

Abstract
Clinical and experimental evidence suggests that spreading of malignant cells from a localized tumor (metastasis) is directly related to the number of microvessels in the primary tumor. This tumor angiogenesis is thought to be mediated by tumor-cell-derived growth factors. However, most tumor cells express a multitude of candidate angiogenesis factors and it is difficult to decipher which of these are rate-limiting factors in vivo. Herein we use ribozyme targeting of pleiotrophin (PTN) in metastatic human melanoma cells to assess the significance of this secreted growth factor for angiogenesis and metastasis. As a model we used human melanoma cells (1205LU) that express high levels of PTN and metastasize from subcutaneous tumors to the lungs of experimental animals. In these melanoma cells, we reduced PTN mRNA and growth factor activity by transfection with PTN-targeted ribozymes and generated cell lines expressing different levels of PTN. We found that the reduction of PTN does not affect growth of the melanoma cells in vitro. In nude mice, however, tumor growth and angiogenesis were decreased in parallel with the reduced PTN levels and apoptosis in the tumors was increased. Concomitantly, the metastatic spread of the tumors from the subcutaneous site to the lungs was prevented. These studies support a direct link between tumor angiogenesis and metastasis through a secreted growth factor and identify PTN as a candidate factor that may be rate-limiting for human melanoma metastasis.
The significance of blood vessel formation (angiogenesis) for the growth of solid tumors and ultimately their metastatic spread has gained wide acceptance. The close relationship between tumor angiogenesis and metastasis has been demonstrated in a series of correlative clinical studies published in the past 5 years after an initial report by Weidner et al. (1) in 1991. In general, these angiogenesis studies showed that the numbers of blood vessels in a given primary tumor specimen is indicative of the rate of metastasis of the respective tumor and provides an independent parameter that helps predict the clinical outcome of the disease. So far a number of different tumor types have been studied and a direct correlation between blood vessel density in primary tumors and their metastasis has been reported for cancers of the breast, lung, prostate, head and neck, ovary, and stomach, as well as for melanoma (for review, see ref. 2).
Since angiogenesis is such an important feature of tumor biology, the factors driving this process need to be understood for further diagnostic or therapeutic exploitation (for review, see, e.g., refs. 3 and 4). It is conceivable that all angiogenic factors present in a given tumor contribute to and are required for the angiogenic and metastatic phenotype of the particular tumor. On the other hand, it is also possible that very few crucial gene products will emerge as rate-limiting factors and that metastasis of a given tumor is ultimately dependent on the production of one major angiogenic growth factor. One of the difficulties in distinguishing between these two concepts is due to the fact that most tumor cell lines constitutively express a multitude of candidate angiogenesis factors in vitro and tumor specimen contain additional growth factor proteins derived from stromal sources (see, e.g., ref. 3). Furthermore, very few specific and efficacious inhibitors have been generated to assess in vivo which of the gene products found in a tumor are only an innocent marker and which contribute significantly to angiogenesis and metastasis. For example, recent studies with blocking antibodies support the notion that vascular endothelial growth factor/vascular permeability factor (VEGF/VPF) (5, 6) can contribute to metastatic seeding of tumor cells. In vivo treatment with anti-VEGF/VPF antibodies blocked human HT-1080 fibrosarcoma seeding in the lungs after intravenous injection (7) and also reduced liver metastasis from intrasplenic injection of human colon cancer cells (8).
In our experiments, we studied the significance of pleiotrophin (PTN) for melanoma angiogenesis and metastasis. PTN is a secreted polypeptide growth factor (9) that is expressed in a variety of established tumor cell lines and primary human tumor specimen (10) and is a mitogen for fibroblasts (9), epithelial cells derived from bovine lens (11) or human adrenal carcinoma (10, 12), and endothelial cells (10, 11). Furthermore, PTN can induce the release of active proteolytic enzymes from endothelial cells (13), can induce tube formation of endothelial cells in vitro (11), and thus, may be able to serve as a tumor angiogenesis factor. Among several tumor types expressing PTN, melanoma seemed most suitable to study the significance of an individual angiogenic factor for tumor metastasis because these tumors grow rapidly and are fatal due to their early metastatic spread in the body. Furthermore, we had observed that a number of melanoma cell lines but not melanocytes expressed this gene product (10).
