

Abstract
The influence of acetohydroxy acid synthase (AHAS) on l-lysine production by Corynebacterium glutamicum was investigated. An AHAS with a deleted C-terminal domain in the regulatory subunit IlvN was engineered by truncating the ilvN gene. Compared to the wild-type AHAS, the newly constructed enzyme showed altered kinetic properties, i.e., (i) an about twofold-lower Km for the substrate pyruvate and an about fourfold-lower Vmax; (ii) a slightly increased Km for the substrate α-ketobutyrate with an about twofold-lower Vmax; and (iii) insensitivity against the inhibitors l-valine, l-isoleucine, and l-leucine (10 mM each). Introduction of the modified AHAS into the l-lysine producers C. glutamicum DM1729 and DM1933 increased l-lysine formation by 43% (30 mM versus 21 mM) and 36% (51 mM versus 37 mM), respectively, suggesting that decreased AHAS activity is linked to increased l-lysine formation. Complete inactivation of the AHAS in C. glutamicum DM1729 and DM1933 by deletion of the ilvB gene, encoding the catalytic subunit of AHAS, led to l-valine, l-isoleucine, and l-leucine auxotrophy and to further-improved l-lysine production. In batch fermentations, C. glutamicum DM1729 ΔilvB produced about 85% more l-lysine (70 mM versus 38 mM) and showed an 85%-higher substrate-specific product yield (0.180 versus 0.098 mol C/mol C) than C. glutamicum DM1729. Comparative transcriptome analysis of C. glutamicum DM1729 and C. glutamicum DM1729 ΔilvB indicated transcriptional differences for about 50 genes, although not for those encoding enzymes involved in the l-lysine biosynthetic pathway.
Corynebacterium glutamicum, a gram-positive soil bacterium that grows on a variety of sugars and organic acids, is the workhorse for the fermentative production of the amino acids l-glutamate (1.5 × 106 tons/year) and l-lysine (0.9 × 106 tons/year) (25, 27, 53). Due to the growing world market and steadily decreasing market prices (24), great efforts have been made to develop more powerful and efficient production strains (8, 20, 24, 42). Since the yields and productivities of the production strains still are below the expected theoretical values, there is a large interest to further improve the performance of bacterial production strains (53).
In C. glutamicum, l-lysine is synthesized from oxaloacetate and pyruvate to the branch of l-piperideine-2,6-dicarboxylate, which is converted to d,l-diaminopimelate either by diaminopimelate dehydrogenase, when ammonium is available in excess, or by the tetrahydrodipicolinate succinylase pathway, when ammonium availability is low (46, 52). The key enzyme for l-lysine synthesis is aspartate kinase, which in wild-type (WT) C. glutamicum catalyzes phosphorylation of aspartate and is strongly feedback inhibited by l-lysine plus l-threonine (33, 48). Overexpression of the respective lysC gene, and especially overexpressing alleles encoding feedback-resistant aspartate kinase, strongly improved l-lysine formation (7, 11, 47). Aside from tailoring the biosynthetic pathway for l-lysine overproduction, carbon flux analysis highlighted the importance of the NADPH supply for efficient l-lysine production with the pentose phosphate pathway (PPP) as the predominant route for NADPH supply during growth on glucose (28). Increased flux from glycolysis into the PPP was achieved by inactivation of phosphoglucose isomerase (29), by introduction of a mutant allele encoding a feedback-resistant 6-phosphogluconate dehydrogenase (34), or by overexpression of fructose 1,6-biphosphatase (1, 15). Also, the expression of the membrane-bound transhydrogenase genes from Escherichia coli in C. glutamicum increased the NADPH supply and thus improved l-lysine production (21). A number of studies indicated the extraordinary role of the pyruvate and/or oxaloacetate supply for l-lysine production by inactivation of the pyruvate dehydrogenase complex (PDHC) (4), by overexpression of the pyruvate carboxylase gene (38), by inactivation of the phosphoenolpyruvate (PEP) carboxykinase gene (41), or by disruption of malate:quinone oxidoreductase (31). Furthermore, the inactivation of citrate synthase and methylcitrate synthase was highly beneficial for l-lysine production due to an increased oxaloacetate supply (40). Other studies described a link between increased l-lysine formation and l-leucine auxotrophy in C. glutamicum MH20-22B, DG-52-5, and KK25 ΔleuA strains (36, 47, 51) or with a limited l-leucine supply in the defined l-lysine producer C. glutamicum ADL-3 (19). Acetohydroxy acid synthase (AHAS) is the key enzyme of the pathways for the synthesis of the branched-chain amino acids (BCAAs) l-valine, l-isoleucine, and l-leucine. It catalyzes the formation of either α-acetolactate from two molecules of pyruvate or the formation of α-acetohydroxybutyrate from pyruvate plus α-ketobutyrate. The C. glutamicum AHAS consists of two catalytic and two regulatory subunits, which are encoded by ilvB and ilvN, respectively. Together with the acetohydroxy acid isomeroreductase gene ilvC, the two genes form the ilvBNC operon (6, 10). On the one hand, AHAS is subject to feedback inhibition by the three BCAAs (10, 14). On the other hand, expression of the ilvBNC operon is controlled by an attenuation mechanism, leading to an about twofold-higher expression in response to the shortage of BCAAs (32). Furthermore, it has been shown that α-ketobutyrate in the medium increases expression of the ilvBNC operon by about 10-fold by a so-far-unknown regulatory mechanism (10, 32). In contrast to C. glutamicum, Escherichia coli possesses three AHAS isoenzymes (I, II, and III), differing in their regulation and biochemical properties (54). AHAS III (encoded by ilvIH) exhibits the highest similarity to the AHAS of C. glutamicum. The regulatory subunit of E. coli (163 amino acids, encoded by ilvH) shares 39% identity with that of C. glutamicum (172 amino acids, encoded by ilvN) (35) and contains a characteristic N-terminal ATC domain, which has been shown to be responsible for l-valine binding (23). However, deletion of the C-terminal domain of ilvH in E. coli resulted in a functional AHAS III that has a higher Vmax, has a higher catalytic efficiency, and is insensitive for l-valine inhibition (30). In a similar manner, the deletion of the C-terminal ATC domain of the l-serine-sensitive 3-phosphoglycerate dehydrogenase in C. glutamicum resulted in a fully functional enzyme that is insensitive to l-serine and that improved l-serine production (37, 39).
In the present work, we engineered a feedback-resistant AHAS, insensitive to the BCAAs by deletion of the C-terminal domain of IlvN. Originally we intended to use the modified enzyme for improvement of l-valine production by C. glutamicum. However, the kinetic properties of the newly constructed AHAS prompted us to test the enzyme for an effect on l-lysine production, leading to the finding that decreased AHAS activity is linked to increased l-lysine formation.
MATERIALS AND METHODS
Bacterial strains and plasmids.
All bacterial strains and plasmids and their relevant characteristics and sources are listed in Table 1. The oligonucleotides used and their sequences are also listed in Table 1.
TABLE 1.
