

Abstract
Genomic studies on pathogenic and environmental mycobacteria are of growing interest for understanding of their evolution, distribution, adaptation, and host-pathogen interaction. Since most mycobacteria are slow growers, material from in vitro cultures is usually scarce. The robust mycobacterial cell wall hinders both experimental cell lysis and efficient DNA extraction. Here, we compare elements of several DNA preparation protocols and describe a method that is economical and practical and reliably yields large amounts—usually 10-fold increased compared to earlier protocols—of highly pure genomic DNA for sophisticated downstream applications. This method was optimized for cultures of a variety of pathogenic and environmental mycobacterial species and proven to be suitable for direct mycobacterial DNA extraction from infected insect specimens.
Mycobacterial diseases are a major health concern for humans (i.e., Mycobacterium tuberculosis, M. leprae, M. ulcerans, M. avium, and M. paratuberculosis) (4, 13, 18, 29, 30), livestock (M. bovis and M. avium subsp. paratuberculosis) (1, 3), fisheries (M. marinum) (25), and wildlife (M. avium, M. pinnipedii, M. microti, M. caprae, and other species) (13, 20). Efficient methods for DNA preparation are required both for the identification and genotyping of such pathogens and for population genomics, which is developing into an important tool to study bacterial evolution, virulence, and epidemiology.
Extraction of mycobacterial genomic DNA is especially demanding since (i) many mycobacterial species are among the most extreme slow growers, accounting for small amounts of starting material, and (ii) a robust and waxy cell wall renders mycobacteria difficult to lyse. Published protocols for mycobacterial DNA preparations and commercially available extraction kits are mainly designed for the isolation of small amounts of genomic material suitable for conventional PCR application (2, 7, 9, 11, 14, 15, 23, 24, 27, 28, 33), such as for testing of potentially contaminated milk (6, 8, 17). However, such DNA quantities and qualities are usually not sufficient for more sophisticated molecular analyses.
M. ulcerans, the causative agent of the devastating human skin disease Buruli ulcer, is one of the slowest growers among mycobacterial species, and the development of molecular tools is crucial for studying its transmission and microepidemiology. The objective of this study was to develop an optimized extraction protocol for DNA of both high quantity and quality from scarce material of in vitro-cultivated M. ulcerans disease isolates. We compared elements of several protocols and developed a DNA preparation method that is optimized in each individual step and thus ready to use for virtually all mycobacterial species to yield a maximum of pure genetic material. In addition, we applied the established method to cultures of a variety of pathogenic and environmental mycobacterial species and tested it by isolating DNA from insects experimentally infected with M. ulcerans.
MATERIALS AND METHODS
Mycobacterial strains and preparation of mycobacterial cell suspensions.
The strains used for this investigation and their origins are as follows: M. ulcerans Agy99, Malaysia 1615, and Japan 753 (21); Ghana IFIK1066089, Ghana Nm50/04, Ghana Nm51/04, Ghana Nm53/04, Ghana Nm74/02, Ghana Nm97/02, Ghana Nm98/02, Ghana Nm103/02, and Mexico IFIK 973880 (this study); Ghana Nm18/02, Ghana Nm21/02, Ghana Nm31/04, Ghana Nm38/02, and Ghana Nm59/02 (10); and Japan ITM 8756, French Guiana ITM 7922, and Surinam ITM 842 (22); M. marinum M, ATCC 927, CC240299, and DL240490 and M. pseudoshottsii L15 (21); M. liflandii XT128 (32); M. tuberculosis Pasteur 14001.0001 (5); and M. bovis BCG ATCC35734 (5). Mycobacteria were obtained from cultures as described earlier (19, 31), resuspended in phosphate-buffered saline (PBS; pH 7.4), and heat inactivated at 95°C for 60 min. Note that pathogenic mycobacteria need to be processed under appropriate biosafety containment. To avoid cross-contamination, 1.5-ml screw-cap tubes were used. Samples were centrifuged for 5 min at 2,500 × g to remove residual PBS.
Extraction of mycobacterial DNA from pellets.
