

Abstract
In Dictyostelium, sporulation occurs synchronously as prespore cells approach the apex of the aerial stalk during culmination. Each prespore cell becomes surrounded by its own coat comprised of a core of crystalline cellulose and a branched heteropolysaccharide sandwiched between heterogeneous cysteine-rich glycoproteins. The function of the heteropolysaccharide, which consists of galactose and N-acetylgalactosamine, is unknown. Two glycosyltransferase-like genes encoding multifunctional proteins, each with predicted features of a heteropolysaccharide synthase, were identified in the Dictyostelium discoideum genome. pgtB and pgtC transcripts were modestly upregulated during early development, and pgtB was further intensely upregulated at the time of heteropolysaccharide accumulation. Disruption of either gene reduced synthase-like activity and blocked heteropolysaccharide formation, based on loss of cytological labeling with a lectin and absence of component sugars after acid hydrolysis. Cell mixing experiments showed that heteropolysaccharide expression is spore cell autonomous, suggesting a physical association with other coat molecules during assembly. Mutant coats expressed reduced levels of crystalline cellulose based on chemical analysis after acid degradation, and cellulose was heterogeneously affected based on flow cytometry and electron microscopy. Mutant coats also contained elevated levels of selected coat proteins but not others and were sensitive to shear. Mutant spores were unusually susceptible to hypertonic collapse and damage by detergent or hypertonic stress. Thus, the heteropolysaccharide is essential for spore integrity, which can be explained by a role in the formation of crystalline cellulose and regulation of the protein content of the coat.
In Dictyostelium, spores are the only surviving cell type produced by starvation-induced multicellular development. During this process, solitary amoebae aggregate to form a migratory slug composed of prespore and prestalk cells, which then culminate to form a fruiting body consisting of spores perched on top of a 1 to 2 mm tall cellular stalk. As each prespore cell becomes a spore, it dehydrates, accumulates trehalose, and encloses itself in a specialized cell wall (37). This physicochemical barrier protects the enclosed amoeba from external stress and probably actively regulates terminal sporulation and spore germination. The spore coat is formed de novo from four known sources: (i) an early-formed pool of proteins and (ii) a galactose-rich polysaccharide (GPS) stored together in prespore vesicles (PSVs) of the slug, (iii) a late-formed pool of protein(s), including SP65 (23), and (iv) cellulose formed de novo at the cell surface. These components are separately deposited at the cell surface, where they organize into an asymmetrical trilaminar “sandwich” with proteins on either side enclosing the polysaccharides in the interior (see Fig. 5B). Cellulose is the primary structural component of the middle layer and is required for organization of the protein layers (47). More than 10 coat structural proteins are known and mutational studies have revealed essential roles for outer layer proteins in coat permeability (23, 37). An inner layer protein, SP85, forms direct contacts with cellulose and the coat protein SP65 and is important for morphogenesis of both cellulose and the outer layer (22).
FIG. 5.

Mutant spores produce less cellulose. (A) Flow cytometry of normal (Ax3) and mutant (pgtB, pgtC, and pgtB/pgtC) spores incubated in 0.01% Calcofluor White ST (Calc). The distribution of forward light scattering and fluorescence are shown. (B) Transmission electron microscopy of conventional thin sections of normal and mutant (pgtC) spores decorated with lead citrate and uranyl acetate. The position of cellulose and the GPS in normal spores is indicated; the inner protein layer is not contrasted in this image. The range of variation of coats of better-preserved spores, from sections approximately normal to the plasma membrane, is shown. (C) Sugar composition analysis of spores and ISM. Fractions were first heated in 2 M TFA to hydrolyze amorphous glucans, and the insoluble material was subjected to trifluoroacetolysis, followed by acid hydrolysis to degrade crystalline cellulose. Fuc, fucose; GalN, galactosamine, GlcN, glucosamine; Gal, galactose; Glc, glucose; Man, mannose. The results are typical of two independent trials.
Cellulose-based cell walls also surround somatic cells of vascular plants, algae, and oomycetes, and cysts of Acanthamoeba, Achlya, Hartmanella, Naegleria, and Schizopyrenus (37). The nonplant walls are poorly characterized, but a general feature is the presence of polysaccharides in addition to cellulose. The function of polysaccharides is challenging to study genetically because, in contrast to proteins, genes direct polysaccharide formation indirectly. Although plant cell wall architecture is better understood (7), it is not known whether wall polysaccharides directly influence cellulose deposition, in part because the glycosyltransferase (GT) repertoire that underlies heteropolysaccharide synthesis is complex and not fully explored.
Previous studies in Dictyostelium highlighted the role of cellulose in spore cell wall formation (2, 47), but the role of the GPS, the only known heteropolysaccharide of the wall, has not been examined. The GPS was originally identified based on its antigenicity, and the GPS antigen and an assay for its biosynthesis were early historical markers for prespore cell differentiation (32). Partially purified GPS contains Gal and GalNAc (6, 44), which are uncommon sugars in other Dictyostelium glycoconjugates (10, 13). In the related species D. mucoroides, the major GPS fraction from slugs was shown by nuclear magnetic resonance to consist of the repeating branched trisaccharide 3Galβ1,3(Galβ1,6)GalNAcα1- (27). The D. mucoroides GPS is heterogeneous with respect to charge, size, and lectin affinity and might represent a family of polysaccharides or a common core with heterogeneous modifications. GPSs from D. mucoroides and D. discoideum are related based on cross-reactivity with antisera and lectins and on accumulation in spore coats.
The function of cell wall components can be examined advantageously in Dictyostelium because wall composition is simpler and its formation is facultative. The GPS is formed in advance in the Golgi (14, 34) and stored in PSVs, based on immunocytochemical probing of prespore cells with fluorescent antibodies (35) or colloidal gold conjugates of the Gal/GalNAc-specific plant lectins (10). GPS is secreted with other PSV-associated coat proteins during the first step in coat assembly and incorporated into both the coat and the interspore matrix (ISM) that accumulates between spores (38, 44; the present study). Ultrastructural analysis shows that the GPS is associated with the middle layer of the spore coat with cellulose and with the inner layer. Cellulose and the GPS may interact because they both resist extraction with protein denaturants (48).
Bioinformatics studies have provided an approach to examine GPS function. A recent Dictyostelium genomics analysis identified two GT-like genes predicted to function as GPS synthases (41). pgtB and pgtC appear to each encode proteins with separate GT domains predicted to retain and invert, respectively, the anomeric configuration of the donor sugar, as required if these two domains were to function together to form the disaccharide repeat of the GPS backbone as described in D. mucoroides. A function for these two genes in GPS biosynthesis is documented here, and this has in turn revealed that the GPS plays an important role in cellulose deposition, protein incorporation, and the resistance of spores to stress. The effect on spore stress resistance is profound: while less severe than disruption of the dcsA cellulose synthase gene itself (47), it is more severe than coat structural protein mutants examined up to now (23, 30, 37).
MATERIALS AND METHODS
Cells.
Cells were grown axenically in HL-5 growth medium, or in association with Klebsiella aerogenes on SM agar (31). Development was induced by washing HL-5-grown cells in KP buffer (10 mM potassium phosphate [pH 6.5]) and either incubating them at 2 × 107/ml in aggregation buffer for up to 8 h, depositing them on non-nutrient agar plates, or simply allowing the cells to exhaust the bacteria on SM agar. Spores were picked from sori into KP buffer and examined in a hemacytometer after an eightfold dilution into Vectashield mounting medium (99.2% [wt/wt] glycerol) with DAPI (4′,6′-diamidino-2-phenylindole; Vector Labs, Burlingame, CA). For germination, manually picked 2-day-old spores were incubated in 0.2% (vol/vol) NP-40 in H2O for 5 min, washed by centrifugation, counted in a hemacytometer, and serially diluted with K. aerogenes onto SM agar. The plating efficiency was calculated as the colony number at day 4 divided by initial spore number and then multiplied by 100.