Strains, plasmids, and oligonucleotides used in this study
| Strain, plasmid, or oligonucleotide | Relevant characteristics or sequence | Source, reference, or purpose |
|---|---|---|
| Strains | ||
| E. coli DH5α | supE44 hsdR17 recA1 endA1 gyrA96 thi-1 relA1 | 17 |
| WT C. glutamicum | WT strain ATCC 13032, biotin-auxotrophic | American Type Culture Collection |
| C. glutamicum DM1729 | pyc(P458S) hom(V59A) lysC(T311I), derived from WT C. glutamicum | For construction, see Table S1 in the supplemental material |
| C. glutamicum DM1729 ΔilvB | Strain DM1729 with deletion of ilvB, encoding the large subunit of the acetolactate synthase (AHAS) | This work |
| C. glutamicum DM1729 ΔC-T ilvN | Strain DM1729 with deletion of the last 83 amino acids of the C-terminal domain of ilvN, encoding the small subunit of the AHAS | This work |
| C. glutamicum DM1933 | Δpck pyc(P458S) hom(V59A), 2 copies of lysC(T311I), 2 copies of asd, 2 copies of dapA, 2 copies of dapB, 2 copies of ddh, 2 copies of lysA, 2 copies of lysE derived from WT C. glutamicum | For construction, see Table S1 in the supplemental material |
| C. glutamicum DM1933 ΔilvB | Strain DM1933 with deletion of ilvB, encoding the large subunit of the AHAS | This work |
| C. glutamicum DM1933 ΔC-T ilvN | Strain DM1933 with deletion of the last 83 amino acids of the C-terminal domain of ilvN, encoding the small subunit of the AHAS | This work |
| Plasmids | ||
| pK19mobsacB | Kmr, mobilizable (oriT), oriV | 44 |
| pK19mobsacB ΔilvB | pK19mobsacB carrying a truncated ilvB gene | 32 |
| pK19mobsacB ΔC-T ilvN | pK18mobsacB carrying a truncated ilvN gene (shortened by 249 bp) | This work |
| Oligonucleotides | ||
| P1 | 5′-CCCAAGCTTGCTGTTTCCAGATGACCAACC-3′ | Primer for deletion of ΔC-T ilvN |
| P2 | 5′-GGCGATAGTGGTCTCTTCATCAAGTCGCACGACTTTGAGC-3′ | Primer for deletion of ΔC-T ilvN; crossover overlap underlined |
| P3 | 5′-GAAGAGACCACTATCGCCACAGCAATTAATCTGATTGC-3′ | Primer for deletion of ΔC-T ilvN; crossover overlap underlined |
| P4 | 5′-CGCGGATCCCGTTCAGGTTTGGCTCGATG-3′ | Primer for deletion of ΔC-T ilvN and to verify ilvB deletion |
| PD1 | 5′-CCAAGATGGCTAATTCTGACGTCACC-3′ | Primer to verify ΔC-T ilvN deletion |
| PD2 | 5′-GACTAGTCACATTTATGCAGCAGGTGC-3′ | Primer to verify ΔC-T ilvN deletion |
| PilvB | 5′-GCAACAGACATCTGTCGC-3′ | Primer to verify ilvB deletion |
DNA preparation and transformation.
The isolation of plasmids from E. coli was performed as described previously (13). Plasmid DNA transfer into C. glutamicum was carried out by electroporation, and the recombinant strains were selected on LB brain heart infusion agar plates containing kanamycin (50 μg ml−1) (55). The isolation of chromosomal DNA from C. glutamicum was performed as described previously (13). Electroporation of E. coli was carried out with competent cells according to the method of Dower et al. (9).
Culture conditions.
E. coli was grown aerobically in 2× tryptone-yeast extract (TY) complex medium (43) at 37°C as 50-ml cultures in 500-ml baffled Erlenmeyer flasks on a rotary shaker at 120 rpm. Precultures of WT C. glutamicum and C. glutamicum ΔC-T ilvN were grown in 2× TY medium. C. glutamicum DM1729, C. glutamicum DM1933, and their derivatives were grown in 3.7% (wt/vol) brain heart infusion medium (Merck). For amino acid fermentations in shake flasks, the cells of an overnight preculture were washed with 0.9% (wt/vol) NaCl and inoculated into CGXII minimal medium (12) containing 4% (wt/vol) glucose to give an initial optical density at 600 nm (OD600) of about 1. As indicated in Results, 0.5% (wt/wt) corn steep liquor (CSL; Roquette) or l-valine, l-isoleucine, and/or l-leucine (2 mM each) were added to the medium. C. glutamicum was grown aerobically at 30°C in 50-ml cultures in 500-ml baffled Erlenmeyer flasks on a rotary shaker at 120 rpm. Batch fermentations were performed at 30°C in 200-ml cultures in a fedbatch pro fermentation system from DASGIP (Jülich, Germany). The fermentation conditions for aeration and pH control were described by Blombach et al. (4).
Construction of C. glutamicum ΔC-T ilvN and C. glutamicum ΔilvB.
Chromosomal inactivation of the C-terminal domain of IlvN in C. glutamicum was performed using crossover PCR and the suicide vector pK19mobsacB. DNA fragments were generated using the primer pairs P1/P2 and P3/P4, respectively. The two fragments were purified, mixed in equal amounts, and subjected to crossover PCR using primers P1 and P4. The resulting fusion product (containing the ilvN gene shortened by 249 bp) was ligated into BamHI/HindIII-restricted plasmid pK19mobsacB and transformed into E. coli. After isolation and sequencing (MWG Biotech), the recombinant plasmid was electroporated into WT C. glutamicum, C. glutamicum DM1729, and C. glutamicum DM1933. By application of the method described by Schäfer et al. (44), the intact chromosomal ilvN gene was replaced by the truncated ilvN gene via homologous recombination (double crossover). The screening of the ilvN mutants was done on 2× TY agar plates containing 10% (wt/vol) sucrose. The replacement at the chromosomal locus was verified by PCR using primers PD1/PD2.
Inactivation of the chromosomal ilvB gene in C. glutamicum DM1729 and C. glutamicum DM1933 was performed as described previously for WT C. glutamicum ΔilvB (32), using the suicide vector pK19mobsacB ΔilvB. The deletion at the chromosomal locus was verified by PCR using primers PilvB/P4.
Analytical methods.
For quantification of substrate consumption and product formation, 1-ml samples were taken from the cultures and centrifuged at 15,000 × g (10 min), and the supernatant was used for determination of amino acid, glucose, and/or organic acid concentrations in the culture fluid. The amino acid concentrations were determined by reversed-phase high-pressure liquid chromatography as described previously (3). Glucose, acetate, and l-lactate concentrations were determined by enzymatic tests (Roche Diagnostics). The pyruvate concentrations were determined enzymatically according to Bergmeyer (2). α-Ketobutyrate concentrations were determined by reversed-phase high-pressure liquid chromatography with fluorimetric detection (excitation at 361 nm; emission at 448 nm) after precolumn derivatization with 1,2-diamino-4,5-dimethoxybenzene (18). Separation was carried out at 40°C on a Multohyp octadecyl silane column (particle size, 5 μm; 125 by 4 mm) (CS-Chromatographie, Langerwehe, Germany). The elution buffer consisted of a polar phase (water) and a nonpolar phase (methanol). Quantification was done by calculation of the concentration using an internal standard (α-ketovalerate at 100 μM) and by a five-point-calibration curve for α-ketobutyrate.
Online analysis of the oxygen and carbon dioxide contents of the exhaust gas was performed using the GA4 gas analyzer from DASGIP (Jülich, Germany). The carbon evolution rate [CER; given in moles/(liters·hours)] was determined by using the following equation:
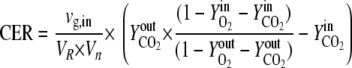 |
Vn is the mole volume of the ideal gas (liters/moles) at standard conditions, VR is the working volume of the bioreactor (liters), vg,in is the volumetric inlet airflow (liters/hours) at standard conditions, and 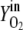 ,
, 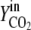 ,
, 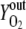 , and
, and 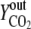 are molecular fractions of oxygen and carbon dioxide in the inlet and outlet air, respectively. The total carbon dioxide concentration (moles/liters) was calculated by integration of the CER over the fermentation time.
are molecular fractions of oxygen and carbon dioxide in the inlet and outlet air, respectively. The total carbon dioxide concentration (moles/liters) was calculated by integration of the CER over the fermentation time.
Determination of AHAS activities and kinetic parameters.