Mycobacterial pellets were resuspended in 300 μl of lysis buffer A, B, or C (buffer A contained 5% monosodium glutamate, 50 mM Tris [pH 8.5], and 25 mM EDTA; buffer B contained 15% sucrose, 50 mM Tris [pH 8.5], and 50 mM EDTA; and buffer C contained 4 M guanidine isothiocyanate [GITC] and 50 mM Tris [pH 7.2]) and then incubated with different amounts of lysozyme. After incubation at 37°C for 1 h, sodium dodecyl sulfate (SDS) and proteinase K (PK) were added at different end concentrations and the mixture was further incubated at 37°C for 1 h, followed by enzyme inactivation at 70°C for 5 min. Some pellets were preincubated with chloroform-methanol (MeOH) at a 2:1 ratio for delipidation. Various matrix materials (200 μl of 0.1-mm zirconia beads [BioSpec Products, Bartlesville, OK] or 0.5-mm or 1.4-mm ceramic or glass beads [Bertin Technologies, Montigny-le-Bretonneux, France]) were used, and samples were homogenized with a mechanical bead beater device, the Mikro-Dismembrator S (B. Braun Biotech International, Melsungen, Germany) for 2 to 7 min at 2,000 to 3,000 rpm or the Precellys 24 (Bertin Technologies, Montigny-le-Bretonneux, France) at conditions ranging from 2 × 40 s at 5,000 rpm to 3 × 30 s at 6,800 rpm. Supernatants were transferred to new 1.5-ml reaction tubes and subjected to phenol-chloroform (Fluka, Buchs, Switzerland) extraction and chloroform purification (Fluka, Buchs, Switzerland). For this, addition of 500 μl of phenol-chloroform or chloroform was followed by a vortexing step and centrifugation at room temperature for 5 min at 4,000 rpm. After isopropanol or ethanol (EtOH) precipitation at −70°C for >30 min, DNA pellets were resuspended in 100 μl nuclease-free water. Alternatively, mycobacterial purification kits (Promega Wizard [Promega AG, Dübendorf, Switzerland], Sigma GeneElute [Sigma-Aldrich, Buchs, Switzerland], and the BD GeneOhm lysis kit [Becton Dickinson Biosciences, Allschwil, Switzerland]) were used according to the manufacturers’ protocols, without or in combination with mechanical treatment.
For further purification and quality control, extracted DNA was freed from residual RNA by incubation with 1.5 mg/ml RNase A (Fermentas, St. Leon-Rot, Germany) for 2 min at 37°C and purified from degradation products, residual solvent, and protein contaminants with the QIAmp DNA purification MiniKit (Qiagen AG, Hombrechtikon, Switzerland) according to the manufacturers’ protocols. For genome sequencing applications, DNA was concentrated with a Concentrator 5301 (Vaudaux-Eppendorf AG, Basel, Switzerland).
Experimental infection of aquatic insects and DNA extraction.
Wild-caught insects belonging to the family Naucoridae (Naucoris cimicoides) were collected from swamps in western France. They were housed in an aquarium—filled with water of their natural environment—at 28°C and a photoperiod of 12 h each light and dark without any feeding for 7 days. These insects were then fed with a 15- to 20-day-old grub of Phormia terrae novae (Verminière de l'Ouest, Tremblay, France) that was infected by inoculation with 106 CFU of M. ulcerans in a volume of 30 μl with a 25-gauge needle. Alternatively, Naucoris insects were directly inoculated in the coelomic cavity with 106 CFU of M. ulcerans in a volume of 30 μl with a 25-gauge needle (16). Insects were sacrificed with 70% cold EtOH, transferred to a 1.5-ml screw-cap tube, and processed for DNA extraction as described for mycobacterial pellets.
DNA quantification, amplification, and gel electrophoresis.
DNA concentration was determined with the NanoDrop Spectrophotometer ND-1000 (NanoDrop, Wilmington, DE) by measuring the absorption at 260 nm, and the decontamination of DNA from solvents and proteins was estimated by measuring the absorption at 230 and 280 nm, respectively. Purity and fragmentation of the extracted genomic DNA were assessed by 1% agarose gel electrophoresis. Detection limits of purified genomic materials were assayed by PCR with primers targeting unique regions in the mycobacterial genomes (primers MK810 [TCTGTCAAGACAAGCCGATG], MK811 [GACTCGTGGTGATCGAGGAT], MK60 [ATCGTTTAGCGCATCGATCT], MK61 [CACAGGTCGACCCCAACTAC], MK63 [GTCGATGATCGCCTGTGGT], and MK35 [GTCGGCATCTTGTTGCTCA]). The presence of M. ulcerans in environmental insect specimens was tested with primers MU5 and MU6 (26) for detection of IS2404 and MK289 (GTCGTAGATGTGGGCGAAA) and MK263 (GGTGCGGTTCCATTGAGA) for detection of IS2606. Primers were designed with the Primer3 software (http://frodo.wi.mit.edu/cgi-bin/primer3/primer3_www.cgi). PCR was performed with FirePol 10× buffer and 0.5 μl FirePolTaq polymerase (Solis BioDyne, Tartu, Estonia), 10 ng genomic DNA, 0.6 μM each forward and reverse primer, 1.5 mM MgCl2, and 0.4 mM each deoxynucleoside triphosphate in a total volume of 30 μl. PCRs were run in a GeneAmp PCR System 9700 PCR machine (Perkin-Elmer, Schwerzenbach, Switzerland). The thermal profile for PCR amplification of M. ulcerans genomic DNA included an initial denaturation step of 95°C for 5 min, followed by 32 cycles of 95°C for 20 s, annealing at 58°C for 30 s, and elongation at 72°C for 45 s up to 2 min 20 s. The PCR was finalized by an extension step at 72°C for 10 min, followed by the analysis of the PCR products on 1% agarose gels by gel electrophoresis with ethidium bromide staining and the AlphaImager illuminator (Alpha Innotech, San Leandro, CA).