Real-time PCR.
Total RNA was isolated by using a TRIzol-based method as described previously (28). RNA was treated with RNase-free DNase I and DNase inactivation reagent (Ambion, Austin, TX). Equal amounts of total RNA based on the absorbance were reverse transcribed by using random primers and Superscript III reverse transcriptase (Invitrogen, Carlsbad, CA). cDNAs were diluted 1:10, mixed with PowerSYBR Green PCR master mix (Applied Biosystems, Foster City, CA), and amplified in an ABI 7500 Fast real-time PCR machine (Applied Biosystems) as described previously (25). The efficiency of each primer pair (see Table S1 in the supplemental material) was verified to be within 80 to 120% of theoretical exponential amplification using cDNA dilutions. The threshold cycle (CT) was determined by using the thermal cycler software, and changes in expression levels calculated by a comparative CT method: (Ctarget, time x − Ctarget, time zero) − (Cfpa1, time x − Cfpa1, time zero) = ΔΔCT (20), where fpa1 is a constitutively expressed gene (28). Expression levels at development time x are expressed as the log2 of the fold-change relative to time zero (growth phase), expressed as 2−ΔC′T. Absence of genomic DNA contamination was confirmed by the lack of signal when reverse transcriptase was omitted, and no signal was detected in the absence of added RNA (data not shown).
Disruption of pgtB and pgtC.
For disruption of pgtB by homologous recombination, a 960-nucleotide (nt) targeting DNA was amplified from the 5′ end of the coding region using D2S1 and D2AS1 (Fig. 1, Table S1 in the supplemental material, and Fig. S1 in the supplemental material) and cloned into pCR4-TOPO (Invitrogen) to yield pB5′ (see Fig. 1). An 832-nt targeting DNA was amplified from the 3′ end of the coding region using D2S3 and D2AS2, cloned into pCR4-TOPO, excised with XbaI and SalI, and cloned into pB5′ after predigestion with XbaI and SalI to yield pB5′3′. The bsr cassette was excised from pBSR519 (26) with XbaI and cloned into the XbaI site of pB5′3′ to yield pBbsr. For disruption of pgtC, a 960-nt targeting DNA was amplified from the 5′ end of the coding region using D1S2 and D1AS1, and a 921-nt targeting DNA was amplified from the 3′ end of the coding region using D1S4 and D1AS2. These DNA fragments were assembled as described above, except that a BamHI site was used in place of the XbaI site, yielding pCbsr. To create the double (pgtB/pgtC) disruption strain, the hygromycin resistance cassette from pHygT(plus)/pG7 (a gift from M. Nelson, M. Fukuzawa, and J. G. Williams) was excised using BamHI and cloned into the BamHI site of pC5′3′, yielding pChyg. All disruption DNAs were excised with EcoRI, trimmed with Bal31 exonuclease, and electroporated into Dictyostelium strain Ax3 as described previously (49).
FIG. 1.

Predicted di-GT genes and strategies for their disruption. (A) Predicted PgtB protein; (B) predicted pgtB gene (DDB0231870 at www.dictybase.org); (C) disruption strategy for pgtB; (D) predicted PgtC protein; (E) predicted pgtC gene (DDB0231959); (F) disruption strategy for pgtC. The putative signal anchor and the GT2, GT4, and GT61 domains are depicted in panels A and D. The positions of the oligonucleotide primers are indicated in panels B and E.
The oligonucleotides used for PCR trials to confirm the expected recombination events are shown in Table S1 in the supplemental material, Fig. 1, and Fig. S1 and S2 in the supplemental material.
GPS synthase activity.
Fruiting bodies were scraped from nonnutrient agar plates immediately upon sporulation, resuspended in 20 mM Tris-HCl (pH 7.4), and centrifuged at 5,000 × g for 3 min. The supernatants were frozen at −80°C and subsequently assayed for GPS synthase activity as described previously (24). To measure Gal transferase activity, aliquots were incubated with 8.5 μM UDP-[6-3H]Gal (1.0 μCi), 15 μM UDP-GalNAc, and 47 μg of acceptor polysaccharide (a generous gift from K. Okamoto) in 20 mM Tricine-NaOH (pH 7.8), 14 mM KCl, 10 mM MgCl2, 5 mM spermine, and 0.1% (vol/vol) Triton X-100 for 2 h on ice. To measure the GalNAc-transferase activity, aliquots were incubated in 6.1 μM UDP-[6-3H]GalNAc (0.25 μCi) and 15 μM UDP-Gal (in place of UDP-[6-3H]Gal and UDP-GalNAc) in the cocktail described above. The reaction was stopped by freezing, and incorporation was measured by ethanol precipitation on Whatman no. 2 paper.
Monosaccharide composition analysis.
Spores were picked from 2- to 3-day-old sori with a loop into water and washed twice by centrifugation at 10,000 × g for 15 s. For ISM, sori were mechanically slapped into the lids of inverted agar plates, pipetted into a 1.5-ml microcentrifuge tube using 20 mM NH4HCO3, and centrifuged at 12,000 × g for 1 min. The supernatant was transferred to a fresh tube and dried in a vacuum centrifuge; the sample was rehydrated and dried twice again. A blank agar plate processed in parallel served as a negative control. For spore coats, the spore pellets from the first centrifugation were resuspended in 0.2% (vol/vol) NP-40 in KP buffer, counted in a hemacytometer, centrifuged, resuspended in 8 M urea-50 mM dithiothreitol-20 mM Tris-HCl (pH 7.4), and incubated at 100°C for 3 min. Spores were recovered by centrifugation after dilution in H2O and washed centrifugally in H2O twice. This fraction consists of insoluble spore coats and lacks glycogen (43). Sugars were quantified as described previously (48). Briefly, samples from equivalent numbers of spores were hydrolyzed in 2 M trifluoroacetic acid (TFA) to release sugars from most glycans, including amorphous cellulose, and an aliquot was subsequently subjected to trifluoroacetolysis to release sugar from crystalline cellulose. Hydrolysates were analyzed using a Dionex PA-10 column eluted isocratically with 16 mM NaOH on a Dionex DX-600 HPAEC (high pH anion exchange chromatography) system or 6 mM NaOH to differentiate mannose and xylose, which coelute at 16 mM NaOH. Sugars were identified by pulsed amperometric detection (PAD), based on elution position, and quantified relative to external standards. Blank values were <5% and subtracted.
Flow cytometry.
Spores were manually picked with a loop (to avoid debris) from 2- to 3-day-old sori into H2O and frozen at −80°C. Thawed spores were centrifuged at 10,000 g for 10 s, resuspended in 0.5% (vol/vol) NP-40 in Tris-buffered saline for 5 min, washed twice by centrifugation in H2O, and finally resuspended in 0.2% Calcofluor White ST (American Cyanamid) in H2O. Before flow cytometry, spores were diluted 6.7-fold into 10 mM potassium phosphate (pH 6.5). A total of 10,000 particles were analyzed in an InFlux cell sorter (Cytopeia) at the Flow and Image Cytometry Laboratory at the Oklahoma University Health Sciences Center, using UV excitation at 355 nm and emission at 460 ± 50 nm.
Electron microscopy.
Sori were fixed, embedded, sectioned, and examined by transmission electron microscopy as described previously (22).
Western blotting.