The standard assay for determination of AHAS activities was carried out using the colorimetric single-point method (26). C. glutamicum cells were cultivated in minimal medium containing 4% (wt/vol) glucose to an OD600 of about 5 and were harvested by centrifugation (4,200 × g for 10 min at 4°C). The cells were washed three times with ice-cold 2% (wt/vol) KCl at 4°C, resuspended in disruption buffer (100 mM potassium phosphate buffer [pH 7.3] containing 0.5 mM dithiothreitol and 20% [vol/vol] glycerol), and disrupted with a RyboLyser at 4°C. The reaction mixture (5 ml) contained 100 mM potassium phosphate buffer (pH 7.3), 10 mM MgCl2, 100 μM flavin adenine dinucleotide, and 50 mM pyruvate. The reaction was started by adding 500 μl of diluted cell extract and was stopped by adding 100 μl of 50% (vol/vol) H2SO4 to 1 ml of the reaction mixture. Then, the assay solution was incubated for 30 min at 37°C to allow the conversion of α-acetolactate to acetoin. The concentration of acetoin was determined by the method of Westerfeld (56). The protein concentration was quantified with the BCA protein assay (Pierce) with bovine serum albumin as the standard. Assays were linear over time and proportional to the protein concentration.
For the determination of the Michaelis-Menten constant (Km,P) and the maximal reaction rate (Vmax,P) for pyruvate, 10 pyruvate concentrations ranging from 2.5 to 60 mM were used, and the formation of α-acetolactate was monitored as described above. For the determination of Km,K and Vmax,K for α-ketobutyrate, nine α-ketobutyrate concentrations ranging from 1 to 40 mM at a constant pyruvate concentration (100 mM) were used, and the decrease of α-ketobutyrate was measured as described above. Km and Vmax values were calculated using the Hanes-Wilkinson plot. One micromole of α-acetolactate formed or 1 μmol α-ketobutyrate converted per milligram of protein per minute corresponds to one unit.
RNA preparation and transcriptome analysis.
For RNA isolation, C. glutamicum DM1729 and DM1729 ΔilvB were grown in minimal medium containing 4% (wt/vol) glucose with l-valine, l-isoleucine, and l-leucine (2 mM each) harvested in the exponential growth phase (OD600 of about 20) and treated with 1 volume of ice-cold killing buffer (20 mM Tris-HCl, pH 8.0, 20 mM NaN3, 5 mM MgCl2). The isolation procedure was performed as described previously (45), and aliquots of the RNA were stored at −70°C until use.
DNA microarray analysis, cDNA synthesis, fragmentation, and biotin labeling were carried out as described previously for samples of prokaryotes in the Affymetrix technical support manual (http://www.affymetrix.com/support/technical/manual/expression_manual.affx/). Labeled cDNA samples were hybridized to Affymetrix GeneChip Corynea520112F genome arrays (custom-specific design). This array consists of 3,571 probe sets which can be divided in genes and hypothetical open reading frames (3,221), intergenic probe sets (305), and control probe sets (45). Hybridized arrays were stained with streptavidin-phycoerythrin using the Affymetrix Fluidics station and scanned. The experiment was designed to minimize both false-positive and false-negative results for expressed genes. Two biological and two technical replicates were performed for the analysis of C. glutamicum DM1729 and DM1729 ΔilvB. Statistical expression analysis was performed with Genedata Expressionist 5.0 software on the probe-level data from Affymetrix's CEL files condensed with the MAS 5.0 algorithm. The data quality P value threshold was set to 0.05. To test for significant differences in expression between the strains, one-way analysis of variance was performed at a significance level of 0.001; thus, for every 1,000 genes tested, only one false positive would be expected.
RESULTS
Impact of IlvN modification on growth of C. glutamicum and kinetic properties of the AHAS.
For the E. coli AHAS III, it has been shown that deletion of the C-terminal 80 amino acids in the regulatory subunit led to a functional enzyme released from feedback inhibition by l-valine (30). To test for a similar effect, we deleted the last 249 bp of ilvN in WT C. glutamicum, resulting in C. glutamicum ΔC-T ilvN, and studied the effect of the IlvN modification on growth and AHAS properties. For comparative characterization of growth, we performed shake flask cultivations with WT C. glutamicum and C. glutamicum ΔC-T ilvN in minimal medium containing 4% (wt/vol) glucose with or without different combinations of the BCAAs l-valine, l-isoleucine, and l-leucine (2 mM each) (Fig. 1). Under all conditions tested, WT C. glutamicum showed a growth rate of 0.30 h−1 and reached a final OD600 of about 55. Figure 1 shows a representative growth curve of WT C. glutamicum in minimal medium containing glucose. C. glutamicum ΔC-T ilvN also grew under all conditions to a final OD600 of about 55. However, except when all three BCAAs were added to the medium, the growth rates of the mutant were lower than those of WT C. glutamicum. In minimal medium with glucose, C. glutamicum ΔC-T ilvN grew with a growth rate of 0.22 h−1, the addition of l-leucine resulted in a decreased growth rate of 0.19 h−1, and the addition of l-isoleucine resulted in biphasic growth, with growth rates of 0.15 h−1 in the first and 0.27 h−1 in the second exponential growth phase (Fig. 1). After supplementation of l-valine, an increased growth rate of 0.27 h−1 was observed.
FIG. 1.

Growth of WT C. glutamicum on CGXII medium containing glucose (4%) (+) and C. glutamicum ΔC-T ilvN on CGXII medium containing glucose (4%) (▵) or with 2 mM l-isoleucine (▴), 2 mM l-leucine (○), 2 mM l-valine (•), or all three amino acids (2 mM each) (▪).
The observation that growth of C. glutamicum ΔC-T ilvN was impaired under certain conditions prompted us to compare selected kinetic parameters of AHAS ΔC-T ilvN and the WT AHAS (Table 2). For this purpose, cells were grown in minimal medium with glucose, and the kinetic parameters of the enzyme were determined in crude extracts. AHAS ΔC-T ilvN showed an about twofold-higher affinity for pyruvate (Km,P, 4.7 mM versus 7.8 mM) and an about fourfold-lower Vmax,P (22.3 mU/mg versus 77.6 mU/mg) than that of the WT AHAS. With α-ketobutyrate as the substrate, AHAS ΔC-T ilvN exhibited a slightly higher Km,K (6.9 mM) and an about twofold-lower Vmax,K (56.8 mU/mg) than that of the WT enzyme (Km,K, 5.6 mM; Vmax,K, 113.9 mU/mg). In contrast to that of the WT AHAS, the specific activity of the modified AHAS was not affected by the presence of 10 mM l-valine, l-isoleucine, l-leucine, or all three BCAAs (10 mM each), showing that the C-terminal domain of IlvN in C. glutamicum is responsible for the feedback inhibition by the BCAAs.
TABLE 2.
Km,P, Km,K, Vmax,P, Vmax,K, and the residual activities in the presence of 10 mM l-valine, l-isoleucine, l-leucine, or all three BCAAs (10 mM each) of the WT AHAS or the modified AHAS ΔC-T ilvN
| Enzyme | Values for indicated substrate
|
Residual activity in the presence of 10 mM inhibitor(s) (%)a
|
||||||
|---|---|---|---|---|---|---|---|---|
| Pyruvate + pyruvate
|
α-Ketobutyrate + pyruvate
|
|||||||
| Km,P (mM) | Vmax,P (mU/mg) | Km,K (mM) | Vmax,K (mU/mg) | l-Valine | l-Isoleucine | l-Leucine | All three BCAAs | |
| Wild-type AHAS | 7.8 | 77.6 | 5.6 | 113.9 | 50 | 54 | 65 | 46 |
| AHAS ΔC-T ilvN | 4.7 | 22.3 | 6.9 | 56.8 | 104 | 104 | 104 | 107 |
Activities were measured using 50 mM pyruvate as the substrate. A value of 100% corresponds to 73.5 mU/mg for the WT AHAS and 22.1 mU/mg for AHAS ΔC-T ilvN.
IlvN modification improves l-lysine production with C. glutamicum.
