [lane IκBα (LPS)]. Data in panels A and B are the means ± standard errors of the means from three independent experiments and are presented as the increase in copy number relative to that of normal cells at 0 h (set as an arbitrary unit of one). *, significant difference (P < 0.05). Data in panel C are representative of three independent experiments.
FIG. 3.

RelB is a transcriptional activator of IκBα in LPS-tolerant cells. Cells were transfected with control (nonspecific) or RelB-specific siRNA to test for a functional role for RelB in IκBα transcription. Immediately after transfection, cells were left unstimulated or treated with 1 μg/ml LPS to induce tolerance. After 48 h, cells were harvested, washed, and stimulated for 3 h with 1 μg/ml LPS. (A) Cells transfected with RelB siRNA versus control siRNA had pronounced reductions in RelB protein in total cell extracts, cytosol, and nuclear extracts. LDH, lactate dehydrogenase; sumo, small ubiquitin-related modifier protein. (B) LPS induction of IκBα transcription was markedly reduced in RelB siRNA-transfected cells compared with control siRNA. Results using normal, untransfected cells stimulated with LPS, where there was the expected rapid induction of IκBα transcription, are compared. Real-time PCR results are presented as the increase in mRNA copies relative to that of normal cells at 0 h (set as an arbitrary unit of one), as analyzed by quantitative real-time PCR. (C) ChIP assays confirmed that RelB binding to the IκBα promoter was diminished concomitantly with a reduction in IκBα transcription following RelB siRNA knockdown and loss of RelB protein. Data are the means ± standard errors of the means of three independent experiments. *, significant difference (P < 0.05).
Peripheral blood leukocyte (PBL) isolation from SSI patients and healthy participants.
The selection of patients with SSI was based on the modified physiologic criteria of Bone (3) as previously described (18). Based on these criteria, 95% of patients with sepsis were reproducibly shown to have reduced IL-1β or TNF-α production in response to LPS (19). The three sepsis patients enrolled in the present study were found positive for gram-positive and/or gram-negative bacteria. SSI in humans is not limited, however, to infection and may be found in cases of severe trauma, burns, and other pathologies. Patients with known human immunodeficiency virus infection, hematological malignancies affecting leukocyte counts, current cytotoxic chemotherapy, and high-dose glucocorticoid treatment were excluded. The internal review board endorsed the study for clinical research associated with the general clinical research center of the medical center.
PBLs were isolated as previously described (15, 19). Briefly, a venous or arterial heparinized blood sample (up to 30 ml) was drawn from each patient within 1 to 4 days after the onset of sepsis and from each healthy control and immediately processed. PBLs were isolated by Isolymph sedimentation (Gallard-Schlesinger), followed by centrifugation for 5 min at 200 × g at room temperature. The remaining erythrocytes were lysed in LPS-free H2O for 20 s before isotonicity was restored with 3.6% NaCl. PBLs isolated during sepsis have a profile of tolerance of inflammatory genes similar to that of neutrophils or mononuclear cells isolated during sepsis (19). PBLs were washed and counted in phosphate-buffered saline (PBS). The proportions of polymorphonuclear leukocytes (PMN) in PBLs vary (85 to 95%) compared with those in healthy individuals (60 to 80%). In order to more closely approximate the cell populations in healthy and septic participants prior to chromatin immunoprecipitation (ChIP), PBLs from healthy participants were further separated into PMN (90 to 95%) by a second centrifugation over 3 ml of Isolymph for 30 min at 400 × g. The PMN pellet was washed and counted in PBS. A sample of 5 × 106 cells/condition was used for ChIP as described below. Thus, PBL preparations from patients and controls were partially matched for cell types. Gene tolerance is similar in myeloid cell-derived leukocytes such as PMN and monocytes (15).
ChIP assay.