RESULTS AND DISCUSSION
Our first aim was to reduce the DNA extraction volume to 1.5-ml centrifugation tubes to facilitate rapid processing of large numbers of samples, in comparison to the commonly used volumes of 10 to 50 ml (2, 24). Extraction of DNA from samples with a wet weight of up to 20 mg in a volume of 1.5 ml turned out to be highly economical with respect to time and reagent consumption, while the use of more starting material did not result in a proportional increase in the amount of extracted DNA (Table 1).
TABLE 1.
Comparative advantages and/or disadvantages of various elements of DNA extraction protocolsa
| Protocol step and option(s) | Advantage(s) and/or disadvantage(s) |
|---|---|
| Reaction vol | |
| 1.5, 2, or 10 ml | Minimized vol reduces time and reagent consumption, round bottom of 2-ml tubes complicates separation of EtOH precipitate from supernatant |
| Starting material | |
| 5 to 40 mg (wet wt) pellets | Maximum DNA yield with 20 mg, no increase with more material in 1.5-ml reaction mixture vol |
| Delipidation pretreatment | |
| Chloroform-MeOH treatment preceding lysis | Enhanced purity but substantial loss of material |
| Lysis buffer | |
| A: 5% monosodium glutamate, 50 mM Tris (pH 8.5), | |
| 25 mM EDTA | GITC lysis buffer less efficient; otherwise, no differences encountered |
| B: 15% sucrose, 50 mM (Tris pH 8.5), 50 mM EDTA | |
| C: 4 M GITC, 50 mM Tris (pH 7.2) | |
| Disruption/digestion | |
| 1 vs 10 mg/ml lysozyme, 100 vs 200 μg/ml PK | No differences encountered |
| SDS concn, 0.05 to 4% | Highest concn yields best result |
| Mechanical vs chemical lysis (BD GeneOhm kit) | Higher yields with mechanical lysis |
| High-velocity mechanical treatment | Yields efficiently increased |
| Mikro-Dismembrator S vs Precellys 24 homogenization device | Slightly enhanced yields with Precellys 24 |
| Glass, zirconia, or ceramic beads | Higher yields with glass and zirconia beads |
| 0.1-mm vs 0.5-mm beads | Higher yields with 0.1-mm beads |
| Homogenization conditions: 2-7 min at 2,000-3,000 rpm | |
| (Mikro-Dismembrator S), 2 × 40 s at 5,000 rpm to 3 × 30 s at | |
| 6,800 rpm (Precellys 24) | Harshest conditions result in highest yields |
| Purification | |
| Phenol-chloroform extraction | Purity efficiently increased |
| 2 phenol-chloroform extraction steps followed by 1 chloroform | |
| purification step only | Purity further increased |
| Phenol-chloroform vs column purification | Higher yields and purity with phenol-chloroform but less labor intensive when using columns |
| Isopropanol vs EtOH precipitation | Higher yields with isopropanol but better to handle pellet with EtOH |
| Second round of phenol-chloroform extraction and EtOH | |
| precipitation | Higher DNA quality but substantial loss of DNA material |
These comparisons were made with M. ulcerans strain IFIK1066089. For details of the methodology, see Materials and Methods.
Next, we adopted and evaluated elements of several protocols of mycobacterial DNA extraction (2, 9, 15, 23, 24) for a comprehensive method optimization (Table 1). It was reported earlier that removal of lipids and lipid-like surface proteins by chloroform-MeOH treatment preceding cell lysis improved DNA purity, especially of large-size DNA (2, 9, 12). We confirmed this but also experienced substantial loss of genomic material. In contrast to an earlier study (15), the type of lysis buffer used had no marked influence on the DNA yield. Likewise, changes in lysozyme and PK concentrations made no significant differences. However, we found that an increase in the concentration of SDS resulted in increasing DNA yields. A commercially available kit designed to chemically lyse mycobacteria for PCR purposes yielded only small amounts of DNA suitable for PCR. In contrast, high-velocity mechanical treatment turned out to be very efficient in cell wall disruption, as already reported in several studies (2, 17, 33). When comparing matrix materials for bead beater treatment, good results were obtained with both zirconia and glass beads, and cell solubilization was best when using beads of small diameter. The DNA yields tended to be higher when using harsh conditions, i.e., highest speed conditions and homogenization times of the respective device (Table 1) (17). However, since high-velocity treatment jeopardizes shearing of DNA (2, 33), we recommend the performance of quality control by agarose gel electrophoresis after purification. Figure 1 shows that a combination of mechanical disruption and chemical solubilization of the waxy lipid and mycolic acid-containing cell wall renders the mycobacteria accessible for enzymatic lysis. Incubation with 4% SDS followed by mechanical disruption, a combination that was (to our knowledge) not used in any previous protocol, was here found to be essential for a high DNA yield (sample D). All protocols lacking one of these two treatments yielded strikingly smaller amounts of DNA (samples B and C, respectively). When both steps were omitted, no DNA was extracted at all (sample A).