As a model we selected human melanoma cells (1205LU) that constitutively express high levels of PTN mRNA, grow subcutaneous tumors in nude mice, and metastasize to the lungs from the subcutaneous tumor site. PTN production in these cells was reduced with specific ribozymes that cleave PTN mRNA and, thus, can decrease the amount of endogenous PTN mRNA and secreted protein (14). We speculated that a reduction of PTN production in the melanoma cells might reduce angiogenesis in the primary tumors growing at the subcutaneous injection site and that this reduced angiogenesis might in turn reduce the opportunity of the tumor cells to gain access to the blood stream and form hematogenous lung metastases. To address this hypothesis, we decided to generate a panel of tumor cell lines that produce different residual levels of their endogenous PTN after stable transfection with PTN-targeted ribozymes (14) and then study their in vitro and in vivo phenotype. By “dosing” the endogenous PTN expression to different levels, we also hoped to find out how far PTN expression would have to be reduced to show any effect on tumor growth, angiogenesis, and metastasis in vivo. Promising data from this study would imply to what extent targeting of PTN may be successful in gene therapy applications of our approach.
The results obtained in the present study show a direct link among spontaneous expression of a secreted polypeptide growth factor (PTN) in human melanoma cells in vivo, tumor angiogenesis, and metastasis. Furthermore, our data suggest that a partial reduction of PTN in a fraction of the tumor cells has a significant effect on in vivo tumor growth.
MATERIALS AND METHODS
Generation of Constructs.
Construction of the ribozyme expression vectors pRz66 and pRz261 was described earlier (14). In brief, to generate the ribozyme constructs, synthetic sense and antisense oligonucleotides containing the catalytic center and the flanking regions of each of the ribozymes were annealed and ligated into the HindIII/NotI sites of the pRc/CMV plasmid (Invitrogen) that also contains a G418 drug-resistance gene and expresses the ribozymes under the control of a cytomegalovirus (CMV) promoter (10). Both ribozyme expression vectors contain antisense flanking regions of 12 and 11 nt or 9 and 10 nt, respectively, that target the ribozyme transcripts to their respective cleavage site 3′ of nt 66 or 261 in the open reading frame of the human PTN mRNA (GenBank accession no. M53799M53799).
Cell Lines, Transfections, and Growth Assays.
Human melanoma cells (1205LU, gift from M. Heerlyn, Wistar Institute, Philadelphia) were transfected with 10 μg of empty vectors or pRz66 or pRz261 using LipofectAmine (Life Technologies, Gaithersburg, MD). Briefly, cells at 50% confluence were incubated for 5 h with plasmid DNA mixed with 70 μl of LipofectAmine reagant in serum-free medium (Optimem, Life Technologies) at 37°C in 5% CO2/95% air. The transfection medium was then replaced with fresh medium and 36 h later G418 (1500 μg/ml) was added to select stably transfected cells during another 4–6 weeks of culture in the presence of the drug (pRz66-mass). Clonal cell lines were obtained by limited dilution of a suspension of freshly transfected cells and subsequent growth of individual clones in the presence of G418 (clones pRz66-4, -6, and -10 and pRz261-1 and -6). To assess the amount of growth factor activity released into the supernatants of transfected tumor cells, soft agar colony formation of SW-13 cells was used as described (12). In brief, conditioned medium from the differently transfected cells was concentrated and partially purified by heparin affinity chromatography. The 0.9 M NaCl eluates from the columns contained the peak amount of PTN as assessed by immunoassay (see below) and an aliquot was assayed for soft agar colony-stimulating activity of the indicator cell line SW-13.
Northern Blots and Immunoassay for PTN.