Samples were processed for sodium dodecyl sulfate-polyacrylamide gel electrophoresis and subjected to Western blot analysis as described previously (42). Lanes contained 5 × 105 spores, slug cells, or the equivalent amount of ISM. SP85 was detected using monoclonal antibodies (MAbs) 5F5 (19), SP96, SP80, and SP75 were detected using MAb 83.5 (33), and SP87 was detected by using the PL3 antiserum (46), a gift from D. Blumberg.
RESULTS
Candidate GPS GTs.
A GPS from the related species D. mucoroides contains a repeating disaccharide backbone consisting of d-sugars connected via alternating α- and β-linkages (see the introduction). The synthesis of related glycosaminoglycan (GAG) polysaccharides in prokaryotes (9) and eukaryotes (18, 21) is associated with enzyme proteins having two GT domains. Of 77 D. discoideum GT-like genes (41), 3 exhibit related domain architecture and a potential Golgi targeting sequence. Two, pgtB (designated DDB0231870 at www.dictybase.org, on chromosome 5) and pgtC (DDB0231959 on chromosome 4), are predicted to encode proteins of 1,338 amino acids (Mr = 153,422) and 1,065 amino acids (Mr = 124,011), respectively (see Fig. S1 and S2 in the supplemental material), and are potentially N glycosylated based on eight and two predicted N glycosylation sites, respectively. As shown in the sequence alignments (see Fig. S3 in the supplemental material), the N-terminal region of each encodes a family GT2-like domain, expected to invert the configuration of the transferred sugar (8). Their GT2 domains are 36% identical and 68% similar over 201 amino acids comprising the majority of the putative catalytic domain (Table 1). In comparison, this region is only 23% identical and 55% similar to the corresponding domain of Dictyostelium pgtA, a bifunctional di-GT in the cytoplasm (36), and to a bacterial β4-GalT and a bacterial β3-GalNAcT. Downstream, each contains a family GT4-like domain (see Fig. S4 in the supplemental material) expected to retain the sugar configuration (8). The family GT4 domains are 37% identical and 67% similar to each other, with slightly less similarity to a putative bacterial GT and much less similarity to another GT4 sequence from Dictyostelium, PigA (Table 1). A GT2 family inverting domain and a GT4 family retaining domain have the potential to form the alternating α- and β-linkages of the GPS backbone as for GAG synthases cited above. In addition, PgtB is predicted to have a large C-terminal region of 480 amino acids, which BLAST analysis suggests includes a domain with remote similarity to family GT61 plant β2-xylosyltransferases (data not shown). PgtB and PgtC appear to be related by a gene duplication in the amoebozoa lineage, because their GT2 and GT4 domains are more similar to each other than any other sequences in the nonredundant database at the National Center for Biotechnology Information.
TABLE 1.
Sequence similarity of the GT-like domainsa
| Domain (no. of residues) | Organism (activity) | Accession no.b | % Identity/% similarityc
|
||||||
|---|---|---|---|---|---|---|---|---|---|
| PgtC | PgtA | Cj | LgtD | PgtC (356) | Pa (356) | Dd-PigA (301) | |||
| GT2 (201) | |||||||||
| PgtB | D. discoideum | DDB0231870 | 36/68 | 18/50 | 24/55 | 25/58 | |||
| PgtC | D. discoideum | DDB0231959 | 29/59 | 26/58 | 26/59 | ||||
| PgtA | D. discoideum (β3-GalT) | DDB0191458 | 24/62 | 24/61 | |||||
| Cj | C. jejuni (β3-GalT) | GB:EAQ72078.1 | 21/54 | ||||||
| LgtD | N. gonorrhoeae (β4-GalNAcT) | GI:59802452 | |||||||
| GT4 (356 or 301) | |||||||||
| PgtB | D. discoideum | DDB0231870 | 37/67 | 35/63 | 16/41 | ||||
| PgtC | D. discoideum | DDB0231959 | 33/66 | 16/47 | |||||
| Pa | P. aeruginosa (putative GT) | GI:15596588 | 16/45 | ||||||
| PigA | D. discoideum (GPI α6-GlcNAcT) | GI:DDB0231698 | |||||||
The alignments on which these calculations are based are shown in Fig. S3 and S4 in the supplemental material.
The DDB accession numbers are found at www.dictybase.org; the other numbers are from GenBank (National Center for Biotechnology Information).
The numbers of amino acids compared are indicated in parentheses.
Based on N-terminal signal anchor-like sequences (see Fig. S1 and S2 in the supplemental material), the encoded proteins are predicted to be type 2 membrane proteins with their catalytic domains oriented toward the lumen, as is typical for Golgi GTs (5). The predicted luminal domains have 12 Cys residues each and, although their positions are not perfectly conserved, some degree of conservation of disulfide bonding is implied. PgtB has a short predicted cytoplasmic domain (7 amino acids), whereas PgtC is predicted to have an Asn-rich cytoplasmic domain, frequently observed in Dictyostelium cytoplasmic proteins (19), of 123 amino acids.
Expression of pgtB and pgtC.
pgtB is expressed from mid to late development based on numerous expressed sequence tags, and pgtC expression is supported by a single slug 5′ expressed sequence tag (www.dictybase.org). These are stages during which the GPS is synthesized (14, 34). Real-time reverse transcription-PCR showed that pgtB and pgtC are expressed during growth and weakly induced by starvation. In addition, pgtB is strongly induced an additional 100-fold at the time of GPS synthesis between 13 and 16 h. The specificity of the primers for pgtB and pgtC was confirmed by the absence of detectable products in 16-hr RNA from the pgtB- and pgtC-disruption strains described below, respectively, and the double disruption pgtB/pgtC mutant strain. In addition, pgtB was expressed normally in the pgtC mutant strain, pgtC was expressed normally in the pgtB mutant strain, and fpa1 was expressed normally in all three single- and double-disruption strains.
Disruption of the pgtB and pgtC.
To examine function, pgtB and pgtC were disrupted by using homologous recombination. The disruption DNAs, shown in Fig. 1C and F, were designed to replace 880 and 507 nt of the central coding regions of pgtB and pgtC, respectively, with either Blasticidin-S (bsr) or hygromycin (hygr) resistance cassettes. bsr DNAs were used for the single disruption mutants, and the pgtB/pgtC double mutant was created by electroporating the pgtB mutant with the pgtC(hygr) DNA. Recombination at the pgtB locus was confirmed by using a PCR containing a primer within the 5′-targeting sequence (D2S2, see Fig. 1B) and a reverse primer 3′ to the targeting DNA (D2AS3). The PCR product had the expected length of 1.4 kb in the unmodified gene and the expected length of 2.2 kb resulting from insertion of the bsr cassette in both the pgtB mutant and the double mutant (Fig. 3A, upper panel). The expected recombination event for pgtC was confirmed using a primer within the 5′-targeting DNA (D1S3) and a reverse primer 3′ to the targeting DNA (D1AS3). The product had the expected length of 1.8 kb from the unmodified gene and 2.2 kb when pgtC was disrupted using the bsr cassette (middle panel). Neither band was observed in the double mutant in which the hygr cassette was used, suggesting that the gene was altered but that the distinct sequence interfered with amplification. Modification of the gene was confirmed in a second PCR in which a primer 5′ to the disruption DNA (D1S1) was used with a reverse primer from the hygromycin cassette (hygrAS1), yielding the expected 1.6-kp product only in the double mutant (lower panel). As described above (Fig. 2), these disruptions led to the expected absence of detectable RNA in the respective strains.
FIG. 3.