To assess p65, p50, RelB, and p52 binding to the IκBα promoter in LPS-tolerant and normal cells, ChIP assays (Upstate Biotechnology) were performed according to the manufacturer's instructions, with the following modifications. Cells (5 × 106 cells/condition) were fixed by adding formaldehyde (HCHO, from a 37% HCHO-10% methanol stock; Calbiochem) into the medium for a final formaldehyde concentration of 1% and incubated at room temperature for 10 min with gentle shaking. The chromatin was sheared by sonication using a Diagenode Bioruptor sonicator (model UCD-200TM-EX; Tosho Denk1 Co., Ltd). Sonication at a high-level power setting (30 s on and 30 s off for 23 min) generated DNA fragments of ∼0.5 to 1.5 kb. Each condition was divided into two samples, providing an “input” sample that was not incubated with antibodies. The other portion was incubated overnight with antibodies specific for p65 (SC-372), p50 (SC-7178), RelB (SC-226), and p52/p100 (SC-298) (Santa Cruz Biotechnology) and immunoglobulin G (SC-2027) (Upstate Biotechnology) for use as a negative control. Purified DNA was resuspended in 10 μl distilled H2O.
Standard PCR.
A standard PCR mixture (total 25 μl) contained 2 μl ChIP DNA, 1 μM of each primer, 2 mM MgCl2, 0.2 M of each deoxynucleoside triphosphate (dNTP), and 0.03 U/μl AmpliTaq Gold DNA polymerase (Applied Biosystems). The procedure was 1 cycle at 94°C for 5 min, 35 cycles at 55°C for 30 s, and 72°C for 30 s each, plus a final cycle at 72°C for 10 min. The PCR products were visualized on 1.5% agarose gels, and images were captured using a Quantity One imager (Bio-Rad, Hercules, CA). The IκBα promoter contains three functional NF-κB sites, where the κB1 site of the proximal promoter is the most important κB site for transactivation and transcription can be autoregulated by the NF-κB proteins (13). The primers used to amplify the IκBα promoter were designed with Primer Express R version 3.0 software (Applied Biosystems) to amplify a sequence from the human IκBα promoter region containing the κB1 site at −96 bp relative to the transcription start site, as follows: Primer1, 5′-AGCAGAGGACGAAGCCAGTTCT-3′; and Primer2, 5′-GACTGCTGTGGGCTCTGCAG-3′.
Real-time PCR.
The IκBα promoter in the ChIP DNA was quantified by real-time PCR. Input or ChIP DNA was used for each reaction. The same primers for the IκBα promoter described above were used with an internal fluorogenic probe (5′-FAM-TCTGGTCTGACTGGCTT-TAMRA-3′ [FAM, 6-carboxyfluorescein; TAMRA, 6-carboxytetramethylrhodamine]; Applied Biosystems) designed with Primer Express R version 3.0 software (Applied Biosystems). The real-time PCR mixture (total 25 μl) contained 3 μl DNA, 12.5 μl 2× TaqMan Universal Master Mix, 300 nM each primer, and 100 nM of each dNTP. The real-time PCR procedure was 2 min at 50°C and 10 min at 95°C, followed by 40 cycles of 15 s at 95°C and 1 min at 60°C, using an ABI Prism 7000 sequence detection system (Applied Biosystems). Data were normalized to the input DNA and are presented as the changes in DNA levels relative to DNA from untreated cells.
To measure IκBα mRNA, total RNA was isolated at various times after LPS stimulation of normal and LPS-tolerant cells by using RNA Stat-60, according to the manufacturer's protocol (Tel-Test, Friendswood, TX). For IκBα mRNA half-life determination, normal cells and tolerant cells were treated with LPS for 6 h and transcriptional arrest was induced with actinomycin D (1 μg/ml). One microgram of RNA was reverse transcribed to cDNA in a 20-μl volume containing 5 mM MgCl2, 1 mM of each dNTP, 2.5 μM oligo d(T), and 2.5 U/μl murine leukemia virus reverse transcriptase (Applied Biosystems). The PCR was performed using 4 μl cDNA and GAPDH (glyceraldehyde-3-phosphate dehydrogenase) predesigned TaqMan primer/probe kits (Applied Biosystems), under the conditions described above. Data were normalized to those for GAPDH mRNA and are presented as the increase in mRNA levels relative to mRNA from untreated cells.
Western blotting.