FIG. 1.

DNA yields (bars) and purity (line) achieved when testing different combinations of DNA extraction, purification, and precipitation procedures. Twenty-milligram (wet weight) pellets of M. ulcerans strain IFIK1066089 were used. (Superscript 1) SDS was applied to a final concentration of 4%. (Superscript 2) Mechanical disruption was performed with a Precellys 24 homogenizer at 6,800 rpm for 3 × 30 s with glass beads. (Superscript 3) Second round, after EtOH precipitation, of three-step phenol-chloroform (Phe-Chl) extraction. (Superscript 4) Promega Wizard. (Superscript 5) Sigma GeneElute. For details, see Materials and Methods.
For purification, the properties of DNA binding to silica in the presence of chaotropic salts are generally used to circumvent the use of phenol-chloroform. However, silica-based commercial purification kits reached neither our elevated quantity nor quality requirements, even when applied after mechanical solubilization (Table 1; Fig. 1, samples G and H). Thus, although column use facilitates handling, we decided to use conventional phenol-chloroform extraction. Two steps of phenol-chloroform extraction were found to be crucial for removing protein and lipid contaminations from the genomic DNA, and an additional purification step with only chloroform helped in removing residual phenol (Table 1). A second round of this three-step phenol-chloroform extraction after EtOH precipitation led to enhanced DNA purity but reduced the overall yield (Fig. 1, sample E). Although the highest DNA yield and purity were represented by sample F (Fig. 1), where DNA was precipitated with isopropanol instead of EtOH, this protocol resulted, in most cases, in a yellowish, slimy pellet that was difficult to resuspend and thus prone to material loss during handling. In conclusion, when a maximum of extracted genetic material is required, the protocol described for sample D should be applied, which had a 260/280-nm ratio (∼1.6) sufficient for most downstream applications. When a higher purity of DNA is required, a protocol including chloroform-MeOH pretreatment and/or a second round of phenol-chloroform extraction and EtOH precipitation is indicated, although it is associated with lower yields.
From these results, we derived an improved standard protocol (Table 2) that involves (i) the use of 20 mg (wet weight) of pellets; (ii) pretreatment with 10 mg/ml lysozyme, 200 μg/ml PK, and 4% SDS; (iii) mechanical disruption with 0.1-mm zirconia beads; (iv) two phenol-chloroform extractions, followed by chloroform purification; and (v) EtOH precipitation with two EtOH washing steps, followed by resuspension of the DNA pellet in 100 μl of nuclease-free water.
TABLE 2.