The assumption that the lower specific activity of the AHAS ΔC-T ilvN may lead to a reduction in the carbon flux toward the BCAAs, in combination with the previous findings that l-leucine limitation or a l-leucine auxotrophy seems to be beneficial for l-lysine biosynthesis (see Introduction), prompted us to test the relevance of the ΔC-T ilvN mutation for l-lysine production by C. glutamicum. For this purpose, we introduced the ΔC-T ilvN mutation into the chromosomal ilvN locus of the two l-lysine producers C. glutamicum DM1729 and C. glutamicum DM1933, resulting in C. glutamicum DM1729 ΔC-T ilvN and C. glutamicum DM1933 ΔC-T ilvN, and performed shake flask fermentations with the mutants and the parental strains in minimal medium containing glucose (Table 3). C. glutamicum DM1729 showed a growth rate of 0.25 h−1, reached a final OD600 of 43, and accumulated 21.0 mM l-lysine. C. glutamicum DM1729 ΔC-T ilvN grew with a lower growth rate (0.17 h−1) to about the same final OD600; however, it produced 43% more l-lysine (29.7 mM). In the presence of l-valine, l-isoleucine, l-leucine, and 4% glucose, both C. glutamicum DM1729 and C. glutamicum DM1729 ΔC-T ilvN grew with identical growth rates of 0.24 h−1 to OD600 values of 31 and produced within 48 h about 25 mM and 29 mM l-lysine, respectively (Table 3). C. glutamicum DM1933 ΔC-T ilvN was unable to grow in minimal medium with glucose; therefore, we added 0.5% (wt/vol) CSL to the medium. Under these conditions, C. glutamicum DM1933 ΔC-T ilvN grew with a lower growth rate (0.19 h−1) than strain DM1933; however, the mutant produced 36% more l-lysine (50.5 mM versus 37.0 mM) (Table 3).
TABLE 3.
Final OD600 values, growth rates, and l-lysine concentrations of C. glutamicum DM1729, C. glutamicum DM1933, and recombinant derivatives grown in shake flasks in minimal medium containing 4% glucose with or without different supplements after 48 ha
| C. glutamicum strain | Supplement(s) | Final OD600 | μ (h−1) | Concn of l-lysine (mM) |
|---|---|---|---|---|
| DM1729 | 43 ± 4 | 0.25 ± 0.01 | 21.0 ± 0.9 | |
| DM1729 ΔC-T ilvN | 44 ± 2 | 0.17 ± 0.01 | 29.7 ± 2.5 | |
| DM1729 | 2 mM l-valine, l-isoleucine, and l-leucine | 31 ± 4 | 0.24 ± 0.01 | 24.7 ± 0.6 |
| DM1729 ΔC-T ilvN | 2 mM l-valine, l-isoleucine, and l-leucine | 31 ± 2 | 0.24 ± 0.01 | 29.0 ± 1.0 |
| DM1933 | 40 ± 4 | 0.23 ± 0.01 | 32.6 ± 2.3 | |
| DM1933 ΔC-T ilvN | NGb | |||
| DM1933 | 0.5% (wt/vol) CSL | 51 ± 2 | 0.25 ± 0.03 | 37.0 ± 2.0 |
| DM1933 ΔC-T ilvN | 0.5% (wt/vol) CSL | 41 ± 2 | 0.19 ± 0.01 | 50.5 ± 0.4 |
| DM1729 ΔilvB | 2 mM l-valine, l-isoleucine, and l-leucine | 21 ± 3 | 0.20 ± 0.01 | 51.3 ± 1.3 |
| DM1933 | 2 mM l-valine, l-isoleucine, and l-leucine | 34 ± 2 | 0.19 ± 0.01 | 40.5 ± 1.3 |
| DM1933 ΔilvB | 2 mM l-valine, l-isoleucine, and l-leucine | 14 ± 3 | 0.18 ± 0.01 | 62.8 ± 4.2 |
All values are means ± standard deviations of at least three independent experiments.
NG, no growth.
Inactivation of the AHAS further improves l-lysine production with C. glutamicum.
The results described above led us conclude that increased l-lysine formation might be linked to decreased AHAS activity. To test this hypothesis, we inactivated the AHAS completely by deletion of ilvB in C. glutamicum DM1729 and DM1933, resulting in C. glutamicum DM1729 ΔilvB and DM1933 ΔilvB. As expected, both of these mutants showed no AHAS activity (i.e., <0.2 mU [mg protein]−1) and were unable to grow in minimal medium unless supplemented with l-valine, l-isoleucine, and l-leucine (data not shown). In minimal medium containing all three amino acids (2 mM each) and 4% glucose, C. glutamicum DM1729 grew with a growth rate of 0.24 h−1 to a maximal OD600 of 43, which then decreased to an OD600 of 31 after 48 h, and produced 24.7 mM l-lysine (Fig. 2). C. glutamicum DM1729 ΔilvB showed a lower growth rate of 0.20 h−1 and reached a maximal OD600 of 43, which rapidly dropped after complete consumption of the glucose to a final value of 21 after 48 h. However, C. glutamicum DM1729 ΔilvB produced more than twice as much l-lysine as the parental strain (51.3 mM versus 24.7 mM). For C. glutamicum DM1933, inactivation of the AHAS also increased l-lysine formation, although to a somewhat lesser extent of 55% (62.8 mM versus 40.5 mM) (Table 3). In glucose minimal medium containing 0.5% (wt/vol) CSL instead of 2 mM l-valine, l-isoleucine, and l-leucine, C. glutamicum DM1933 ΔilvB grew only to an OD600 of about 6 (data not shown), indicating that 0.5% of CSL cannot completely substitute the three BCAAs.
FIG. 2.

Growth, substrate consumption, and product accumulation during a representative shake flask batch cultivation of C. glutamicum DM1729 (A) and C. glutamicum DM1729 ΔilvB (B) on CGXII medium containing glucose (4%) and l-valine, l-isoleucine, and l-leucine (2 mM each). ⋄, growth; ▪, glucose; ×, l-lysine. Three independent fermentations were performed, all three showing comparable results.
To test for suitability of DM1729 ΔilvB for improved l-valine production on a larger scale, we performed comparative batch cultivations in a parallel fermentation system. These fermentations with C. glutamicum DM1729 and DM1729 ΔilvB were carried out in CGXII medium containing 0.5% (wt/vol) CSL, 7% (wt/vol) glucose, and l-valine, l-isoleucine, and l-leucine (4 mM each). Growth, substrate consumption, product and by-product accumulation, and carbon dioxide formation were monitored throughout the course of the experiment. Under these conditions, both strains grew with identical growth rates of 0.19 h−1 to maximal OD600 values of about 61 (DM1729) and 64 (DM1729 ΔilvB). As shown in Table 4, C. glutamicum DM1729 ΔilvB showed about 85%-higher l-lysine formation, an 85%-higher substrate-specific product yield (YP/S), and 10%-higher productivity than the parental strain DM1729. Neither strain secreted pyruvate, l-lactate, or acetate; however, C. glutamicum DM1729 ΔilvB accumulated about 10 mM l-glutamate. Additionally, C. glutamicum DM1729 ΔilvB showed a substrate-specific carbon dioxide yield (YCO2/S) of 0.424 mol C/mol C, which is about 20% lower than the YCO2/S of C. glutamicum DM1729 (0.526 mol C/mol C) (Table 4). This result indicates that improved l-lysine formation of C. glutamicum DM1729 ΔilvB is due to reduced carbon dioxide formation.
TABLE 4.
Maximal ODs, growth rates, l-lysine concentrations, substrate-specific product yields (YP/S), substrate-specific carbon dioxide yields (YCO2/S), and productivities of C. glutamicum DM1729 and DM1729 ΔilvBa
| C. glutamicum strain | Maximal OD600 | μ (h−1) | Concn of l-lysine (mM) | YP/S (mol C/mol C) | YCO2/S (mol C/mol C) | Productivity [mmol/(g CDW × h)]b |
|---|---|---|---|---|---|---|
| DM1729 | 61 | 0.19 | 38 | 0.098 | 0.526 | 0.074 |
| DM1729 ΔilvB | 64 | 0.19 | 70 | 0.180 | 0.424 | 0.082 |
Two independent batch fermentations were performed, both showing comparable results. C. glutamicum DM1729 and DM1729 ΔilvB were grown in batch culture in minimal medium containing 7% glucose, 0.5% CSL, and 4 mM each of l-valine, l-isoleucine, and l-leucine.