Total, nuclear, and cytoplasmic RelB protein levels in normal and tolerant cells were assayed by Western blotting. For whole-cell protein extraction, 1.5 × 106 cells/sample were collected and washed twice with PBS. Cells were resuspended in 100 μl RIPA buffer (cell lysis buffer containing 0.45% NaCl, 0.5% deoxycholate, 0.5% Triton X-100, 0.05% sodium dodecyl sulfate [SDS], 0.005 M Tris) and incubated on ice for 20 min. For nuclear and cytoplasmic protein extraction, 5 × 106 cells/sample were collected and washed twice with PBS and then suspended in 50 μl buffer A (10 mM HEPES, 1.5 mM MgCl2, 10 mM KCl, 0.25 mM EDTA) and incubated on ice for 15 min. One milliliter of buffer A contains 10 μl Sigma 100× inhibitor cocktail, 20 μl 1 M NaF, 2 μl 0.5 M Na4P2O7, 1 μl 1 M Na3VO4, and 1 μl 1 M dithiothreitol to which 0.8 μl 25% Triton X-100 was added. After being vortexed for 10 s and spun at 10,000 rpm for 5 min, supernatant for cytoplasmic protein was saved, and the pellet was suspended in 100 μl RIPA (1% NP-40, 0.5% sodium deoxycholate, 0.1% SDS in PBS). Supernatant for nuclear protein was collected after being spun for nuclear protein. Twenty micrograms of whole-cell protein extract or 30 μg of nuclear/cytoplasmic protein was separated by SDS-polyacrylamide gel electrophoresis and transferred to an Immunoblot polyvinylidene difluoride membrane (Bio-Rad). RelB antibody (SC-266; Santa Cruz Biotechnology) was used to visualize and quantify protein levels by using ImageQuant software (Molecular Dynamics and Amersham Pharmaceutical Biotech).
RNA interference.
Cells (5 × 106) were transfected by electroporation with 8 μl (0.5 μM) of control small interfering RNA (siRNA; catalog no. SC-37007) or RelB siRNA (catalog no. SC-36402) (Santa Cruz Biotechnology) in 100 μl of transfection medium (Nucleofector; Amaxa, Gaithersburg, MD) as previously described (6, 29). Immediately after transfection, cells were left unstimulated or treated with 1 μg/ml LPS to induce tolerance. After 48 h, cells were harvested, washed, and stimulated for 3 h with 1 μg/ml LPS. Cells were harvested, and RNA was isolated and used to determine the IκBα mRNA expression levels.
Statistical analysis.
Statistical analysis and graphic presentations were performed using Microsoft Excel XP software. Results shown are the averages of three ChIP assays. Significance was taken at a P value of <0.05 (Student's t test).
RESULTS
LPS induces sustained transcription of IκBα during LPS-mediated tolerance.
Our previous report indicated that IκBα protein levels are sustained in LPS-tolerant THP-1 cells with repressed transcription of IL-1β, suggesting persistent transcription of IκBα in contrast to repressed transcription of acute proinflammatory genes (15). To test whether IκBα transcription persists following LPS-induced tolerance, we measured IκBα mRNA levels in LPS-naïve and LPS-tolerant cells, as shown in Fig. 1A. As assessed by real-time PCR, IκBα mRNA levels in normal cells stimulated with LPS rapidly and dramatically increased eightfold, diminished somewhat, and then remained substantially elevated over the 12-h test. IκBα mRNA levels remained elevated at the 12-h time point (e.g., the 0-h time point of tolerant cells is shown in Fig. 1A). Following the second LPS stimulation, IκBα mRNA slightly but significantly increased in tolerant THP-1 cells, after which levels remained elevated. To ensure that transcription was the major mechanism controlling the steady-state level of IκBα mRNA, we also measured the IκBα mRNA half-lives in normal and tolerant cells and found no significant differences (Fig. 1B). Together, the data support sustained transcription of IκBα RNA during LPS-mediated gene tolerance.
We previously reported that IκBα protein, which is prominent in unstimulated normal and LPS-tolerant THP-1 cells, is rapidly degraded and resynthesized following LPS stimulation of both LPS-responsive and LPS-tolerant cells (29). We affirmed this paradigm in this study, as shown in Fig. 1C. Protein production persisted in unstimulated normal and tolerant cells (Fig. 1C, lane 2). Using actinomycin D to block de novo IκBα synthesis and to follow decay, we confirmed the high turnover rate of IκBα in unstimulated normal and tolerant THP-1 cells (data not shown). When normal cells were stimulated by LPS, IκBα protein levels diminished quickly and then increased. In tolerant THP-1 cells, the IκBα level following LPS stimulation appeared to diminish somewhat less than that observed in normal cells and increased thereafter (Fig. 1C). The steady-state levels did not closely parallel the increases in IκBα mRNA noted after LPS stimulation (Fig. 1A and C, lane 4), supporting the constantly high turnover rate of the protein in the basal free state (21). Together, the IκBα mRNA and protein data support sustained expression of the IκBα gene during LPS-mediated gene reprogramming.