Optimized protocol for preparation of mycobacterial DNA
| Process and step | Vol (μl) |
|---|---|
| Cell wall disruption and digestion | |
| Transfer 20-mg (wet wt) pellet or environmental sample into a 1.5-ml screw-cap tube and wash with PBS if needed | ∼10 |
| Resuspend in lysis buffer (15% sucrose, 0.05 M Tris [pH 8.5], 0.05 M EDTA) | 300 |
| Add lysozyme (stock, 100 mg/ml; end concn, 10 mg/ml): | 50 |
| Incubate for 1 h at 37°C | |
| Add SDS (stock, 20%; end concn, 4%) | 100 |
| Add PK (stock, 2.5 mg/ml; end concn, 0.2 mg/ml) | 40 |
| Total | 500 |
| Incubate for 1 h at 37°C | |
| Incubate for 5 min at 70°C | |
| Add matrix material (vol of ∼150 μl glass or zirconia beads of 0.1-mm diam) and homogenize with mechanical treatment device | |
| Centrifuge at 14,000 rpm for 2 min | |
| Transfer supernatant to 1.5-ml reaction tube | |
| Phenol-chloroform extraction and EtOH precipitation | |
| Add phenol-chloroform-isoamyl alcohol (Tris saturated) | 500 |
| Vortex, centrifuge at 14,000 rpm for 5 min | |
| Collect supernatant | |
| Repeat phenol-chloroform extraction step | |
| Add chloroform-isoamyl alcohol | 500 |
| Vortex, centrifuge at 14,000 rpm for 5 min | |
| Collect supernatant | ∼200 |
| Add 3 M sodium acetate | 20 |
| Add 100% EtOH | 500 |
| Freeze for >30 min at −70°C, centrifuge at 14,000 rpm for 30 min at 4°C | |
| Wash with 70% EtOH | 700 |
| Centrifuge at 14,000 rpm for 10 min at 4°C | |
| Repeat EtOH wash step | |
| Dry at 50°C, resuspend in nuclease-free H2O | 100 |
| Quality control | |
| Measure DNA concn and purity with a spectrophotometer | |
| DNA concn: >100 ng/μl; protein: 260/280 nm, >1.8; detergent: 260/230 nm, >1.8 | |
| Monitor quality on 1% agarose gel |
We applied the established DNA extraction method to cell pellets of a panel of M. ulcerans strains and other mycobacterial species including M. marinum, M. pseudoshottsii, M. liflandii, M. tuberculosis, and M. bovis BCG (Table 3). A mean yield of 713 ng genomic DNA per mg (wet weight) cells with a 260/280-nm ratio of 1.58 (standard deviation, 0.09) was obtained. This represents a 10- to 20-fold increase in DNA yield in our experiments compared to previous protocols, elements of which we combined for optimization. We performed detailed quality control for DNA extractions from two strains, M. ulcerans Agy99 and Japan ITM 8756, representing the two distinct lineages of M. ulcerans (Fig. 2). Genomic DNA yields from 20-mg (wet weight) cell pellets were sufficient in quantity (typically, >5 μg; Fig. 2) and quality for whole-genome microarray hybridization and whole-genome sequencing analyses. Single-copy gene sequences of >2 kb could be easily amplified by PCR with 10 ng as the template (Fig. 2). Subsequent RNase treatment and genomic DNA column purification decontaminated the samples from RNA and small DNA fragments resulting from shearing of genomic DNA (Fig. 2).
TABLE 3.
DNA yields from a variety of mycobacterial speciesa
| Species and lineage/ecotype/strain | No. of isolates | Mean DNA yield (μg) (SD) | Mean DNA quality (260/280 nm ratio) (SD) |
|---|---|---|---|
| M. ulcerans classical lineage | 34 | 19.83 (9.63) | 1.63 (0.25) |