Cell dry weight (CDW) was calculated from the OD600, using a ratio of 0.3 g CDW liter−1 per OD600 (4).
Comparison of the transcriptomes of C. glutamicum DM1729 and C. glutamicum DM1729 ΔilvB.
The finding that decreased or abolished AHAS activity leads to increased l-lysine formation might be due to a change in transcription of relevant genes in response to a diminished or abolished flux toward the BCAAs. To compare the genome-wide transcriptional profiles of C. glutamicum DM1729 ΔilvB and C. glutamicum DM1729, both strains were cultivated in minimal medium with glucose plus l-valine, l-isoleucine, and l-leucine, total RNA was prepared, and labeled cDNA samples were hybridized to Affymetrix GeneChip arrays. The analysis revealed 49 genes with different mRNA levels (more than twofold) in C. glutamicum DM1729 ΔilvB compared to the parental strain C. glutamicum DM1729 (Table 5). Among these, 18 genes showed a higher mRNA level, including genes of acetate metabolism (aceA, aceB, pta, ack) and two genes annotated as predicted transcriptional regulators (cg2320 and cg3303), as well as ilvN (about sevenfold) and ilvC (about threefold). The group of genes with significantly lower mRNA levels consisted of 31 candidates, including genes of l-leucine (leuC), l-arginine (argBCDF), and l-methionine (metE) biosynthesis, as well as several genes encoding enzymes involved in Fe-S cluster assembly (sufBCDR) and iron acquisition (cg0771, cg0924, cg0926, cg0927, cg0928, cg2445, and cg3404). Furthermore, the genes for a putative transcriptional regulator (cg0156), for SufR (probable regulator of the suf operon), for the l-arginine repressor ArgR, and for the l-leucine and l-tryptophan biosynthesis regulator LtbR showed lower mRNA levels in C. glutamicum DM1729 ΔilvB. Interestingly, no significant changes were observed in the mRNA levels of genes encoding enzymes involved in the PPP, at the pyruvate-oxaloacetate node (e.g., pyruvate carboxylase, PEP carboxylase, PEP carboxykinase, and PDHC) or in l-lysine biosynthesis.
TABLE 5.
Comparison of gene expression in C. glutamicum DM1729 ΔilvB to that in DM1729
| Locus tag | mRNA ratio | Gene | Function(s)a |
|---|---|---|---|
| Amino acid biosynthesis | |||
| cg1290 | 0.40 | metE | Homocysteine methyltransferase |
| cg1436 | 7.43 | ilvN | AHAS, small subunit |
| cg1437 | 2.68 | ilvC | Acetohydroxy acid isomeroreductase |
| cg1487 | 0.35 | leuC | 3-Isopropylmalate dehydratase, large subunit |
| cg1580 | 0.47 | argC | N-Acetyl-gamma-glutamyl-phosphate reductase |
| cg1582 | 0.38 | argB | Acetylglutamate kinase |
| cg1583 | 0.43 | argD | Acetylornithine aminotransferase |
| cg1584 | 0.32 | argF | Ornithine carbamoyltransferase |
| cg1739 | 0.39 | Glutamine amidotransferase domain | |
| Central metabolism | |||
| cg2559 | 3.25 | aceB | Malate synthase |
| cg2560 | 2.30 | aceA | Isocitrate lyase |
| cg3047 | 2.47 | ackA | Acetate kinase |
| cg3048 | 2.27 | pta | Phosphotransacetylase |
| Transcriptional | |||
| cg0156 | 0.46 | Bacterial regulatory protein, Crp family | |
| cg1486 | 0.38 | ltbR | l-Leucine and l-tryptophan biosynthesis regulator, IclR family |
| cg1585 | 0.27 | argR | l-Arginine repressor |
| cg1765 | 0.44 | sufR | Predicted transcriptional regulator for the suf operon |
| cg2320 | 2.20 | Predicted transcriptional regulator | |
| cg3303 | 2.35 | Transcriptional regulator, PadR-like family | |
| Iron metabolism | |||
| cg0771 | 0.34 | DtxR/iron-regulated lipoprotein | |
| cg0924 | 0.20 | ABC-type cobalamin/Fe3+-siderophore transport system | |
| cg0926 | 0.39 | ABC-type cobalamin/Fe3+-siderophore transport system | |
| cg0927 | 0.29 | ABC-type cobalamin/Fe3+-siderophore transport system | |
| cg0928 | 0.37 | ABC-type cobalamin/Fe3+-siderophore transport system | |
| cg1762 | 0.46 | sufC | Iron-regulated ABC transporter, ATPase subunit |
| cg1763 | 0.35 | sufD | Component of an uncharacterized iron-regulated ABC-type transporter |
| cg1764 | 0.35 | sufB | Component of an uncharacterized iron-regulated ABC-type transporter |
| cg2445 | 0.45 | Probable heme oxygenase | |
| cg3404 | 0.25 | ABC-type cobalamin/Fe3+-siderophore transport system | |
| Others | |||
| cg0277 | 0.49 | Sodium sulfate symporter transmembrane component | |
| cg0503 | 0.45 | Probable 3-dehydroquinate dehydratase | |
| cg0569 | 3.30 | Cation-transporting ATPase | |
| cg0607 | 2.95 | Hypothetical secreted protein | |
| cg0963 | 2.38 | Hypothetical protein | |
| cg1043 | 2.09 | Thiol-disulfide isomerase and thioredoxins | |
| cg1049 | 2.15 | Enoyl-coenzyme A hydratase/carnithine racemase | |
| cg1085 | 2.68 | Hypothetical protein predicted by Glimmer criteria | |
| cg1087 | 2.21 | Putative membrane protein | |
| cg1090 | 2.71 | Probable γ-glutamyltranspeptidase | |
| cg1229 | 0.45 | ABC-type cobalt transport system, permease component CbiQ | |
| cg1279 | 0.44 | Putative secreted protein | |
| cg1365 | 0.47 | atpH | H+-ATPase, δ subunit |
| cg1367 | 0.47 | atpG | ATP synthase, γ subunit |
| cg1419 | 0.49 | Putative Na+-dependent transporter | |
| cg1998 | 0.49 | cglIIR | Restriction endonuclease CGLIIR protein |
| cg2095 | 0.42 | Putative membrane protein | |
| cg2438 | 2.39 | Hypothetical protein predicted by Glimmer criteria | |
| cg2477 | 2.13 | Conserved hypothetical protein | |
| cg3256 | 0.43 | Alkanal monooxygenase, alpha chain |
See reference 22.
DISCUSSION
In the present work, we engineered a modified AHAS by deletion of the C-terminal domain in the regulatory subunit IlvN. The newly constructed enzyme showed a twofold-lower Km for the substrate pyruvate, a slightly higher Km for the substrate α-ketobutyrate, and a lower Vmax for both pyruvate and pyruvate plus α-ketobutyrate. Furthermore, the modified AHAS was completely insensitive against the inhibitors l-valine, l-isoleucine, and l-leucine. These results indicate (i) that the C-terminal domain of the regulatory subunit IlvN of the C. glutamicum AHAS is responsible for inhibitor (l-valine, l-leucine, and l-valine) binding and/or inhibitor response and (ii) that the C-terminal domain is essential for maximal AHAS activity. The former result is in accordance with results obtained with the E. coli AHAS III, namely, with the findings that an AHAS with a deletion of 80 amino acids from the C terminus of the regulatory IlvH subunit is feedback resistant to l-valine and also shows an about twofold-higher affinity for the substrate pyruvate (30). However, in contrast to the C. glutamicum ΔC-T ilvN AHAS, the truncated AHAS III from E. coli even showed 40%-higher activity than the original AHAS III, indicating that in this enzyme, the C terminus of the regulatory subunit is not involved in recognition and activation of the catalytic subunit IlvI (30).