Disruption of pgtB or pgtC results in loss of GPS. (A) PCR analysis of disruption strains. gDNA from Ax3 (normal), a pgtB clone, a pgtC clone, and a clone from the double mutant was used as the template for PCRs using the indicated primers specific for the gene indicated. Reactions were resolved on a 1.2% agarose gel and stained with ethidium bromide. Size standards in kilobases are to the left, and apparent sizes in kilobases of PCR products are on the right. (B) GPS synthase assay. ISM collected at the time of sporulation was assayed for transfer of 3H from UDP-[3H]Gal or UDP-[3H]GalNAc in a reaction containing GPS acceptor. Values obtained in the absence of acceptor are subtracted for the UDP-[3H]Gal reaction but not the UDP-[3H]GalNAc reaction (see the text). (C) Sugar composition analysis. Equal numbers of parental (Ax3) and mutant spores were subjected to acid hydrolysis (4 M TFA, 4 h, 100°C). Released material was chromatographed on a CarboPac PA-1 HPAEC column with identification by PAD. (D) Lectin probing of parental (Ax3) and mutant (pgtB) spores. Spores were treated with boiling 8 M urea-β-mercaptoethanol and labeled with FITC-RCA-I to detect the GPS. (E) Similar analysis of parental spores and spores formed from a mixture of parental and mutant cells which were codeveloped. The results are representative of at least two independent trials.
FIG. 2.

Developmental regulation of pgtB and pgtC mRNA accumulation. Axenically grown normal strain Ax3 cells were starved for the indicated number of hours. RNA was isolated, and equivalent amounts were converted to cDNA and subjected to quantitative real-time PCR using gene-specific primers for pgtB, pgtC, and fpa1. The log2 of the ratio of cDNA at each time point relative to the zero hour (growth phase), normalized to the value for constitutively expressed fpa1, was determined by using a comparative CT method. The specificity of the primer pairs for pgtB and pgtC was established by the absence of detectable product in RNA at 16 h from their respective gene disruption strains described in Fig. 3A.
All three mutant strains formed normal appearing fruiting bodies with typical ellipsoid spores (see below). GPS synthase is normally secreted during sporulation after GPS biosynthesis is concluded (33). To determine whether GPS synthase activity is affected in the mutants, nascent fruiting bodies were harvested and the cellular supernatant (ISM) was assayed for transfer of 3H from UDP-[3H]Gal or UDP-[3H]GalNAc to a polysaccharide acceptor isolated from a D. mucoroides mutant (24). Incorporation of [3H]Gal, which was time and acceptor-dependent (data not shown), was reduced to < 25% of wild-type levels in both single mutants and the double mutant (Fig. 3B). Incorporation of [3H]GalNAc was similarly reduced although, for this activity, incorporation into normal (Ax3) extracts was not acceptor dependent (data not shown), as if endogenous acceptor for this activity was present. Microsomes from normal and mutant cells exhibited similar and very high levels of incorporation of 3H from these sugar nucleotides or possibly their epimers in the presence or absence of acceptor (unpublished data), which indicated the presence of other GT activities that might be the origin of the residual activity in the mutant ISMs. The data are consistent with a loss of GPS synthase activity in the single and double mutant cells.
Mutant spores lack GPS-like material.
Spores were examined microscopically for GPS expression using the fluorescent lectin fluorescein isothiocyanate (FITC)-RCA-I. To expose the GPS, masked by the outer layer permeability barrier, spores were extracted with hot urea-β-mercaptoethanol. In contrast to normal spores, which fluoresced brightly, pgtB mutant (Fig. 3D), pgtC mutant, and double mutant (not shown) spores were fluorescence negative, suggesting that the lectin-reactive polysaccharide was absent. This was confirmed by sugar composition analysis after acid hydrolysis. Analysis of whole spores showed a near-complete absence of Gal and galactosamine (derived from GalNAc during hydrolysis) based on HPAEC (Fig. 3C), whereas fucose, glucosamine, and mannose levels were unaffected. The molar ratio of Gal to galactosamine in normal spores, which ranged from 1.5 to 2.0, is consistent with the composition of the D. mucoroides GPS.
Coat incorporation of the GPS is cell autonomous.
To examine whether secreted GPS can be a precursor of the coats of adjacent spores, normal and mutant cells were codeveloped. Whereas normal sori contained almost exclusively FITC-RCA-I-positive spores, sori from the mixture consisted of fluorescent and nonfluorescent spores (Fig. 3E). Evidently, GPS was not transferred from normal to mutant spores, despite the fact that the GPS is also secreted into the ISM, where it would be expected to be available to nearby spores. Like cellulose (47) and some coat proteins, but in contrast to other coat protein precursors (39), the GPS appears to be incorporated only into the coats of cells in which is it is synthesized.
Mutant spores are less resistant to stress.
Spores are normally impervious to treatment with nonionic detergent. However, fewer than 4% of mutant spores, identified by morphological criteria, survived pretreatment with NP-40 as determined by plating efficiency on bacterial plates (Fig. 4A). Although difficult to precisely quantify because of the possibility of contaminating amoebae, the plating efficiency of untreated Ax3 and mutant spores was, in contrast, similar (data not shown). Therefore, the mutations cause a loss of normal resistance to nonionic detergent.
FIG. 4.

Sensitivity of mutant spores to stress. (A) Plating efficiency of 2-day-old normal and mutant spores exposed to 0.2% NP-40 nonionic detergent and seeded onto lawns of K. aerogenes. The results are representative of two independent trials. (B) One-day-old spores were incubated in the DNA dye DAPI in 88% glycerol for 1 to 2 h and photographed under phase contrast or epifluorescence illumination. (C) Autofluorescence of untreated spores in water, visualized in the fluorescein channel. The phase-contrast images of normal and mutant spores were similar when viewed directly in the microscope.
In a second test, spores were diluted 1:8 in glycerol. Almost instantaneously, Ax3 spores shrank modestly in response to the hypertonicity but gradually returned to their normal shape over a few hours (Fig. 4B), suggesting osmotic equilibration of glycerol across the plasma membrane. The majority of pgtB mutant spores responded similarly, except that shrinkage was more extreme. Over the course of 2 days in glycerol, however, these spores became phase dark, and 75 to 80% stained with the membrane impermeant DNA dye DAPI, suggesting that their plasma membranes were compromised. The remaining (25 to 50%) mutant spores became phase dark immediately and did not shrink. The nuclei of these osmotically inactive cells were stained with DAPI, confirming that their plasma membranes were compromised. Similar results were observed for pgtC mutant and double mutant spores (data not shown). Although occasional Ax3 spores became phase dark after 2 days in glycerol, fewer than 1% were DAPI positive. Therefore, mutant spores are unable to survive long-term hypertonic stress but were heterogeneous in their resistance.
Although nonstressed parental Ax3 and mutant spores were similar based on phase-contrast microscopy, 33 to 60% mutant spores (single and double mutants were similar) exhibited abnormal cytoplasmic autofluorescence in the DAPI, fluorescein, and rhodamine filter channels (Fig. 4C). In comparison, <1% of normal 2-day-old spores were autofluorescent. Autofluorescence was suppressed by glycerol, so it could not be ascertained whether autofluorescent spores correspond to the rapidly osmotically incompetent mutant spores. Although the reason for autofluorescence is not known, its existence indicates that some mutant spores are altered in the absence of intentional stress.
Analysis of cellulose.