| M. ulcerans ancestral lineage | 7 | 12.14 (7.72) | 1.74 (0.24) |
| M. marinum human isolates | 3 | 18.38 (6.58) | 1.73 (0.03) |
| M. marinum fish isolates | 4 | 19.99 (19.09) | 1.40 (0.09) |
| M. pseudoshottsii L15 | 4 | 19.36 (11.44) | 1.63 (0.14) |
| M. liflandii XT128 | 3 | 7.26 (5.86) | 1.49 (0.21) |
| M. tuberculosis Pasteur 14001.0001 | 1 | 6.22 (NA)b | 1.30 (NA) |
| M. bovis BCG ATCC 35734 | 1 | 10.78 (NA) | 1.70 (NA) |
| Overall | 57 | 14.25 (4.88) | 1.58 (0.09) |
The optimized protocol was applied to 20 mg(wet weight) of pellets.
NA, not applicable.
FIG. 2.
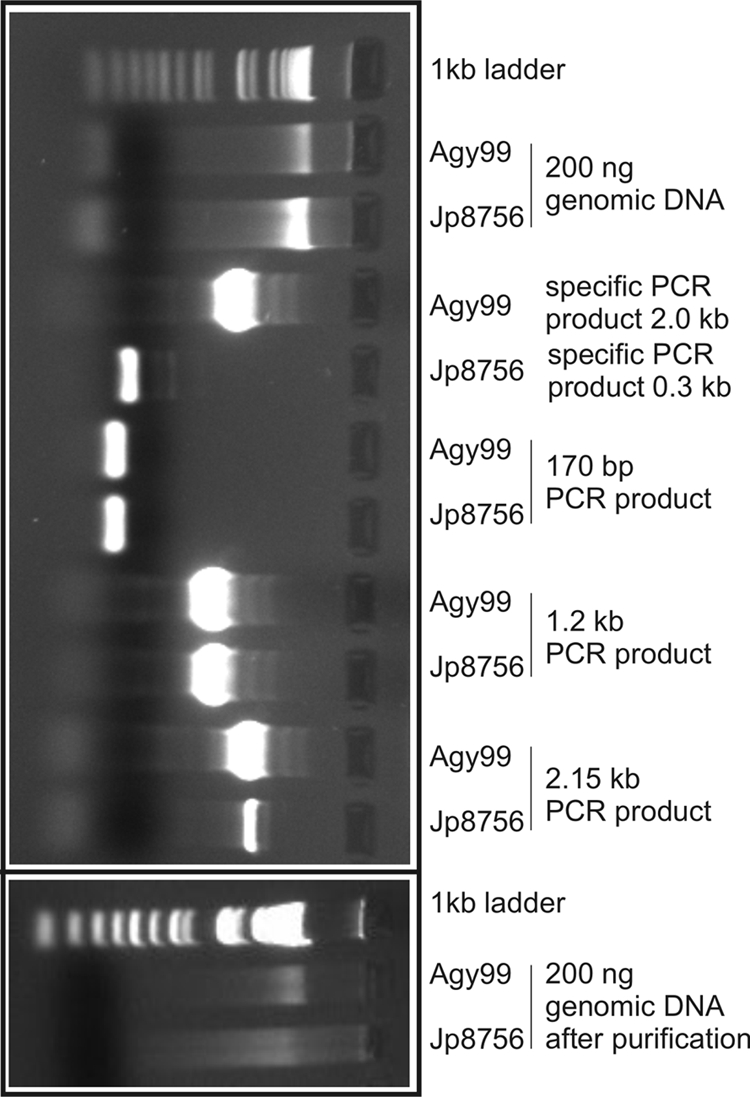
Quality control of DNA preparations from two M. ulcerans strains (Agy99 and Japan ITM 8756 [Jp8756]) by agarose gel electrophoresis. Shown are ethidium bromide stainings of isolated genomic DNA, four PCR products thereof, and RNase-treated and column-purified aliquots (right panel). Primer pair MK810/MK811 was designed to yield PCR product sizes specific for either Agy99 (2 kb) or Jp8756 (0.3 kb). Primer pairs MK60/MK61, MK60/MK63, and MK60/MK35 were designed to yield identical PCR products in both strains with increasing sizes (0.17, 1.2, and 2.15 kb, respectively).
In addition, we applied the optimized DNA extraction method to aquatic insects experimentally infected with M. ulcerans. Genomic DNA was sufficient in quantity and purity to specifically detect M. ulcerans in infected insects (Table 4), showing that the established method is suitable for direct processing of environmental samples.
TABLE 4.
Detection of M. ulcerans genomic DNA after extraction from infected aquatic insectsa
| N. cimicoides sample | Total/no. (%) PCR positive
|
|
|---|---|---|
| IS2404 | IS2606 | |
| Uninfected control | 0/10 (0) | 0/10 (0) |
| Inoculated | 8/8 (100) | 8/8 (100) |
| Fed | 4/7 (57) | 4/7 (57) |
N. cimicoides insects were subjected to DNA extraction after experimental inoculation or feeding with M. ulcerans bacteria along with control insects (noninfected or sampled from the natural environment). M. ulcerans was identified by using two different specific loci.
In conclusion, we envision this protocol to facilitate the investigation of pathogenic and nonpathogenic mycobacteria sampled from both infected tissue and the environment. In combining and optimizing crucial elements of established DNA extraction methods, our ready-to-use protocol meets the challenging characteristics of slow growth and distinct cell wall composition of mycobacteria and greatly enhances both the yield and the purity of mycobacterial DNA preparations.
Acknowledgments
We are grateful to Pamela C. Small for provision of strains (M. marinum CC240299 and DL240490, M. pseudoshottsii L15, and M. liflandii XT128), Thomas Bodmer for cultivation of strain M. ulcerans IFIK1066089, and Dorothy Yeboah-Manu for M. ulcerans strains from Ghana.
M. Käser was supported by a grant from the Deutsche Forschungsgemeinschaft (KA 1842/1-1). L. Marsollier was supported by the Institut National de la Santé et de la Recherche Médicale (INSERM) and the Foundation Raoul Follereau.
Footnotes
Published ahead of print on 1 December 2008.