For the Northern blots, 20 μg of total RNA from each cell line was separated in an agarose gel and blotted onto nitrocellulose, hybridized with a PTN cDNA probe, and autoradiographed for 48 h (10). For quantitation, a PhosphorImager was used. To correct for different loading among different lanes, blots were stripped and probed for β-actin. For the immunoassays, conditioned medium from the different transfected cells was concentrated and partially purified by heparin affinity chromatography (12) and eluates were assayed for PTN protein by using an immunoassay with rabbit antiserum as described (10).
Tumor Growth and Metastasis in Animals.
One million tumor cells in 0.1 ml of growth medium were injected into two subcutaneous sites on the flanks of athymic nude mice (female NCr nu/nu; National Cancer Institute) and observed for up to 3 months for tumor growth. Tumor sizes were estimated from the product of the perpendicular diameters of the tumors. In five studies, tumors grown from control cells reached a mean size of 150 mm2 within 3 weeks after initial inoculation. Transfection of an empty vector did not affect this tumor growth. Unless indicated otherwise, tumors carried by an animal were resected surgically when they reached between 50 and 150 mm2. Lung metastasis was assessed at different time points (see Results) after tumor cell inoculation by macroscopic inspection of the lungs. Paraffin embedding and hematoxylin/eosin staining of at least three sections per lung was used to check for micrometastases. Examples of the stainings are shown in Fig. 4.
Figure 4.
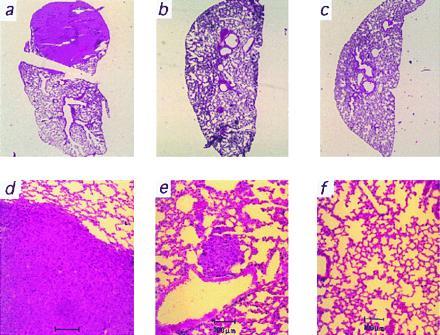
Representative microscopic analysis of lung metastases growing from melanoma cells producing different amounts of PTN mRNA. Hematoxylin/eosin-stained whole mount sections (a–c) and microscopic views (d–f) of representative lungs from the groups producing high (a and d), medium (b and e), and low levels (c and f) of PTN are shown. For quantitative data, see Fig. 2d and Results.
Mouse Anti-CD31 (PECAM-1) Immunostaining for Capillaries.
Tissues were fixed in 10% buffered formalin and embedded in paraffin blocks. Five-micrometer sections were cut, and the sections were deparaffinized and rehydrated by passage through decreasing ethanol concentrations. The slides were then incubated for 30 min in 0.3% hydrogen peroxide solution (Mallinckrodt), washed twice in phosphate-buffered saline (PBS) (Sigma) and blocked with a 1:66 dilution of normal horse serum (Vector Laboratories) for 30 min. The blocking serum was then removed by tapping, replaced by a 1:20 dilution of primary anti-mouse CD31 monoclonal rat antibody (PharMingen) in blocking serum, and incubated for 2 h at 4°C in a humid chamber. The slides were then washed twice with PBS for 5 min, incubated with a 1:200 dilution of secondary peroxidase-coupled rabbit anti-rat (mouse adsorbed) serum (Vector Laboratories) for 30 min. After two 5-min washes in PBS, immunocomplexes were highlighted using the ABC kit (Biomedia, Foster City, CA) with diaminobenzidine (Sigma) as a substrate. Finally, slides were counterstained using Harris hematoxylin (Lerner Laboratories, New Haven, CT), dehydrated, and mounted. Stainable capillaries were counted at ×400 magnification by independent investigators who were not informed about the origin of individual samples.
Evaluation of Apoptosis.
The extent of apoptosis was assessed in tumor tissues by an ELISA (cell death detection; Boehringer Mannheim) that quantitates apoptotic DNA fragmentation in cells by detecting DNA–histone complexes (15). From each tumor studied, three separate biopsies were used for the assay. The same tumor samples were stained for apoptotic nuclei using TdT(terminal deoxynucleotidyltransferase)-mediated dUTP nick end labeling (TUNEL; ref. 16) to confirm the ELISA results and rule out necrosis in the tumors.
Data Analysis.