Differential binding of NF-κB p65 and RelB transcription factors to the IκBα core promoter occurs in normal and LPS-tolerant cells.
A previous report showed that p65, p50, and RelB may differentially alter LPS-induced transcription in dendritic cells through a process of dimer exchange, wherein the rapidly formed p65/p50 dimer is replaced by slowly formed RelB/p50 or RelB/p52 (22). To test whether such a process might occur in the LPS-tolerant THP-1 cell model of SSI, we used a ChIP assay to assess the binding of p65, p50, p52, and RelB to the κB1 site at the core promoter, the functionally important NF-κB site for the activation of IκBα transcription (13). We observed no detectable nonspecific binding of matched immunoglobulin G to the IκBα promoter (not shown). Real-time PCR and standard PCR analyses of ChIP DNA revealed that NF-κB p65 was enhanced at the IκBα promoter following LPS stimulation in LPS-responsive cells. The binding was increased threefold by 60 min and then diminished slightly over 3 h (Fig. 2A). The elevated NF-κB p65 binding correlated with the increased IκBα mRNA transcription with LPS stimulation in normal cells, as shown in Fig. 1A. p65 binding was also detected in LPS-tolerant cells prior to stimulation with a second dose of LPS. In contrast to control cells, LPS stimulation of tolerant cells resulted in a significant reduction in p65 binding to the IκBα promoter (Fig. 2A). NF-κB p50 binding to the IκBα promoter remained constant in unstimulated and LPS-stimulated normal cells. LPS stimulation of LPS-tolerant cells resulted in an increase in p50 binding by 3 h (Fig. 2B). Limited RelB binding to the IκBα core promoter was detected in normal cells for up to 3 h, without or with LPS stimulation (Fig. 2C). This was expected, since RelB is minimally expressed in LPS-naïve THP-1 cells until 6 h (31). RelB promoter binding was increased fourfold in LPS-tolerant cells 16 h after the initial LPS dose and prior to the secondary LPS stimulation. LPS secondary stimulation of tolerant cells induced rapid and transient increases in RelB promoter binding at 30 min and 1 h after LPS stimulation, which occurred coincident with reduced p65 binding. RelB promoter binding then returned to a level similar to that in unstimulated LPS-tolerant cells (Fig. 2C). We did not detect binding of p52 to the IκBα promoter (not shown). Together, these data supported an exchange of the p65/p50 dimer with RelB/p50.
FIG. 2.

Differential binding of the NF-κB p65, p50, and RelB transcription factors to the IκBα promoter in normal and LPS-tolerant cells. Normal and tolerant THP-1 cells were incubated with 1 μg/ml LPS for 0, 0.5, 1, and 3 h. DNA was isolated from samples before and after ChIP assay with anti-p65 (A), anti-p50 (B), and anti-RelB (C) antibodies. Results are shown for standard and real-time PCR. (A) p65 binding to the IκBα promoter increased rapidly after LPS stimulation of normal cells. In contrast, p65 binding decreased rapidly and remained low after 3 h in tolerant cells following LPS stimulation. (B) No change in p50 binding was seen in normal cells. In contrast, p50 binding increased in tolerant cells at 3 h. (C) RelB binding increased significantly in tolerant cells with secondary LPS stimulation compared with that of normal cells. Data for real-time PCR are the means ± standard errors of the means for three independent experiments and are presented as the increase in binding relative to that of normal cells at 0 h (set as an arbitrary unit of one). *, significant difference (P < 0.05). Standard PCR results of ChIP samples with the same primer sets as shown above the graphed data are representative of three independent experiments.
IκBα transcription in tolerant THP-1 cells requires RelB.