As an analogy to the above-mentioned findings with the E. coli AHAS III, the C. glutamicum ΔC-T ilvN AHAS originally was expected to be feedback resistant and also highly active. Since the AHAS reaction is one of the bottlenecks for efficient l-valine production (3, 5, 14), we therefore originally intended to use the ΔC-T ilvN AHAS for improvement of l-valine production strains (3, 5). Having tested the kinetic properties and having found the nearly fourfold-reduced Vmax,P values of the mutated AHAS, we realized that chromosomal introduction of the AHAS ΔC-T ilvN into l-valine producer strains most probably does not lead to improved l-valine production. It might be that multicopy introduction (plasmid-bound introduction) of the AHAS ΔC-T ilvN allele will lead to a higher carbon flux toward l-valine and/or the other BCAAs and their precursors; however, so far we did not test this possibility. Instead, we tested the newly constructed AHAS for its effect on l-lysine production. Chromosomal introduction of the AHAS ΔC-T ilvN into two l-lysine-producing strains of C. glutamicum and analysis of the resulting strains led us to conclude that a decrease in AHAS activity causes an increase in l-lysine formation. This conclusion is corroborated by the findings that (i) addition of the AHAS inhibitors l-valine, l-isoleucine, and l-leucine to the medium also resulted in increased l-lysine formation by both parental strains, C. glutamicum DM1729 (24.7 mM versus 21.0 mM) and DM1933 (40.5 mM versus 32.6 mM) (Table 3), and (ii) deletion of the ilvB gene (encoding the catalytic subunit of AHAS) and thus complete inactivation of the AHAS in both l-lysine producers resulted in even higher l-lysine production compared to only partial inactivation in C. glutamicum AHAS ΔC-T ilvN. Thus, this work identified the AHAS as a novel and promising target to improve l-lysine production with C. glutamicum.
The positive effect of decreased/inactivated AHAS activity for l-lysine production might at least partially be due to a reduced or abolished carbon flux toward the BCAAs and thus to an increase of the intracellular pyruvate availability. Pyruvate and oxaloacetate are central metabolic precursors for l-lysine formation, and their supply should be balanced for optimal l-lysine production. Several previous studies already indicated positive effects of increasing the pyruvate precursor supply on l-lysine production. Shiio et al. (49, 50) found that undefined mutants of C. glutamicum (formerly “Brevibacterium flavum”) with either low citrate synthase or PDHC activity showed higher l-lysine production than their respective parental strains. Moreover, we recently showed that complete inactivation of the PDHC in C. glutamicum DM1729 led to improved l-lysine production (4). However, the PDHC-deficient C. glutamicum DM1729 BB1 did not accumulate as much l-lysine as C. glutamicum DM1729 ΔilvB (30.0 mM versus 51.3 mM) and even excreted pyruvate and l-alanine (4). On the one hand, these observations indicate that l-lysine production in C. glutamicum DM1729 BB1 is not limited by pyruvate; on the other hand, they suggest that the positive effect of decreased/inactivated AHAS activity for l-lysine production cannot be explained exclusively by an increased pyruvate supply.
Hayashi et al. (19) recently showed that introduction of a leuC mutation, and thus introduction of a partial l-leucine auxotrophy in C. glutamicum, leads to an increased (14%) l-lysine production. The authors also reported that in the leuC mutant (strain ADL-3), many different amino acid biosynthetic genes were upregulated, including the lysC-asd operon (encoding the l-lysine biosynthetic enzymes aspartate kinase and aspartate semialdehyde dehydrogenase, respectively), and they speculated that increased expression of lysC-asd is responsible for increased l-lysine production (19). As an analogy to these observations, we speculated that the positive effect of decreased or abolished AHAS activity (and thus, reduced or abolished carbon flux toward the BCAAs) on l-lysine formation might also at least partially be due to a change in transcription of relevant genes; e.g., of those coding for enzymes involved in the l-lysine biosynthetic pathway. To examine this possibility and to identify candidate genes, we carried out comparative transcriptome analysis of C. glutamicum DM1729 and C. glutamicum DM1729 ΔilvB. This analysis revealed 49 genes with at least twofold-altered mRNA levels in C. glutamicum DM1729 ΔilvB. However, in contrast to the leuC mutant C. glutamicum ADL-3, C. glutamicum DM1729 ΔilvB showed no significant changes in the mRNA levels of l-lysine biosynthetic pathway genes. Also in contrast to the leuC mutant, C. glutamicum DM1729 ΔilvB showed different mRNA levels of some central metabolic pathway genes, some (putative) regulator genes, and genes involved in iron metabolism (Table 5). These results clearly show that effects other than those observed in the leuC mutant C. glutamicum ADL-3 must be the reason for improved l-lysine production by C. glutamicum DM1729 ΔilvB.
None of the genes with different mRNA levels in C. glutamicum DM1729 ΔilvB is directly linked to the pathways from glucose to l-lysine, to NADPH supply, or to known expression regulation of l-lysine synthesis. It might be speculated that higher expression of the isocitrate lyase and malate synthase genes (aceA and aceB, respectively) (Table 5) may lead to a higher glyoxylate cycle flux and thus to a higher oxaloacetate availability for l-lysine formation. The 2.3- and 3.25-fold increases in aceA and aceB mRNA levels are rather low in comparison to the 28.6- and 8.4-fold increases in aceA- and aceB-specific mRNA observed when C. glutamicum cells were grown on acetate instead of on glucose (16). However, a higher glyoxylate cycle activity and thus an at least partial bypass of the CO2-releasing reactions of the tricarboxylic acid cycle (i.e., isocitrate dehydrogenase and 2-oxoglutarate dehydrogenase complex reactions) are in agreement with the low substrate-specific CO2 yield of C. glutamicum DM1729 ΔilvB compared to that of the parental strain DM1729. In fact, the lower substrate-specific CO2 yield (YCO2/S lowered by 0.102 mol C/mol C) approximately corresponds to the increase in substrate-specific product yield (0.082 mol C/mol C). The reduced respiration in combination with a lower tricarboxylic acid cycle activity possibly leads to more balanced oxaloacetate and pyruvate supplies for l-lysine production.
It should be kept in mind that all mRNA changes observed in C. glutamicum DM1729 ΔilvB might be direct or indirect effects in response to abolished AHAS activity but might not be directly connected to l-lysine productivity. Further studies are necessary to mechanistically clarify why inactivation of ilvB in C. glutamicum obviously leads to different mRNA levels (different levels of expression) of so many genes (Table 5). The higher mRNA levels of the ilvN and ilvC genes can be explained by transcriptional activation of the truncated ilvBNC operon (ilvB deleted) by α-ketobutyrate (10, 32), which probably accumulates in C. glutamicum DM1729 ΔilvB. However, it remains unclear whether α-ketobutyrate triggers expression of any of the other genes listed in Table 5.
Supplementary Material
Acknowledgments
We thank Lothar Eggeling for providing plasmid pK19mobsacB ΔilvB.
The support of the Fachagentur Nachwachsende Rohstoffe of the BMVEL (grant 04NR004/22000404) is gratefully acknowledged.
Footnotes
Published ahead of print on 1 December 2008.
Supplemental material for this article may be found at http://aem.asm.org/.