REFERENCES
- 1.Ayele, W. Y., S. D. Neill, J. Zinsstag, M. G. Weiss, and I. Pavlik. 2004. Bovine tuberculosis: an old disease but a new threat to Africa. Int. J. Tuberc. Lung Dis. 8:924-937. [PubMed] [Google Scholar]
- 2.Belisle, J. T., and M. G. Sonnenberg. 1998. Isolation of genomic DNA from mycobacteria. Methods Mol. Biol. 101:31-44. [DOI] [PubMed] [Google Scholar]
- 3.Chacon, O., L. E. Bermudez, and R. G. Barletta. 2004. Johne's disease, inflammatory bowel disease, and Mycobacterium paratuberculosis. Annu. Rev. Microbiol. 58:329-363. [DOI] [PubMed] [Google Scholar]
- 4.Cole, S. T., K. Eiglmeier, J. Parkhill, K. D. James, N. R. Thomson, P. R. Wheeler, N. Honore, T. Garnier, C. Churcher, D. Harris, K. Mungall, D. Basham, D. Brown, T. Chillingworth, R. Connor, R. M. Davies, K. Devlin, S. Duthoy, T. Feltwell, A. Fraser, N. Hamlin, S. Holroyd, T. Hornsby, K. Jagels, C. Lacroix, J. Maclean, S. Moule, L. Murphy, K. Oliver, M. A. Quail, M. A. Rajandream, K. M. Rutherford, S. Rutter, K. Seeger, S. Simon, M. Simmonds, J. Skelton, R. Squares, S. Squares, K. Stevens, K. Taylor, S. Whitehead, J. R. Woodward, and B. G. Barrell. 2001. Massive gene decay in the leprosy bacillus. Nature 409:1007-1011. [DOI] [PubMed] [Google Scholar]
- 5.Diaz, D., H. Dobeli, D. Yeboah-Manu, E. Mensah-Quainoo, A. Friedlein, N. Soder, S. Rondini, T. Bodmer, and G. Pluschke. 2006. Use of the immunodominant 18-kilodalton small heat shock protein as a serological marker for exposure to Mycobacterium ulcerans. Clin. Vaccine Immunol. 13:1314-1321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Donaghy, J. A., M. T. Rowe, J. L. Rademaker, P. Hammer, L. Herman, V. De Jonghe, B. Blanchard, K. Duhem, and E. Vindel. 2008. An interlaboratory ring trial for the detection and isolation of Mycobacterium avium subsp. paratuberculosis from raw milk artificially contaminated with naturally infected faeces. Food Microbiol. 25:128-135. [DOI] [PubMed] [Google Scholar]
- 7.Fyfe, J. A., C. J. Lavender, P. D. Johnson, M. Globan, A. Sievers, J. Azuolas, and T. P. Stinear. 2007. Development and application of two multiplex real-time PCR assays for the detection of Mycobacterium ulcerans in clinical and environmental samples. Appl. Environ. Microbiol. 73:4733-4740. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Gao, A., L. Mutharia, M. Raymond, and J. Odumeru. 2007. Improved template DNA preparation procedure for detection of Mycobacterium avium subsp. paratuberculosis in milk by PCR. J. Microbiol. Methods 69:417-420. [DOI] [PubMed] [Google Scholar]
- 9.González-y-Merchand, J. A., I. Estrada-Garcia, M. J. Colston, and R. A. Cox. 1996. A novel method for the isolation of mycobacterial DNA. FEMS Microbiol. Lett. 135:71-77. [DOI] [PubMed] [Google Scholar]
- 10.Hilty, M., D. Yeboah-Manu, D. Boakye, E. Mensah-Quainoo, S. Rondini, E. Schelling, D. Ofori-Adjei, F. Portaels, J. Zinsstag, and G. Pluschke. 2006. Genetic diversity in Mycobacterium ulcerans isolates from Ghana revealed by a newly identified locus containing a variable number of tandem repeats. J. Bacteriol. 188:1462-1465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Honoré-Bouakline, S., J. P. Vincensini, V. Giacuzzo, P. H. Lagrange, and J. L. Herrmann. 2003. Rapid diagnosis of extrapulmonary tuberculosis by PCR: impact of sample preparation and DNA extraction. J. Clin. Microbiol. 41:2323-2329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Imai, T., K. Ohta, H. Kigawa, H. Kanoh, T. Taniguchi, and J. Tobari. 1994. Preparation of high-molecular-weight DNA: application to mycobacterial cells. Anal. Biochem. 222:479-482. [DOI] [PubMed] [Google Scholar]
- 13.Katoch, V. M. 2004. Infections due to non-tuberculous mycobacteria (NTM). Indian J. Med. Res. 120:290-304. [PubMed] [Google Scholar]
- 14.Khan, I. U., and J. S. Yadav. 2004. Development of a single-tube, cell lysis-based, genus-specific PCR method for rapid identification of mycobacteria: optimization of cell lysis, PCR primers and conditions, and restriction pattern analysis. J. Clin. Microbiol. 42:453-457. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Kotlowski, R., A. Martin, A. Ablordey, K. Chemlal, P. A. Fonteyne, and F. Portaels. 2004. One-tube cell lysis and DNA extraction procedure for PCR-based detection of Mycobacterium ulcerans in aquatic insects, molluscs and fish. J. Med. Microbiol. 53:927-933. [DOI] [PubMed] [Google Scholar]