Our ChIP and mRNA data suggested that RelB might function as a transcription activator at the IκBα gene promoter. To test this, we used RelB siRNA to deplete RelB in LPS-tolerant cells. Western blot analysis confirmed that RelB siRNA knockdown virtually eliminated RelB protein in whole-cell, cytosol, and nuclear extracts of LPS-tolerant cells (Fig. 3A). The functional effects of the removal of RelB from LPS-tolerant cells are shown in Fig. 3B. LPS-stimulated normal cells that were not transfected with either siRNA had the expected rapid increase in IκBα mRNA (compare results for normal cells in Fig. 1A and 3B). The levels of IκBα mRNA increased as expected in the LPS-tolerant cells at the 0-h time point in both control siRNA- and RelB siRNA-treated cells (i.e., before they received a second dose of LPS), since the cells had received a primary dose of LPS that increased steady-state IκBα mRNA prior to the induction of RelB. In contrast to normal LPS-naïve cells, there was a striking reduction in IκBα mRNA following the secondary LPS stimulation of tolerant cells transfected with RelB siRNA versus control siRNA (Fig. 3B).
We then used ChIP analysis to confirm that siRNA knockdown of RelB protein in tolerant cells resulted in a reduction in RelB binding to the IκBα promoter (Fig. 3C), further supporting RelB as a transcription activator in tolerant cells.
RelB binding at the IκBα core promoter is increased in PBLs of SSI patients.
Our report of studies of PBLs obtained from patients with SSI shows that RelB protein expression is markedly increased but that little or no RelB is detectable in normal PBLs (31). To test whether RelB might participate in sustaining activation of IκBα in human SSI, in support of the concept for the paradigm observed for LPS-mediated tolerance of THP-1 cells, we used ChIP to assess in vivo RelB binding to the IκBα promoter in normal PBLs and in PBLs from patients with SSI. We found prominent RelB binding to the IκBα promoter in PBLs obtained from three patients with SSI (Fig. 4).
FIG. 4.
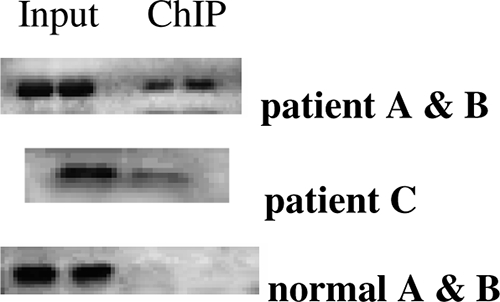
RelB binds to the IκBα proximal promoter in PBLs of SSI patients with sepsis. DNA was isolated from PBLs of healthy subjects and of three patients with SSI caused by sepsis. Cells without LPS stimulation were subjected to a ChIP assay using anti-RelB antibodies. Standard PCR was used to assess RelB binding to the IκBα promoter. Results for patients A and B versus patient C were studied on different days. Results for representative healthy subjects are shown (normal A and B). RelB binding to the IκBα proximal promoter was increased in all patients.
DISCUSSION
Our general conclusion from this study is that de novo induction of RelB is required to sustain transcription of IκBα following LPS-induced tolerance of gene expression in THP-1 promonocytic cells, a model of changes that occur in circulating leukocytes during SSI. This interpretation is supported by several findings. First, persistent transcription and translation of IκBα mRNA occurred following LPS tolerance, and IκBα mRNA further increased after secondary LPS stimulation of cells in which RelB had been induced. Although changes in IκBα protein levels did not precisely parallel levels of mRNA, the rapid repletion of the protein following its LPS-induced proteosome degradation in normal or tolerant cells supports the fact that a persistent state of active transcription existed. The observation that levels of IκBα protein did not precisely follow levels of IκBα mRNA was not surprising, since we have reported that transcriptional and translational events may be under distinct controls in LPS-tolerant cells (16).
A second level of support for our concept relates to the changes that occur in the NF-κB binding to the IκBα promoter. LPS stimulation of normal cells resulted in rapid formation of the transcriptionally active p65/p50 dimer at the IκBα core promoter, which supported an initial wave of increased transcription. During this time, there was little RelB available. After induction of RelB by the initial LPS dose which made the cells tolerant, there was an accumulation of RelB at the IκBα proximal promoter, during which time there was sustained transcription. When LPS-tolerant cells were stimulated with LPS a second time, there was rapid reduction of p65 binding (i.e., p65/p50 dimer formation), during which time RelB binding to the IκBα promoter further increased. This was associated with an apparent RelB/p50 dimer exchange for p65/p50.