Spore coat cellulose was first examined by using Calcofluor White ST, which fluoresces in the presence of crystalline or noncrystalline cellulose and other β-linked glycans (3). Labeling is specific for cellulose based on negative labeling of a dcsA-null strain (47). Microscopic analysis suggested reduced fluorescence intensity of mutant spores (not shown). Flow cytometry showed two fluorescence populations of double-mutant spores: a minor population that fluoresced at a level similar to that of parental spores and a major population that fluoresced at a substantially lower level (Fig. 5A) but was still cellulose positive based on comparison to untreated controls. Similar results were observed for the single mutants (data not shown). Forward light scatter analysis revealed a slight diminution of size or change of shape in the mutant spores (Fig. 5A), but gating on spores with similar forward scatter values revealed the same difference in fluorescence values (data not shown).
Since cellulose fibrils are a major component of the middle region of the spore coat, thin sections were compared by transmission electron microscopy. pgtC mutant spores were heterogeneously swollen and partially lysed (data not shown). Only 1 of 87 mutant spores had the normal appearance presented by the great majority of normal spores. In images of better-preserved mutant spores, the electron-dense outer layer of the coat appeared to be normal, but the cellulose-rich middle layer was thinner and slightly more electron dense than normal (Ax3) spores (Fig. 5B). Similar results were observed for pgtB mutant spores (not shown). Although it was not apparent which spores corresponded to the Calcofluor-normal population detected by flow cytometry (Fig. 5A), the images were consistent with reduced levels or altered packing of cellulose, either of which might explain the reduced stress resistance observed for nearly all spores.
The total cellulose content of the coat was quantitated based on glucose recovered after acid degradation. Attempts to purify spore coats by conventional density gradient centrifugation were unsuccessful because mutant coats could not be detected after shearing in the French pressure cell. Therefore, a crude coat preparation was generated as the insoluble material remaining after extraction of spores with hot 8 M urea-dithiothreitol, which removes glycogen and other solubilizable components. In addition, the ISM, which includes extracellular material not incorporated into coats during formation, was examined. The crude coat and ISM fractions were first subjected to conventional TFA treatment, which hydrolyzes amorphous cellulose and other glycans, followed by trifluoroacetolysis and subsequent acid hydrolysis, which degrades crystalline cellulose (43). Sugars were quantitated by HPAEC-PAD as described above. Similar levels of glucose were released by 2 M TFA from normal and double mutant coats (Fig. 5C, upper left), suggesting that amorphous cellulose was not affected. Values for the other sugars confirmed the results from whole spores above. Treatment of the aqueous TFA-resistant residue by trifluoroacetolysis yielded only glucose (Fig. 5C, upper right), as expected, and the level recovered was reduced by 45% in mutant relative to normal coats. The missing cellulose was not found in the ISM (Fig. 5C, lower panels). The reduced level of glucose confirms the results from Calcofluor fluorescence and transmission electron microscopy and indicates that the effect on cellulose is selective for the crystalline isoform.
Analysis of coat proteins.
To determine whether loss of the GPS has effects on protein, coat protein levels were analyzed by Western blotting. As shown in Fig. 6A to C (left panels), similar amounts of the outer layer proteins SP96 and SP75 were detected in parental (Ax3) and both single-mutant and double-mutant spores, a finding consistent with the normal appearance of the outer layer in Fig. 5B. In contrast, larger amounts of SP85, SP87, and SP80 were recovered from the mutant spores compared to normal spores. Reciprocal amounts were not observed in the ISM (Fig. 6A to C, right panels) where unincorporated proteins accumulate, indicating that the increased levels did not result from diversion from the ISM. Comparison with slugs, where the coat protein precursors are stored, also showed an elevation of the three proteins (Fig. 6D and data not shown). This suggests that the absence of the GPS leads to a compensatory increase in the level of coat protein precursors with which the GPS is normally packaged prior to sporulation.
FIG. 6.
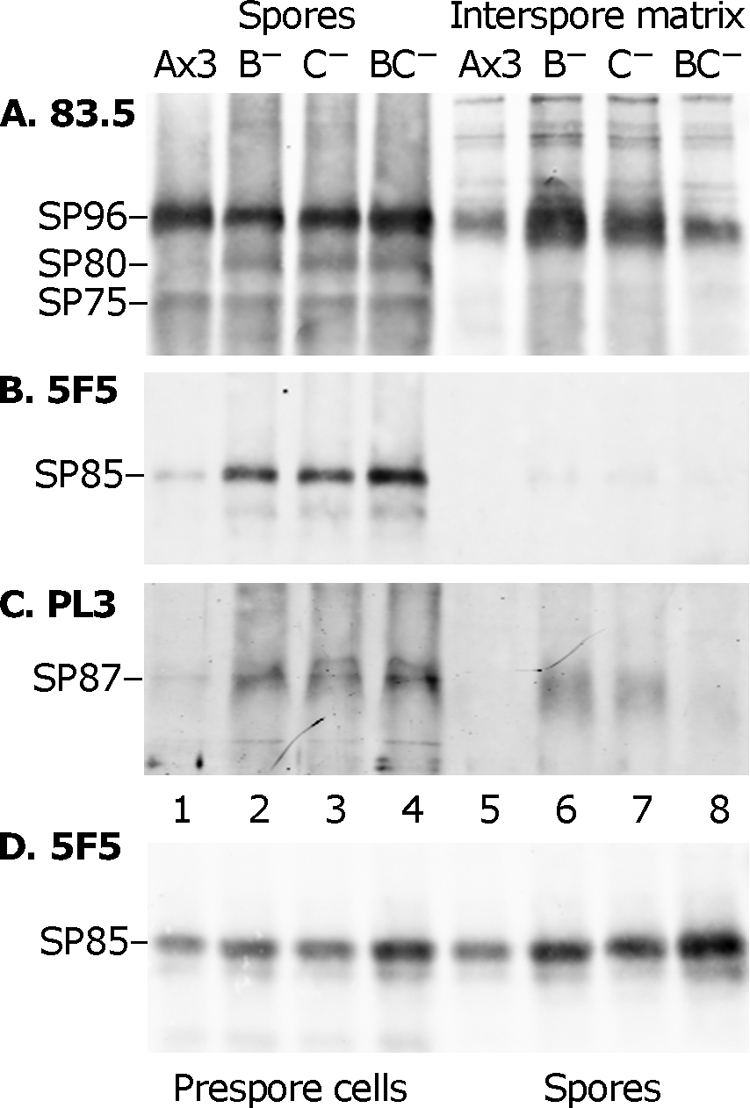
Western blot analysis of spore coat proteins. (A to C) Sori were picked with a loop into water, and spores and ISM were separated by centrifugation and analyzed by sodium dodecyl sulfate-polyacrylamide gel electrophoresis and Western blotting. Spore samples are on the left (lanes 1 to 4); ISM samples are on the right (lanes 5 to 8). Parallel blots were probed with the indicated antibodies. Protein bands are identified at the left. All blots were subsequently probed with MAb 83.5 to verify constant loading. Ax3, parental strain; B−, pgtB mutant; C−, pgtC mutant; BC−, pgtB/pgtC double mutant. The results are representative of two independent trials, except that in the other trial the level of SP96 and SP87 was similar between the single and double mutants (data not shown). (D) Similar to panels A to C, except that slug cells, in lanes 1 to 4, are compared to spores, in lanes 5 to 8.
DISCUSSION
The disruption of either of two genes predicted to encode multifunctional Golgi body-associated GTs resulted in both marked reduction of GPS synthase-like activity and the absence of detectable GPS. This was associated with a reduction in the level of crystalline cellulose and increased amounts of three proteins in the coat. The absence of the GPS was also associated with severe loss of coat physical integrity and sensitivity of the enclosed spore to detergent and hypertonic stress. GPS appears to be synthesized in the Golgi body and plays a larger role than that of any individual coat protein whose function has also been tested by gene disruption.
pgtB and pgtC encode GPS GTs.