Mean ± SEM values are depicted unless indicated otherwise. Student’s t test was used for comparisons between data sets and P < 0.05 was considered significant.
RESULTS
For our studies we chose 1205LU human melanoma cells as a model. These cells express high levels of PTN and metastasize to the lungs after subcutaneous inoculation in athymic nude mice (see below). We used ribozyme targeting of PTN in these tumor cells to generate a panel of cell lines expressing different levels of the growth factor and then subjected these cell lines to further in vitro and in vivo studies.
Ribozyme Targeting of Endogenous PTN mRNA in 1205LU Melanoma Cells.
Expression of PTN mRNA in the 1205LU human melanoma cell line used in our studies is shown in a Northern blot (Fig. 1a, lane 1). Obviously, this cell line expresses easily detectable amounts of PTN mRNA and was one of the highest expressors among all of the cell lines studied by us (see also refs. 10 and 14). Ribozyme targeting of PTN in the 1205LU cells was applied to generate a panel of derivative cell lines that express different extents of residual PTN. Expression vectors for hammerhead ribozymes that are targeted against two separate sites in the PTN transcript were used (14). The efficacy and specificity of the PTN-targeted ribozymes transcribed from these vectors was shown earlier against spontaneously expressed PTN in WM852 melanoma cells and in cotransfection studies with a PTN expression vector in PTN-negative SW-13 cells (14).
Figure 1.
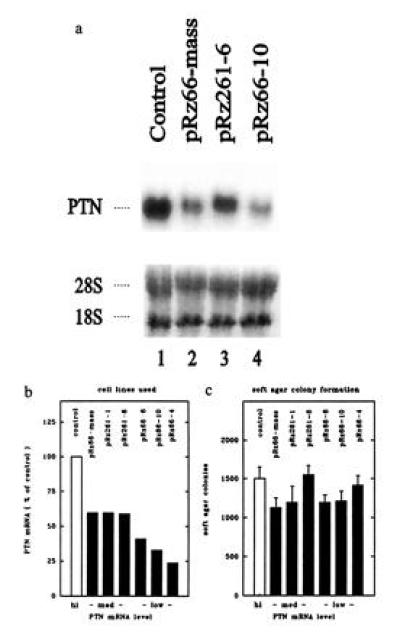
In vitro characteristics of 1205LU melanoma cells after stable transfection with PTN-targeted ribozymes. (a) Northern blot. (b) PTN mRNA levels. (c) Colony growth of melanoma cells in soft agar. (a) Representative Northern blot of some of the cell lines used. The PTN hybridization signal (Upper) and the ethidium bromide stain (Lower) of the respective gel are shown. 28S and 18S indicate the ribosomal RNA bands. (b) PTN mRNA levels of all cell lines used in the studies. After transfection of 1205LU cells with different ribozyme constructs, PTN mRNA levels of G418-resistant clonal or mass-transfected cell lines were examined by Northern blot analysis with β-actin as a loading control. Values are expressed relative to control. Clonal cell lines (pRz66-4, pRz66-6, pRz66-10, pRz261-1, and pRz261-6) and a pool of G418-resistant cells (pRz66-mass) were used. The cell lines are grouped according to their PTN mRNA levels into high (= 100%), medium (>50%), and low (<50%). (c) Soft agar colony growth of melanoma cells. The number of colonies (> 60 μm) grown after 10 days of incubation is shown.
After transfection of 1205LU melanoma cells with the ribozyme expression constructs, we used G418 drug selection to generate stable mass-transfected and clonal derivative cell lines. We assessed the amount of residual PTN mRNA in different cell lines by Northern blot analysis. An example of a Northern blot with some of the cell lines is shown in Fig. 1a and the quantitative results from the seven cell lines used in the further studies is depicted in Fig. 1b. We chose cell lines that express high, medium, and low levels of PTN mRNA (Fig. 1b) and the data presented throughout this paper are organized accordingly. We wish to emphasize that mass-transfected and clonal cell lines as well as cell lines transfected with either of the ribozyme constructs (Rz66 and Rz261) are represented in this set.