REFERENCES
- 1.Becker, J., C. Klopprogge, O. Zelder, E. Heinzle, and C. Wittmann. 2005. Amplified expression of fructose 1,6-bisphosphatase in Corynebacterium glutamicum increases in vivo flux through the pentose phosphate pathway and lysine production on different carbon sources. Appl. Environ. Microbiol. 71:8587-8596. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Bergmeyer, H. U. 1983. Methods of enzymatic analysis, 3rd ed., vol. VI, p. 59-66. Verlag Chemie, Weinheim, Germany. [Google Scholar]
- 3.Blombach, B., M. E. Schreiner, J. Holatko, T. Bartek, M. Oldiges, and B. J. Eikmanns. 2007. l-Valine production with pyruvate dehydrogenase complex-deficient Corynebacterium glutamicum. Appl. Environ. Microbiol. 73:2079-2084. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Blombach, B., M. E. Schreiner, M. Moch, M. Oldiges, and B. J. Eikmanns. 2007. Effect of pyruvate dehydrogenase complex deficiency on l-lysine production with Corynebacterium glutamicum. Appl. Microbiol. Biotechnol. 76:615-623. [DOI] [PubMed] [Google Scholar]
- 5.Blombach, B., M. E. Schreiner, T. Bartek, M. Oldiges, and B. J. Eikmanns. 2008. Corynebacterium glutamicum tailored for high-yield l-valine production. Appl. Microbiol. Biotechnol. 79:471-479. [DOI] [PubMed] [Google Scholar]
- 6.Cordes, C., B. Möckel, L. Eggeling, and H. Sahm. 1992. Cloning, organization and functional analysis of ilvA, ilvB and ilvC genes from Corynebacterium glutamicum. Gene 112:113-116. [DOI] [PubMed] [Google Scholar]
- 7.Cremer, J., L. Eggeling, and H. Sahm. 1991. Control of the lysine biosynthesis sequence in Corynebacterium glutamicum as analyzed by overexpression of the individual corresponding genes. Appl. Environ. Microbiol. 57:1746-1752. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.de Graaf, A. A., L. Eggeling, and H. Sahm. 2001. Metabolic engineering for l-lysine production by Corynebacterium glutamicum. Adv. Biochem. Eng. Biotechnol. 73:9-29. [DOI] [PubMed] [Google Scholar]
- 9.Dower, W. J., J. F. Miller, and C. W. Ragsdale. 1988. High efficiency transformation of E. coli by high voltage electroporation. Nucleic Acids Res. 16:6127-6145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Eggeling, I., C. Cordes, L. Eggeling, and H. Sahm. 1987. Regulation of acetohydroxy acid synthase in Corynebacterium glutamicum during fermentation of α-ketobutyrate to l-isoleucine. Appl. Microbiol. Biotechnol. 25:346-351. [Google Scholar]
- 11.Eggeling, L., S. Oberle, and H. Sahm. 1998. Improved l-lysine yield with Corynebacterium glutamicum: use of dapA resulting in increased flux combined with growth limitation. Appl. Microbiol. Biotechnol. 49:24-30. [DOI] [PubMed] [Google Scholar]
- 12.Eikmanns, B. J., M. Metzger, D. Reinscheid, M. Kircher, and H. Sahm. 1991. Amplification of three threonine biosynthesis genes in Corynebacterium glutamicum and its influence on carbon flux in different strains. Appl. Microbiol. Biotechnol. 34:617-622. [DOI] [PubMed] [Google Scholar]
- 13.Eikmanns, B. J., N. Thum-Schmitz, L. Eggeling, K. U. Lüdtke, and H. Sahm. 1994. Nucleotide sequence, expression and transcriptional analysis of the Corynebacterium glutamicum gltA gene encoding citrate synthase. Microbiology 140:1817-1828. [DOI] [PubMed] [Google Scholar]
- 14.Elisáková, V., M. Pátek, J. Holátko, J. Nesvera, D. Leyval, J. L. Goergen, and S. Delaunay. 2005. Feedback-resistant acetohydroxy acid synthase increases valine production in Corynebacterium glutamicum. Appl. Environ. Microbiol. 71:207-213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Georgi, T., D. Rittmann, and V. F. Wendisch. 2005. Lysine and glutamate production by Corynebacterium glutamicum on glucose, fructose and sucrose: roles of malic enzyme and fructose-1,6-bisphosphatase. Metab. Eng. 7:291-301. [DOI] [PubMed] [Google Scholar]
- 16.Gerstmeir, R., V. F. Wendisch, S. Schnicke, H. Ruan, M. Farwick, D. Reinscheid, and B. J. Eikmanns. 2003. Acetate metabolism and its regulation in Corynebacterium glutamicum. J. Biotechnol. 104:99-122. [DOI] [PubMed] [Google Scholar]
- 17.Hanahan, D. 1983. Studies on transformation of Escherichia coli with plasmids. J. Mol. Biol. 166:557-580. [DOI] [PubMed] [Google Scholar]
- 18.Hara, S., Y. Takemori, M. Yamaguchi, and M. Nakamura. 1985. Determination of α-keto acids in serum and urine by high-performance liquid chromatography with fluorescence detection. J. Chromatogr. 344:33-39. [DOI] [PubMed] [Google Scholar]
- 19.Hayashi, M., H. Mizoguchi, J. Ohnishi, S. Mitsuhashi, Y. Yonetani, S. Hashimoto, and M. Ikeda. 2006. A leuC mutation leading to increased l-lysine production and rel-independent global expression changes in Corynebacterium glutamicum. Appl. Microbiol. Biotechnol. 72:783-789. [DOI] [PubMed] [Google Scholar]
- 20.Ikeda, M. 2003. Amino acid production process. Adv. Biochem. Eng. Biotechnol. 79:1-35. [DOI] [PubMed] [Google Scholar]
- 21.Kabus, A., T. Georgi, V. F. Wendisch, and M. Bott. 2007. Expression of the Escherichia coli pntAB genes encoding a membrane-bound transhydrogenase in Corynebacterium glutamicum improves l-lysine. Appl. Microbiol. Biotechnol. 75:47-53. [DOI] [PubMed] [Google Scholar]
- 22.Kalinowski, J., B. Bathe, D. Bartels, N. Bischoff, M. Bott, A. Burkovski, N. Dusch, L. Eggeling, B. J. Eikmanns, L. Gaigalat, A. Goesmann, M. Hartmann, K. Huthmacher, R. Kramer, B. Linke, A. C. McHardy, F. Meyer, B. Möckel, W. Pfefferle, A. Püuhler, D. A. Rey, C. Ruckert, O. Rupp, H. Sahm, V. F. Wendisch, I. Wiegrabe, and A. Tauch. 2003. The complete Corynebacterium glutamicum ATCC 13032 genome sequence and its impact on the production of l-aspartate-derived amino acids and vitamins. J. Biotechnol. 104:5-25. [DOI] [PubMed] [Google Scholar]
- 23.Kaplun, A., M. Vyazmensky, Y. Zherdev, I. Belenky, A. Slutzker, S. Mendel, Z. Barak, D. M. Chipman, and B. Shaanan. 2006. Structure of the regulatory subunit of acetohydroxyacid synthase isoenzyme III from Escherichia coli. J. Mol. Biol. 357:951-963. [DOI] [PubMed] [Google Scholar]
- 24.Kelle, R., T. Hermann, and B. Bathe. 2005. l-Lysine production, p. 465-488. In L. Eggeling and M. Bott (ed.), Handbook of Corynebacterium glutamicum. CRC Press, Boca Raton, FL.
- 25.Leuchtenberger, W., K. Huthmacher, and K. Drauz. 2005. Biotechnological production of amino acids and derivates: current status and prospects. Appl. Microbiol. Biotechnol. 69:1-8. [DOI] [PubMed] [Google Scholar]
- 26.Leyval, D., D. Uy, S. Delaunay, J. L. Goergen, and J. M. Engasser. 2003. Characterisation of the enzyme activities involved in the valine biosynthetic pathway in a valine-producing strain of Corynebacterium glutamicum. J. Biotechnol. 104:241-252. [DOI] [PubMed] [Google Scholar]
- 27.Liebl, W. 2005. Corynebacterium taxonomy, p. 9-34. In L. Eggeling and M. Bott (ed.), Handbook of Corynebacterium glutamicum. CRC Press, Boca Raton, FL.