As reviewed in the introduction, the GPS of D. discoideum is related to the better characterized GPS from D. mucoroides based on similar developmental regulation of expression, localization, and immunological cross-reactivity. The composition of the D. discoideum GPS, inferred from the change in Gal and GalNAc in the pgtB and pgtC disruption strains compared to normal strain, is comparable to that of the major D. mucoroides species. In addition, pgtB- and pgtC-like sequences are highly conserved in the genome of D. purpureum (BLAST Expect = 0; see http://www.jgi.doe.gov/dpurpureum/), a cellular slime mold more diverged from D. discoideum than D. mucoroides. However, further studies are needed to confirm the expected identity of GPS from the two species.
Conclusions from the present study about the role of the GPS depend on the specificity of the gene disruptions. pgtB and pgtC are each predicted to encode type 2 transmembrane Golgi proteins with their GT domains projecting into the lumen, as inferred from the signal anchor-like sequence near its N terminus. Localization in the lumen of the secretory pathway is consistent with the later appearance of enzyme activity in the ISM after sporulation (33), possibly as a result of proteolytic clipping from the membrane anchor and secretion as occurs for many Golgi GTs (see, for example, reference 4). These observations are consistent with the Golgi being the site of biosynthesis of the GPS (14, 34). However, family GT2 and GT4 domains, as found in PgtB and PgtC, are typically located in or oriented toward the cytoplasm (16, 40). If PgtB and PgtC were anchored in the membrane with their catalytic domains oriented in this way, as occurs for prokaryote di-GT GAG synthases and many GTs involved in lipopolysaccharide and capsule formation (9, 45), the polysaccharide must be translocated across the Golgi membrane. Translocation of lipopolysaccharide precursors and group 1 and group 4 capsular polysaccharides in bacteria require a member of the Wzx family of flippases (45), but no related sequences are detected in the genome of D. discoideum (Expect value of >3.0 using various Wzx-like query sequences). Similarly, no sequences related to Wzy, involved in assembly of these lipid-linked polysaccharides, are detectable with an Expect value of <3.0. Group 2 and group 3 capsular polysaccharides are translocated by using ABC-A2-type transporters such as KpsM (45). Although the Dictyostelium genome harbors five genes in this class (1), none match KpsM-like query sequences with an Expect value of <3.0. Thus, if GPS is polymerized on the cytoplasmic face of a membrane, it does not appear to be translocated by a conserved characterized bacterial mechanism. Further studies are required to determine the mechanism of initiation or priming of GPS synthesis and whether GPS is elongated within the Golgi lumen or on the cytoplasmic face of the Golgi membrane coupled with translocation.
The domain organizations of PgtB and PgtC resemble that of the cytoplasmic di-GT PgtA also discovered in Dictyostelium, and each possesses a family GT2 domain. The GT domains in PgtA act processively but not recursively on the same glycan to extend a monosaccharide to a trisaccharide (36) on the cytoplasmic/nuclear glycoprotein Skp1. In other di-GTs, sugar addition reiterates to form a repeating disaccharide (9). Consistent with the latter model, PgtB and PgtC contain a family GT2 sequence, expected to invert the linkage of the sugar nucleotide donor to add a β-linked sugar, and a family GT4 sequence, expected to add an α-linked sugar, as found in the linear backbone of the D. mucoroides GPS (27). PgtB and PgtC appear to be related by a recent gene duplication; in addition, PgtB acquired an additional, C-terminal domain, which might have a separate function such as branch formation. The loss of >75% of GPS synthase-like activity in the ISM (Fig. 3B), and the essentially complete absence of Gal and GalNAc, which are rare in Dictyostelium glycoproteins (13), from mutant spores and spore coat preparations (Fig. 3C to E) are consistent with direct roles of these genes in GPS synthesis. The induction of pgtB transcript expression in the slug (Fig. 2) is consistent with a specific role in GPS formation, and expression at other stages suggest the possibility of a low level of expression below our detection threshold.
The complete loss of GPS resulting from the disruption of either gene suggests that they collaborate in formation of the GPS backbone, which may be heterogeneously modified. The requirement for two genes is reminiscent of the mechanism of animal heparan sulfate GAG (HS-GAG) formation. The repeating disaccharide of HS-GAG appears to require a heterodimer of EXT1 and EXT2, each of which has two apparent GT domains (18, 21). The apparent Mr of 200,000 for secreted GPS synthase (24) is consistent with the size of a heterodimer after cleavage from the membrane anchor. An intriguing property of the partially purified enzyme is its higher activity at 0°C relative to 22°C (24, 32), which might be explained by relief of a negative entropic effect on heterodimer formation in extracts. In this scenario, the noncoordinate expression of pgtB and pgtC transcripts (Fig. 2) implies that PgtB controls heterodimer formation, but nonstoichiometric models should also be considered. For example, the proteins may separately catalyze GPS initiation (primer formation) and elongation, one protein may support the other as a chaperone (15), or the genes may also contribute to the formation of distinct glycans. Although the biochemical functions of the two proteins remain to be elucidated, the similar effects of disruption of either on the spore coat as discussed below make it unlikely that the effects derive from other unknown functions of the genes.
Role of GPS in cellulose synthesis.
Cellulose levels are heterogeneously reduced in mutant coats based on Calcofluor fluorescence, transmission electron microscopy, and sugar analyses (Fig. 5). A comparison of the range of electron miscroscopic images with the flow cytometry profile of Calcofluor-labeled spores suggests that a minority population of spores produce near-normal levels of cellulose that is nevertheless incorrectly packaged or crystallized. The β4-glucan chains of cellulose are polymerized at the cytoplasmic face of the plasma membrane and simultaneously translocated to the cell surface (29). Each β4-glucan chain is apparently produced by a single transmembrane cellulose synthase protein subunit, encoded by a single gene in Dictyostelium (2). An aggregate of synthases, referred to as the terminal complex, facilitates bundling of the nascent glucan chains into crystalline microfibrils (11). Since GPS secretion precedes cellulose synthesis (47), the β4-glucan chains are exposed to the GPS as they exit the terminal complex. The missing cellulose was not recovered in the ISM, suggesting that cellulose synthesis itself is inhibited. The selective inhibition of the synthesis of crystalline cellulose, relative to amorphous cellulose, suggests that the GPS, which represents about one-third the mass of cellulose (43), may normally facilitate its crystallinization and that failure to crystallize negatively affects synthesis. Consistent with this model, a physical association of the GPS with the forming coat is implied because GPS is not shared between neighboring spores (Fig. 3E). An association with cellulose is suggested by colocalization in the mature coat (Fig. 5B) and its resistance to denaturants that extract protein but not cellulose (48).
In vascular plant cell walls, cellulose fibrils are cross-bridged by hemicellulose and embedded in a pectin polysaccharide gel (7). However, the GPS may have a distinct mechanism of action in the spore coat, because it does not have a β4-linked homopolymeric backbone-like hemicelluloses and is apparently not negatively charged like pectins. Indeed, distinct polysaccharides accompany cellulose in nonplant cell walls (37), suggesting alternative mechanisms for regulating and organizing cellulose. Genetic analysis of plant cellulose synthesis shows that several factors, including the korrigan endoglucanase, contribute to efficient fibril formation or crystallinity (29), and here we suggest that a heteropolysaccharide is a second factor important for cellulose organization.
Role of GPS in coat protein accumulation.