Characterization in Vitro of the Melanoma Cell Lines Expressing Different Levels of PTN mRNA.
In vitro growth. Previous studies with SW-13 cells (10) and NIH 3T3 fibroblasts (17) showed that PTN is an autocrine-acting growth factor for these cells. In parallel with this, we demonstrated recently, that the reduction of PTN in WM852 human melanoma cells by ribozyme targeting reduces their colony formation in vitro and coincidently tumor growth in nude mice (14). Obviously, in the WM852 melanoma cells endogenously expressed PTN also serves as an autocrine growth factor. In contrast with these previous studies, PTN does not appear to be a rate-limiting growth factor for 1205LU melanoma cells in vitro. The reduction of PTN did not affect spontaneous colony formation in soft agar of the various 1205LU cell lines (Fig. 1c) or their proliferation on plastic surfaces (data not shown).
Growth factor activity in the supernatants.
Since PTN is a secreted growth factor (12), we also assessed residual growth factor activity in the supernatants of the different 1205LU cell lines by using soft agar colony formation of SW-13 cells as a bioassay (12). In this semiquantitative assay, an average reduction of growth stimulatory activity by 40–75% in the “medium PTN” and by 70–90% in the “low PTN” group was found. In endothelial cell assays, a similar reduction of growth factor activity was observed as with the SW-13 cell assay. This result suggests that PTN is the predominant growth factor activity among a number of growth factors that can stimulate these two indicator cell types and are commonly found expressed in melanoma (18).
Subcutaneous Tumor Growth in Mice of Cell Lines Expressing Different Levels of PTN.
When injected into nude mice, subcutaneous tumor growth of the melanoma cells producing different residual levels of PTN was reduced in parallel with the reduced levels of growth factor mRNA levels (compare Figs. 1b and 2 a and b). Much to our surprise, even a 40% reduction of endogenous PTN mRNA resulted in a dramatic reduction by 65% of the growth rate of the subcutaneous tumors (medium PTN group of cell lines, compare Figs. 1b and 2b; P < 0.01 vs. control). A further reduction of PTN in the tumor cells (low PTN group) resulted in a further reduction of the growth rate of the primary tumors (see Fig. 2b; P < 0.01 vs. control). Tumors in this group took almost three months to grow to a size reached by control tumors in 3 weeks (Fig. 2a). These data suggest that PTN is rate-limiting for the growth of these tumors in vivo. Furthermore, the effect of the PTN reduction on tumor growth in vivo in contrast to the lack of an effect on cell growth in vitro supports the notion that PTN acts as a paracrine growth factor for the tumor stroma (see above and Fig. 1c). This feature distinguishes the role of PTN for this melanoma cell line from WM852 melanoma cells used in our previous studies with PTN-targeted ribozymes (14).
Figure 2.
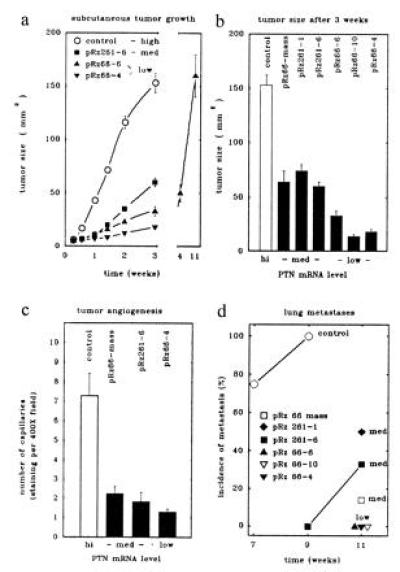
Subcutaneous tumor growth (a and b), angiogenesis (c), and lung metastasis (d) in nude mice of 1205LU melanoma cells producing different levels of PTN mRNA. (a and b) Complete growth curves of subcutaneous tumors from representative cell lines (a) and tumor sizes of all cell lines after 3 weeks (b) are shown. The cell lines producing different levels of PTN mRNA (see Fig. 1b) were injected subcutaneously into nude mice at 106 cells per injection site and two sites per animal (n = 12 animals for control and pRz261-6; n = 5 animals for the other groups). (c) Angiogenesis in subcutaneous tumors grown from representative groups of the melanoma cells expressing different levels of PTN mRNA was quantitated. Capillaries in tumors were highlighted by staining for PECAM (CD31). In three representative tumors from each group, the average number of capillaries in eight high-power fields (×400) was counted. The number of capillaries per field (±SD) of the counts of three investigators not informed about the origins of the samples are shown. (d) Quantitation of lung metastases. Subcutaneous tumors were resected after 3 weeks (controls) or 11 weeks (pRz66-6) or when they reached at least 50 mm2. Lungs were examined macroscopically or after hematoxylin/eosin staining (see Fig. 4). Animals with tumors from high-level PTN-producing cells showed macroscopically visible metastases (see Fig. 4a) whereas metastases in the others groups were only detectable after microscopic inspection of lung sections (see Fig. 4e). Animals with any detectable metastases were scored positive and the relative incidence of animals with lung metastases is shown.