A third level of support for our general conclusion was the fact that significant reductions in IκBα mRNA levels occurred in LPS-tolerant cells following depletion of RelB protein through the siRNA process. The low levels of mRNA observed in LPS-tolerant cells after siRNA knockdown were paralleled by reductions in IκBα protein (not shown) and with a concomitant reduction in RelB binding to the IκBα proximal promoter. These data support a loss of the transactive RelB/p50 dimer when RelB is depleted. Finally, the concept that RelB may function as a transcription activator in human SSI is supported by our finding that RelB also bound to the IκBα proximal promoter in circulating leukocytes obtained from three patients with SSI. RelB binding was not detected on the IκBα promoter of control blood leukocytes.
Taken together with our previous report of RelB-dependent repression of IL-1β and TNF-α (9, 31), our data support the observation that RelB regulated distinct sets of genes in the LPS-tolerant THP-1 model of SSI. Unlike transcriptionally repressed acute proinflammatory IL-1β and TNF-α promoters that bind heterochromatin modifier HP1α in tolerant THP-1 cells (9), no binding of HP1α to the IκBα promoter was found in this study (data not shown). This supports the finding that IκBα persists as euchromatin during gene tolerance, at a time when IL-1β and TNF-α are transformed to silenced heterochromatin. Further support for the euchromatic nature of the IκBα gene in LPS-tolerant cells is our unpublished observation that the histone code mark for active transcription, histone H3 phosphorylation on serine 10, increases following LPS stimulation of tolerant THP-1 cells.
During LPS activation of macrophages, distinct NF-κB factors, such as p65, p50, c-Rel, p52, and RelB, may dynamically interact with the promoter and enhancers of the same gene (23). These transcription factors may bind concomitantly or replace each other by dimer exchange (4). Dimer exchanges between RelB, p65, and promoter-bound p50 occur in dendritic cells following LPS stimulation by a process that can both repress transcription of IL-12p40 and activate transcription of IL-8 (22). Our study supports a similar paradigm contributing to LPS tolerance in the SSI phenotype.
LPS stimulation of normal cells, LPS-tolerant cells, or SSI circulating leukocytes leads to IκBα rapid cytoplasmic degradation with concomitant release and accumulation of p65 and p50 within the nucleus (14). However, the accumulating nuclear p65 cannot bind to the promoters of repressed acute proinflammatory genes in LPS-tolerant cells due to the heterochromatin-based silencing mechanisms (6, 9, 31). Still, the nuclear p65 would be available to rapidly bind to genes that are not repressed and remain as transcriptionally responsive as euchromatin, such as IκBα. The delayed RelB induction by LPS would later support the sustained expression of the IκBα gene through dimer exchange, even when p65 is removed from the promoter and resequestered with the cytoplasmic IκBα (20). Accordingly, the dimer exchange to a RelB/p50 activator could sustain expression of the IκBα gene, since the RelB/p50 dimers cannot be sequestered and rendered inactive by IκBα (22). The occurrence of NF-κB dimer exchange supports the observation that preexisting promoter complexes are disassembling and being reassembled based on available new components. This renders genes sensitive to the expression and concentrations of other NF-κB proteins and cooperating factors.
The mechanisms responsible for the dual RelB functions of activation and repression and its coupling to epigenetic modifications during LPS tolerance are unknown but are of considerable importance. RelB likely mediates different epigenetic modifications at distinct sets of promoters. Promoter specificity could reside in the different DNA structures, the interaction of RelB with distinct coactivators or repressors (8, 12), the nucleosome configurations associated with the genes that render them either euchromatin or heterochromatin, or spatial differences in RelB posttranslational modifications (5).
In summary, we conclude that de novo synthesis of RelB provides a transcription activator that sustains the expression of IκBα when bound to its proximal promoter in the THP-1 cell model of LPS-induced gene tolerance and during human SSI. RelB-dependent transactivation of IκBα occurs concomitantly with RelB-dependent transcription repression of IL-1β and TNF-α. Both processes involve NF-κB dimer exchange between p65 and RelB, wherein one is repressive and one is active. These two functional processes involve changes in chromatin structure. Together, the data support the finding that RelB is a dual-function, gene-specific transcription regulator acting within the same cell type to modulate innate immunity. The observations that distinct classes of genes (e.g., proinflammatory and anti-inflammatory) are regulated differentially following stimulation of the same receptor further support the conclusion that epigenetic gene-specific processes generate the LPS tolerance phenotype associated with SSI.