A second molecular difference in mutant coats was the increased levels of the inner layer protein SP85, and SP80 and SP87 whose locations are unassigned. The outer layer proteins SP96 and SP75 were not affected (Fig. 6), a finding consistent with the normal ultrastructural appearance of the outer layer (Fig. 5B). The increase could be at least partially traced back to the slug, where some coat precursors, including these proteins and the GPS, are stored in PSVs (37). The mechanism by which the levels of protein precursors are affected, possibly in compensation, by absence of the GPS is not known. Previous studies showed that SP85, which binds cellulose in vitro (48), influences cellulose accumulation in vivo. SP85-null cells and strains expressing domain fragments of SP85 produce cellulose with markedly reduced crystalline content (22, 43). The effect of overexpressing normal SP85 is not known but represents an additional potential mechanism for how the loss of GPS affects cellulose.
Role of GPS in spore integrity.
Although pgtB and pgtC mutant spores can germinate, they are unusually sensitive to stress. First, mutant spores are damaged by hypertonic glycerol treatment that does not affect normal spores (Fig. 4). A subpopulation, possibly the intrinsically autofluorescent spores (Fig. 4C), labels almost immediately with DAPI, indicating the loss of membrane impermeability. Most spores collapse to an extreme degree and gradually recover their normal shapes but succumb within 1 to 2 days, becoming phase dark and DAPI positive. The extreme deformation implies that the mutant coat remains firmly bonded to the plasma membrane but is more compliant. Second, mutant coats are so physically delicate that French press shearing of spores obliterates them. Finally, nearly the whole population of mutant spores is sensitive to nonionic detergent, implying a breach in coat barrier function, thought to reside in the outer layer. The increased protein and reduced content of crystalline cellulose might explain the physical and biological spore defects. With a fabric-like arrangement in the coat (12, 17), cellulose microfibrils form a rigid protective shell expected to be compromised by reduced crystallinity. Cellulose and inner layer proteins contribute to the formation of the outer layer permeability barrier (22, 47), expected to be compromised by the altered levels of cellulose and protein, and inner layer proteins contribute to cellulose crystallinity. Further understanding of GPS function will be facilitated by determining the structure(s) of GPS from D. discoideum, understanding the mechanism of GPS initiation, elongation, and branching, including the specific contributions of PgtB and PgtC and a related gene (pgtD), assessment of potential physical interactions between the GPS and cellulose or coat proteins, and further structural studies on the organization of cellulose in the mutants.
Supplementary Material
Acknowledgments
We gratefully acknowledge Keiji Okamoto for providing the polysaccharide acceptor from D. mucoroides, T. K. Johnson and Wendy Ives of the Oklahoma Center for Medical Glycobiology Core Lab for the sugar composition analyses, Mandrin Shima for PCR studies, Tasha Arnett for her early assistance in developing the GalT and GalNAcT assays, Jim Henthorn at the OUHSC Flow and Image Cytometry Laboratory for the flow cytometry, Margaret Nelson for pHygT(plus)/pG7, and Daphne Blumberg for the anti-PL3 antibody.
This study was supported in part by National Science Foundation grant no. MCB-0350516 to C.M.W., NIH grant no. R01-AI069986 to I.J.B., and the SURE program sponsored by the Presbyterian Health Foundation.
Footnotes
Published ahead of print on 7 November 2008.
Supplemental material for this article may be found at http://ec.asm.org/.
REFERENCES
- 1.Anjard, C., W. F. Loomis, et al. 2002. Evolutionary analyses of ABC transporters of Dictyostelium discoideum. Eukaryot. Cell 1643-652. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Blanton, R. L., D. Fuller, N. Iranfar, M. J. Grimson, and W. F. Loomis. 2000. The cellulose synthase gene of Dictyostelium. Proc. Natl. Acad. Sci. USA 972391-2396. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Brown, R. M., Jr., C. H. Haigler, and K. M. Cooper. 1982. Experimental induction of altered non-microfibrillar cellulose. Science 2181141-1142. [DOI] [PubMed] [Google Scholar]
- 4.Cho, S. K., J. C. Yeh, and R. D. Cummings. 1997. Secretion of alpha1,3-galactosyltransferase by cultured cells and presence of enzyme in animal sera. Glycoconj. J. 14809-819. [DOI] [PubMed] [Google Scholar]
- 5.Colley, K. J. 1997. Golgi localization of glycosyltransferases: more questions than answers. Glycobiology 71-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Cooper, D. N., S. C. Lee, and S. H. Barondes. 1983. Discoidin-binding polysaccharide from Dictyostelium discoideum. J. Biol. Chem. 2588745-8750. [PubMed] [Google Scholar]
- 7.Cosgrove, D. J. 2005. Growth of the plant cell wall. Nat. Rev. Mol. Cell. Biol. 6850-861. [DOI] [PubMed] [Google Scholar]
- 8.Coutinho, P. M., E. Deleury, G. J. Davies, and B. Henrissat. 2003. An evolving hierarchical family classification for glycosyltransferases. J. Mol. Biol. 328307-317. [DOI] [PubMed] [Google Scholar]
- 9.DeAngelis, P. L. 2002. Microbial glycosaminoglycan glycosyltransferases. Glycobiology 129R-16R. [DOI] [PubMed] [Google Scholar]
- 10.Erdos, G. W., and C. M. West. 1989. Formation and organization of the spore coat of Dictyostelium discoideum. Exp. Mycol. 13169-182. [Google Scholar]
- 11.Grimson, M. J., C. H. Haigler, and R. L. Blanton. 1996. Cellulose microfibrils, cell motility, and plasma membrane protein organization change in parallel during culmination in Dictyostelium discoideum. J. Cell Sci. 1093079-3087. [DOI] [PubMed] [Google Scholar]
- 12.Hemmes, D. E., E. S. Kojima-Buddenhagen, and H. R. Hohl. 1972. Structural and enzymatic analysis of the spore wall layers in Dictyostelium discoideum. J. Ultrastruct. Res. 41406-417. [DOI] [PubMed] [Google Scholar]
- 13.Hoffman, S., and D. McMahon. 1978. Defective glycoproteins in the plasma membrane of an aggregation minus mutant of Dictyostelium discoideum with abnormal cellular interactions. J. Biol. Chem. 253278-287. [PubMed] [Google Scholar]
- 14.Ikeda, T. 1981. Subcellular distributions of UDP-galactose:polysaccharide transferase and UDP-glucose pyrophosphorylase involved in biosynthesis of prespore-specific acid mucopolysaccharide in Dictyostelium discoideum. Biochim. Biophys. Acta 67569-76. [DOI] [PubMed] [Google Scholar]
- 15.Ju, T., and R. D. Cummings. 2002. A unique molecular chaperone Cosmc required for activity of the mammalian core 1 beta 3-galactosyltransferase. Proc. Natl. Acad. Sci. USA 9916613-16618. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Kapitonov, D., and R. K. Yu. 1999. Conserved domains of glycosyltransferases. Glycobiology 9961-978. [DOI] [PubMed] [Google Scholar]
- 17.Kessin, R. H. 2001. Dictyostelium: evolution, cell biology, and the development of multicellularity, p.212. Cambridge University Press, Cambridge, United Kingdom.