- 28.Marx, A., A. A. de Graaf, W. Wiechert, L. Eggeling, and H. Sahm. 1996. Determination of the fluxes in the central metabolism of Corynebacterium glutamicum by nuclear magnetic resonance spectroscopy combined with metabolite balancing. Biotechnol. Bioeng. 49:111-129. [DOI] [PubMed] [Google Scholar]
- 29.Marx, A., S. Hans, B. Möckel, B. Bathe, and A. A. de Graaf. 2003. Metabolic phenotype of phosphoglucose isomerase mutants of Corynebacterium glutamicum. J. Biotechnol. 104:185-197. [DOI] [PubMed] [Google Scholar]
- 30.Mendel, S., M. Vinogradov, M. Vyazmensky, D. M. Chipman, and Z. Barak. 2003. The N-terminal domain of the regulatory subunit is sufficient for complete activation of acetohydoxyacid synthase III from Escherichia coli. J. Mol. Biol. 325:275-284. [DOI] [PubMed] [Google Scholar]
- 31.Mitsuhashi, S., M. Hayashi, J. Ohnishi, and M. Ikeda. 2006. Disruption of malate:quinone oxidoreductase increases l-lysine production by Corynebacterium glutamicum. Biosci. Biotechnol. Biochem. 70:2803-2806. [DOI] [PubMed] [Google Scholar]
- 32.Morbach, S., C. Junger, H. Sahm, and L. Eggeling. 2000. Attenuation control of ilvBNC in Corynebacterium glutamicum: evidence of leader peptide formation without the presence of a ribosome binding site. J. Biosci. Bioeng. 90:501-507. [DOI] [PubMed] [Google Scholar]
- 33.Nakayama, K., H. Tanaka, H. Hagino, and S. Kinoshita. 1966. Studies on lysine fermentation. V. Concerted feedback inhibition of aspartokinase and the absence of lysine inhibition on aspartic semialdehyde-pyruvate condensation in Micrococcus glutamicus. Agric. Biol. Chem. 30:611-616. [Google Scholar]
- 34.Ohnishi, J., R. Katahira, S. Mitsuhashi, S. Kakita, and M. Ikeda. 2005. A novel gnd mutation leading to increased l-lysine production in Corynebacterium glutamicum. FEMS Microbiol. Lett. 242:265-274. [DOI] [PubMed] [Google Scholar]
- 35.Pátek, M. 2007. Branched-chain amino acids, p. 129-162. In Microbiology monographs, vol. 5. Springer, New York, NY. [Google Scholar]
- 36.Pátek, M., K. Krumbach, L. Eggeling, and H. Sahm. 1994. Leucine synthesis in Corynebacterium glutamicum: enzyme activities, structure of leuA, and effect of leuA inactivation on lysine synthesis. Appl. Environ. Microbiol. 60:133-140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Peters-Wendisch, P., M. Stolz, H. Etterich, N. Kennerknecht, H. Sahm, and L. Eggeling. 2005. Metabolic engineering of Corynebacterium glutamicum for l-serine production. Appl. Environ. Microbiol. 71:7139-7144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Peters-Wendisch, P. G., B. Schiel, V. F. Wendisch, E. Katsoulidis, B. Mockel, H. Sahm, and B. J. Eikmanns. 2001. Pyruvate carboxylase is a major bottleneck for glutamate and lysine production by Corynebacterium glutamicum. J. Mol. Microbiol. Biotechnol. 3:295-300. [PubMed] [Google Scholar]
- 39.Peters-Wendisch, P., R. Netzer, L. Eggeling, and H. Sahm. 2002. 3-Phosphoglycerate dehydrogenase from Corynebacterium glutamicum: the C-terminal domain is not essential for activity but is required for inhibition by L-serine. Appl. Microbiol. Biotechnol. 60:437-441. [DOI] [PubMed] [Google Scholar]
- 40.Radmacher, E., and L. Eggeling. 2007. The three tricarboxylate synthase activities of Corynebacterium glutamicum and increase of l-lysine synthesis. Appl. Microbiol. Biotechnol. 76:587-595. [DOI] [PubMed] [Google Scholar]
- 41.Riedel, C., D. Rittmann, P. Dangel, B. Möckel, H. Sahm, and B. J. Eikmanns. 2001. Characterization, expression, and inactivation of the phosphoenolpyruvate carboxykinase gene from Corynebacterium glutamicum and significance of the enzyme for growth and amino acid production. J. Mol. Microbiol. Biotechnol. 3:573-583. [PubMed] [Google Scholar]
- 42.Sahm, H., L. Eggeling, and A. A. de Graaf. 2000. Pathway analysis and metabolic engineering in Corynebacterium glutamicum. Biol. Chem. 381:899-910. [DOI] [PubMed] [Google Scholar]
- 43.Sambrook, J., and D. W. Russell. 2001. Molecular cloning: a laboratory manual, 3rd ed. Cold Spring Harbor Laboratory, Cold Spring Harbor, NY.
- 44.Schäfer, A., A. Tauch, W. Jäger, J. Kalinowski, G. Thierbach, and A. Pühler. 1994. Small mobilizable multi-purpose cloning vectors derived from the E. coli plasmids pK18 and pK19: selection of defined deletions in the chromosome of Corynebacterium glutamicum. Gene 145:69-73. [DOI] [PubMed] [Google Scholar]
- 45.Schreiner, M. E., D. Fiur, J. Holátko, M. Pátek, and B. J. Eikmanns. 2005. E1 enzyme of the pyruvate dehydrogenase complex in Corynebacterium glutamicum: molecular analysis of the gene and phylogenetic aspects. J. Bacteriol. 187:6005-6018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Schrumpf, B., A. Schwarzer, J. Kalinowski, A. Pühler, L. Eggeling, and H. Sahm. 1991. A functional split pathway for lysine biosynthesis in Corynebacterium glutamicum. J. Bacteriol. 173:4510-4516. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Schrumpf, B., L. Eggeling, and H. Sahm. 1992. Isolation and prominent characteristics of an l-lysine hyperproducing strain of Corynebacterium glutamicum. Appl. Microbiol. Biotechnol. 37:566-571. [Google Scholar]
- 48.Shiio, I., and R. Miyajima. 1969. Concerted inhibition and its reversal by end products of aspartate kinase in Brevibacterium flavum. J. Biochem. (Tokyo) 5:849-859. [DOI] [PubMed] [Google Scholar]
- 49.Shiio, I., H. Ozaki, and K. Ujigawa-Takeda. 1982. Production of aspartic acid and lysine by citrate synthase mutants of Brevibacterium flavum. Agric. Biol. Chem. 46:101-107. [Google Scholar]
- 50.Shiio, I., Y. Toride, and S. Sugimoto. 1984. Production of lysine by pyruvate dehydrogenase mutants of Brevibacterium flavum. Agric. Biol. Chem. 48:3091-3098. [Google Scholar]
- 51.Sindelar, G., and V. F. Wendisch. 2007. Improving lysine production by Corynebacterium glutamicum through DNA microarray-based identification of novel target genes. Appl. Microbiol. Biotechnol. 76:677-689. [DOI] [PubMed] [Google Scholar]
- 52.Sonntag, K., L. Eggeling, A. A. de Graaf, and H. Sahm. 1993. Flux partitioning in the split pathway of lysine synthesis in Corynebacterium glutamicum: quantification by 13C- and 1H-NMR spectroscopy. Eur. J. Biochem. 213:1325-1331. [DOI] [PubMed] [Google Scholar]
- 53.Takors, R., B. Bathe, M. Rieping, S. Hans, R. Kelle, and K. Huthmacher. 2007. Systems biology for industrial strains and fermentation processes-example: amino acids. J. Biotechnol. 129:181-190. [DOI] [PubMed] [Google Scholar]
- 54.Umbarger, H. E. 1996. Biosynthesis of branched-chain amino acids, p. 442-457. In F. C. Neidhardt, R. Curtiss III, J. L. Ingraham, E. C. C. Lin, K. B. Low, B. Magasanik, W. S. Reznikoff, M. Riley, M. Schaechter, and H. E. Umbarger (ed.), Escherichia coli and Salmonella: cellular and molecular biology, 2nd ed. ASM Press, Washington, DC.
- 55.van der Rest, M. E., C. Lange, and D. Molenaar. 1999. A heat shock following electroporation induces highly efficient transformation of Corynebacterium glutamicum with xenogenic plasmid DNA. Appl. Microbiol. Biotechnol. 52:541-545. [DOI] [PubMed] [Google Scholar]
- 56.Westerfeld, W. W. 1945. A colorimetric detection of blood acetoin. J. Biol. Chem. 161:495-502. [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.