Acknowledgments
This work was supported by National Institutes of Health grants RO1AI-09169, RO1AI-065791, and MO1-RR007122 to the Wake Forest University Medical Center General Clinical Research Center.
We thank Michael Seeds for assistance with real-time PCR primer design and Weihong Yin for picture scanning.
Footnotes
Published ahead of print on 19 November 2008.
REFERENCES
- 1.Arenzana-Seisdedos, F., J. Thompson, M. S. Rodriguez, F. Bachelerie, D. Thomas, and R. T. Hay. 1995. Inducible nuclear expression of newly synthesized IκBα negatively regulates DNA-binding and transcriptional activities of NF-κβ. Mol. Cell. Biol. 152689-2696. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Bates, P. W., and S. Miyamoto. 2004. Expanded nuclear roles for IkappaBs. Sci. STKE 25448-51. [DOI] [PubMed] [Google Scholar]
- 3.Bone, R. C. 1996. The sepsis syndrome: definition and general approach to management. Clin. Chest Med. 17175-181. [DOI] [PubMed] [Google Scholar]
- 4.Bosisio, D., I. Marazzi, A. Agresti, N. Shimizu, M. E. Bianchi, and G. Natoli. 2006. A hyper-dynamic equilibrium between promoter-bound and nucleoplasmic dimers controls NF-kappaB-dependent gene activity. EMBO J. 25798-810. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Britanova, L. V., V. J. Makeev, and D. V. Kuprash. 2008. In vitro selection of optimal RelB/p52 DNA-binding motifs. Biochem. Biophys. Res. Commun. 365583-588. [DOI] [PubMed] [Google Scholar]
- 6.Chan, C., L. Li, C. E. McCall, and B. K. Yoza. 2005. Endotoxin tolerance disrupts chromatin remodeling and NF-kappaB transactivation at the IL-1beta promoter. J. Immunol. 175461-468. [DOI] [PubMed] [Google Scholar]
- 7.Cross, A., L. Asher, M. Seguin, L. Yuan, N. Kelly, C. Hammack, J. Sadoff, and P. J. Gemsk. 1995. The importance of a lipopolysaccharide-initiated, cytokine-mediated host defense mechanism in mice against extraintestinally invasive Escherichia coli. J. Clin. Investig. 96676-686. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Eisner, V., C. Quiroga, A. Criollo, J. M. Eltit, M. Chiong, V. Parra, K. Hidalgo, B. Toro, G. Díaz-Araya, and S. Lavandero. 2006. Hyperosmotic stress activates p65/RelB NFkappaB in cultured cardiomyocytes with dichotomic actions on caspase activation and cell death. FEBS Lett. 5803469-3476. [DOI] [PubMed] [Google Scholar]
- 9.El Gazzar, M., B. K. Yoza, J. Y. Hu, S. L. Cousart, and C. E. McCall. 2007. Epigenetic silencing of tumor necrosis factor alpha during endotoxin tolerance. J. Biol. Chem. 28226857-26864. [DOI] [PubMed] [Google Scholar]
- 10.Foster, S. L., D. C. Hargreaves, and R. Medzhitov. 2007. Gene-specific control of inflammation by TLR-induced chromatin modifications. Nature 447972-978. [DOI] [PubMed] [Google Scholar]
- 11.Gerondakis, S., R. Grumont, R. Gugasyan, L. Wong, I. Isomura, W. Ho, and A. Banerjee. 2006. Unravelling the complexities of the NF-kappaB signalling pathway using mouse knockout and transgenic models. Oncogene 256781-6799. [DOI] [PubMed] [Google Scholar]
- 12.Huang, D. B., D. Vu, and G. Ghosh. 2005. NF-kappaB RelB forms an intertwined homodimer. Structure 131365-1373. [DOI] [PubMed] [Google Scholar]
- 13.Ito, C. Y., A. G. Kazantsev, and A. S. Baldwin, Jr. 1994. Three NF-kappa B sites in the Ikappa B-alpha promoter are required for induction of gene expression by TNF alpha. Nucleic Acids Res. 223787-3792. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Karin, M., and Y. Ben-Neriah. 2000. Phosphorylation meets ubiquitination: the control of NF-κB activity. Annu. Rev. Immunol. 18621-663. [DOI] [PubMed] [Google Scholar]