- 18.Kim, B.-T., H. Kitagawa, J. Tanaka, J. Tamura, and K. Sugahara. 2003. In vitro heparan sulfate polymerization: crucial roles of core protein moieties of primer substrates in addition to the EXT1-EXT2 interaction. J. Biol. Chem. 27841618-41623. [DOI] [PubMed] [Google Scholar]
- 19.Kreil, D. P., and G. Kreil. 2000. Asparagine repeats are rare in mammalian proteins. Trends Biochem. Sci. 25270-271. [DOI] [PubMed] [Google Scholar]
- 20.Livak, K. J., and T. D. Schmittgen. 2001. Analysis of relative gene expression data using real-time quantitative PCR and the 2−ΔΔCT method. Methods 25402-408. [DOI] [PubMed] [Google Scholar]
- 21.McCormick, C., G. Duncan, K. T. Goutosos, and F. Tufaro. 2000. The putative tumor suppressors EXT1 and EXT2 form a stable complex that accumulates in the Golgi apparatus and catalyzes the synthesis of heparan sulfate. Proc. Natl. Acad. Sci. USA 97668-673. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Metcalf, T., K. Kelley, G. W. Erdos, L. Kaplan, and C. M. West. 2003. Formation of the outer layer of the spore coat of Dictyostelium depends on the inner layer protein SP85/PsB. Microbiology 149305-317. [DOI] [PubMed] [Google Scholar]
- 23.Metcalf, T., H. van der Wel, R. Escalante, L. Sastre, and C. M. West. 2007. The role of SP65 in assembly of the Dictyostelium spore coat. Eukaryot. Cell 61137-1149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Nakahara, Y., and K. Okamoto. 2004. Unusual properties of the prespore-specific enzyme, UDPgalactose:polysaccharide galactosyl transferase, of Dictyostelium discoideum. J. Basic Microbiol. 44459-470. [DOI] [PubMed] [Google Scholar]
- 25.Phelps, E. D., K. R. Sweeney, and I. J. Blader. 2008. Toxoplasma gondii rhoptry discharge correlates with activation of the EGR2 host cell transcription factor. Infect. Immun., in press. [DOI] [PMC free article] [PubMed]
- 26.Puta, F., and C. Zeng. 1998. Blasticidin resistance cassette in symmetrical polylinkers for insertional inactivation of genes in Dictyostelium. Folia Biol. 44185-188. [PubMed] [Google Scholar]
- 27.Sakurai, M. H., H. Kiyohara, Y. Nakahara, K. Okamoto, and H. Yamada. 2002. Galactose-containing polysaccharides from Dictyostelium mucoroides as possible acceptor molecules for cell-type specific galactosyl transferase. Comp. Biochem. Physiol. B Biochem. Mol. Biol. 132541-549. [DOI] [PubMed] [Google Scholar]
- 28.Sassi, S., M. Sweetinburgh, J. Erogul, P. Zhang, P. Teng-umnuay, and C. M. West. 2001. Analysis of Skp1 glycosylation and nuclear enrichment in Dictyostelium. Glycobiology 11283-295. [DOI] [PubMed] [Google Scholar]
- 29.Somerville, C. 2006. Cellulose synthesis in higher plants. Annu. Rev. Cell Dev. Biol. 2253-78. [DOI] [PubMed] [Google Scholar]
- 30.Srinivasan, S., K. R. Griffiths, V. McGuire, A. Champion, K. L. Williams, and S. Alexander. 2000. The cellulose-binding activity of the PsB multiprotein complex is required for proper assembly of the spore coat and spore viability in Dictyostelium discoideum. Microbiology 1461829-1839. [DOI] [PubMed] [Google Scholar]
- 31.Sussman, M. 1987. Cultivation and synchronous morphogenesis of Dictyostelium under controlled experimental conditions. Methods Cell Biol. 289-29. [DOI] [PubMed] [Google Scholar]
- 32.Sussman, M., and M. J. Osborn. 1964. UDP-galactose polysaccharide transferase I the cellular slime mold Dictyostelium discoideum: appearance and disappearance of activity during cell differentiation. Proc. Natl. Acad. Sci. USA 5281-87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Sussman, M., and N. Lovgren. 1965. Preferential release of the enzyme UDP-galactose polysaccharide transferase during cellular differentiation in the slime mold, Dictyostelium discoideum. Exp. Cell Res. 3897-105. [DOI] [PubMed] [Google Scholar]
- 34.Takemoto, K., A. Yamamoto, and I. Takeuchi. 1985. The origin of prespore vacuoles in Dictyostelium discoideum cells as analysed by electron-microscopic immunocytochemistry and radioautography. J. Cell Sci. 7793-108. [DOI] [PubMed] [Google Scholar]
- 35.Takeuchi, I. 1963. Immunochemical and immunohistochemical studies on the development of the cellular slime mold Dictyostelium mucoroides. Dev. Biol. 81-26. [DOI] [PubMed] [Google Scholar]
- 36.van der Wel, H., S. Z. Fisher, and C. M. West. 2002. A bifunctional diglycosyltransferase forms the Fucα1,2Galβ,3-disaccharide on Skp1 in the cytoplasm of Dictyostelium. J. Biol. Chem. 27746527-46534. [DOI] [PubMed] [Google Scholar]
- 37.West, C. M. 2003. Comparative analysis of spore coat formation, structure, and function in Dictyostelium. Int. Rev. Cytol. 222237-293. [DOI] [PubMed] [Google Scholar]
- 38.West, C. M., and G. W. Erdos. 1988. The expression of glycoproteins in the extracellular matrix of the cellular slime mold Dictyostelium discoideum. Cell Differ. 231-16. [DOI] [PubMed] [Google Scholar]
- 39.West, C. M., and G. W. Erdos. 1992. Incorporation of protein into spore coats is not cell-autonomous in Dictyostelium. J. Cell Biol. 1161291-1300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.West, C. M., H. van der Wel, and E. A. Gaucher. 2002. Complex glycosylation of Skp1 in Dictyostelium: implications for the modification of other eukaryotic cytoplasmic and nuclear proteins. Glycobiology 1217R-27R. [DOI] [PubMed] [Google Scholar]
- 41.West, C. M., H. van der Wel, P. M. Coutinho, and B. Henrissat. 2005. Glycosyltransferase genomics in Dictyostelium discoideum, p.235-264. In W. F. Loomis and A. Kuspa (ed.), Dictyostelium genomics. Horizon Scientific Press, Norfolk, United Kingdom.
- 42.West, C. M., H. van der Wel, and Z. A. Wang. 2007. Prolyl 4-hydroxylase-1 mediates O2-signaling during development of Dictyostelium. Development 1343349-3358. [DOI] [PubMed] [Google Scholar]
- 43.West, C. M., P. Zhang, A. C. McGlynn, and L. Kaplan. 2002. Outside-In signaling of cellulose synthesis by a spore coat protein in Dictyostelium. Eukaryot. Cell 1281-292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.White, G., and M. Sussman. 1963. Polysaccharides involved in slime mold development. II. Water-soluble acid mucopolysaccharides(s). Biochim. Biophys. Acta 74179-187. [DOI] [PubMed] [Google Scholar]
- 45.Whitfield, C. 2006. Biosynthesis and assembly of capsular polysaccharides in Escherichia coli. Annu. Rev. Biochem. 7539-68. [DOI] [PubMed] [Google Scholar]
- 46.Yoder, B. K., J. Mao, G. W. Erdos, C. M. West, and D. D. Blumberg. 1994. Identification of a new spore coat protein gene in the cellular slime mold Dictyostelium discoideum. Develop. Biol. 16349-65. [DOI] [PubMed] [Google Scholar]
- 47.Zhang, P., A. C. McGlynn, W. F. Loomis, R. L. Blanton, and C. M. West. 2001. Spore coat formation and timely sporulation depend on cellulose in Dictyostelium. Differentiation 6772-79. [DOI] [PubMed] [Google Scholar]
- 48.Zhang, Y., R. D. Brown, and C. M. West. 1998. Two proteins of the Dictyostelium spore coat bind to cellulose in vitro. Biochemistry 3710766-10779. [DOI] [PubMed] [Google Scholar]
- 49.Zhang, Y., P. Zhang, and C. M. West. 1999. A linking function for the cellulose-binding protein SP85 in the spore coat of Dictyostelium discoideum. J. Cell Sci. 1124367-4377. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.