- 16.Marsollier, L., R. Robert, J. Aubry, J. P. Saint Andre, H. Kouakou, P. Legras, A. L. Manceau, C. Mahaza, and B. Carbonnelle. 2002. Aquatic insects as a vector for Mycobacterium ulcerans. Appl. Environ. Microbiol. 68:4623-4628. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Odumeru, J., A. Gao, S. Chen, M. Raymond, and L. Mutharia. 2001. Use of the bead beater for preparation of Mycobacterium paratuberculosis template DNA in milk. Can. J. Vet. Res. 65:201-205. [PMC free article] [PubMed] [Google Scholar]
- 18.Onyebujoh, P., and G. A. Rook. 2004. Tuberculosis. Nat. Rev. Microbiol. 2:930-932. [DOI] [PubMed] [Google Scholar]
- 19.Palomino, J. C., and F. Portaels. 1998. Effects of decontamination methods and culture conditions on viability of Mycobacterium ulcerans in the BACTEC system. J. Clin. Microbiol. 36:402-408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Petrini, B. 2006. Non-tuberculous mycobacterial infections. Scand. J. Infect. Dis. 38:246-255. [DOI] [PubMed] [Google Scholar]
- 21.Ranger, B. S., E. A. Mahrous, L. Mosi, S. Adusumilli, R. E. Lee, A. Colorni, M. Rhodes, and P. L. C. Small. 2006. Globally distributed mycobacterial fish pathogens produce a novel plasmid-encoded toxic macrolide, mycolactone F. Infect. Immun. 74:6037-6045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Rondini, S., M. Käser, T. Stinear, M. Tessier, C. Mangold, G. Dernick, M. Naegeli, F. Portaels, U. Certa, and G. Pluschke. 2007. Ongoing genome reduction in Mycobacterium ulcerans. Emerg. Infect. Dis. 13:1008-1015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Rondini, S., E. Mensah-Quainoo, H. Troll, T. Bodmer, and G. Pluschke. 2003. Development and application of real-time PCR assay for quantification of Mycobacterium ulcerans DNA. J. Clin. Microbiol. 41:4231-4237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Ross, B. C., K. Raios, K. Jackson, and B. Dwyer. 1992. Molecular cloning of a highly repeated DNA element from Mycobacterium tuberculosis and its use as an epidemiological tool. J. Clin. Microbiol. 30:942-946. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Stamm, L. M., and E. J. Brown. 2004. Mycobacterium marinum: the generalization and specialization of a pathogenic mycobacterium. Microbes Infect. 6:1418-1428. [DOI] [PubMed] [Google Scholar]
- 26.Stinear, T., B. C. Ross, J. K. Davies, L. Marino, R. M. Robins-Browne, F. Oppedisano, A. Sievers, and P. D. Johnson. 1999. Identification and characterization of IS2404 and IS2606: two distinct repeated sequences for detection of Mycobacterium ulcerans by PCR. J. Clin. Microbiol. 37:1018-1023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Sweeney, R. W., R. H. Whitlock, and S. C. McAdams. 2006. Comparison of three DNA preparation methods for real-time polymerase chain reaction confirmation of Mycobacterium avium subsp. paratuberculosis growth in an automated broth culture system. J. Vet. Diagn. Investig. 18:587-590. [DOI] [PubMed] [Google Scholar]
- 28.Thomson, L. M., H. Traore, H. Yesilkaya, C. Doig, H. Steingrimsdottir, L. Garcia, and K. J. Forbes. 2005. An extremely rapid and simple DNA-release method for detection of M. tuberculosis from clinical specimens. J. Microbiol. Methods 63:95-98. [DOI] [PubMed] [Google Scholar]
- 29.Turenne, C. Y., R. Wallace, Jr., and M. A. Behr. 2007. Mycobacterium avium in the postgenomic era. Clin. Microbiol. Rev. 20:205-229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Wansbrough-Jones, M., and R. Phillips. 2005. Buruli ulcer. BMJ 330:1402-1403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Yeboah-Manu, D., T. Bodmer, E. Mensah-Quainoo, S. Owusu, D. Ofori-Adjei, and G. Pluschke. 2004. Evaluation of decontamination methods and growth media for primary isolation of Mycobacterium ulcerans from surgical specimens. J. Clin. Microbiol. 42:5875-5876. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Yip, M. J., J. L. Porter, J. A. Fyfe, C. J. Lavender, F. Portaels, M. Rhodes, H. Kator, A. Colorni, G. A. Jenkin, and T. Stinear. 2007. Evolution of Mycobacterium ulcerans and other mycolactone-producing mycobacteria from a common Mycobacterium marinum progenitor. J. Bacteriol. 189:2021-2029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Zhang, Z. Q., and M. Ishaque. 1997. Evaluation of methods for isolation of DNA from slowly and rapidly growing mycobacteria. Int. J. Lepr. Other Mycobact. Dis. 65:469-476. [PubMed] [Google Scholar]