- 15.LaRue, K. E., and C. E. McCall. 1994. A labile transcriptional repressor modulates endotoxin tolerance. J. Exp. Med. 1802269-2275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Learn, C. A., S. B. Mizel, and C. E. McCall. 2000. mRNA and protein stability regulate the differential expression of pro- and anti-inflammatory genes in endotoxin-tolerant THP-1 cells. J. Biol. Chem. 27512185-12193. [DOI] [PubMed] [Google Scholar]
- 17.McCall, C. E., and B. K. Yoza. 2007. Gene silencing in severe systemic inflammation. Am. J. Respir. Crit. Care Med. 175763-767. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.McCall, C. E., L. M. Grosso-Wilmoth, K. LaRue, R. N. Guzman, and S. L. Cousart. 1993. Tolerance to endotoxin-induced expression of the interleukin-1β gene in blood neutrophils of humans with the sepsis syndrome. J. Clin. Investig. 91853-861. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Mueller, L. P., B. K. Yoza, K. Neuhaus, C. S. Loeser, S. Cousart, M. C. Chang, J. W. Meredith, L. Li, and C. E. McCall. 2001. Endotoxin-adapted septic shock leukocytes selectively alter production of sIL-1RA and IL-1β. Shock 16430-437. [DOI] [PubMed] [Google Scholar]
- 20.Natoli, G., S. Saccani, D. Bosisio, and I. Marazzi. 2005. Interactions of NF-kappaB with chromatin: the art of being at the right place at the right time. Nat. Immunol. 6439-445. [DOI] [PubMed] [Google Scholar]
- 21.O'Dea, E. L., J. D. Kearns, and A. Hoffmann. 2008. UV as an amplifier rather than inducer of NF-κB activity. Mol. Cell 30632-641. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Saccani, S., S. Pantano, and G. Natoli. 2003. Modulation of NF-kappaB activity by exchange of dimers. Mol. Cell 111563-1574. [DOI] [PubMed] [Google Scholar]
- 23.Schreiber, J., R. G. Jenner, H. L. Murray, G. K. Gerber, D. K. Gifford, and R. A. Young. 2006. Coordinated binding of NF-kappaB family members in the response of human cells to lipopolysaccharide. Proc. Natl. Acad. Sci. USA 1035899-5904. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Singh, H., R. Sen, D. Baltimore, and P. A. A. Sharp. 1986. A nuclear factor that binds to a conserved sequence motif in transcriptional control elements of immunoglobulin genes. Nature 319154-158. [DOI] [PubMed] [Google Scholar]
- 25.Viatour, P., M. P. Merville, V. Bours, and A. Chariot. 2005. Phosphorylation of NF-kappaB and IkappaB proteins: implications in cancer and inflammation. Trends Biochem. Sci. 3043-52. [DOI] [PubMed] [Google Scholar]
- 26.Voboril, R., S. N. Hochwald, J. Li, A. Brank, J. Weberova, F. Wessels, L. L. Moldawer, E. R. Camp, and S. L. MacKay. 2004. Inhibition of NF-kappa B augments sensitivity to 5-fluorouracil/folinic acid in colon cancer. J. Surg. Res. 120178-188. [DOI] [PubMed] [Google Scholar]
- 27.Wang, T., X. Zhang, and J. J. Li. 2002. The role of NF-kappaB in the regulation of cell stress responses. Int. Immunopharmacol. 21509-1520. [DOI] [PubMed] [Google Scholar]
- 28.Weat, M. A., and W. Heagy. 2002. Endotoxin tolerance: a review. Crit. Care Med. 30S64-S73. [PubMed] [Google Scholar]
- 29.Yoza, B., K. LaRue, and C. McCall. 1998. Molecular mechanisms responsible for endotoxin tolerance. Prog. Clin. Biol. Res. 397209-215. [PubMed] [Google Scholar]
- 30.Yoza, B. K., J. Y. Hu, S. L. Cousart, and C. E. McCall. 2000. Endotoxin inducible transcription is repressed in endotoxin reprogrammed cells. Shock 13236-342. [DOI] [PubMed] [Google Scholar]
- 31.Yoza, B. K., J. Y. Hu, S. L. Cousart, L. M. Forrest, and C. E. McCall. 2006. Induction of RelB participates in endotoxin tolerance. J. Immunol. 1774080-4085. [DOI] [PubMed] [Google Scholar]