Subcutaneous Tumor Growth of Mixtures of Melanoma Cells Expressing Different Levels of PTN.
Further analysis of the data presented above shows that tumor growth of heterogeneous mass-transfected cells was no different from tumor growth of clonal homogeneous cell lines that produced comparable levels of PTN (compare pRz66-mass versus pRz261-1 and pRz261-6; Figs. 1b and 2b). From this we hypothesized that higher or lower amounts of PTN produced by different subpopulations of tumor cells is averaged out in the tumor bed in vivo and that tumor growth is dependent on the overall paracrine stimulus generated by the tumor cell mass as a whole. Consequently, individual tumor cells or subpopulations would not gain or loose a selective growth advantage from a higher or lower production of PTN. In support of this hypothesis, we found no in vivo selection for high PTN producer subpopulations in Northern blots with tumors harvested at the end of the study (data not shown).
To test this hypothesis more rigorously, we mixed cells from the “high PTN” and the low PTN group at different ratios and inoculated a fixed total number of cells into nude mice (Fig. 3). Indeed, after 3 weeks the size of the subcutaneous tumors grown from these cell mixtures mimicked the results obtained with individual cell lines expressing different levels of PTN and tumor growth was reduced in parallel with a decreasing portion of high PTN cells (Fig. 3). Furthermore, PTN levels in these tumors assessed by Northern blots followed the levels predicted from the different ratios of cell mixtures (data not shown). From this set of data, it is tempting to speculate that therapeutic targeting of PTN should have an impact even if only a fraction of malignant cells can be reached in a solid tumor by the targeting method.
Figure 3.
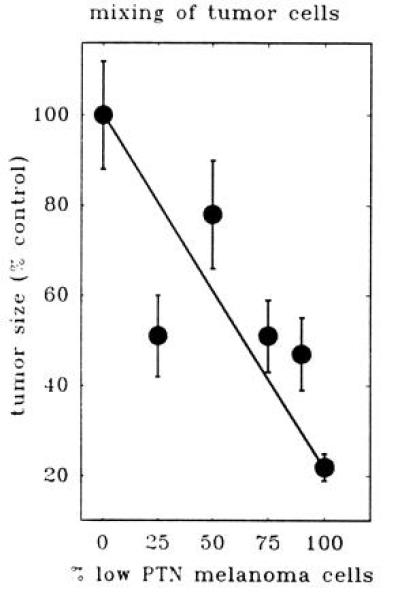
Tumor growth in nude mice of mixtures of melanoma cells producing high and low levels of PTN mRNA. Control cells producing high levels and ribozyme-transfected cells producing low levels of PTN (pRz66-10; see Fig. 1b) were mixed at different ratios. Of these mixtures, 106 cells per site were injected subcutaneously into two sites per mouse (n = 5 animals per group). Tumor sizes after 3 weeks of growth are shown relative to control.
Tumor Angiogenesis and PTN Levels.
To establish whether PTN acts as a mediator of tumor angiogenesis in vivo, we quantitated the number of microvessels in subcutaneous tumors using an endothelial cell marker (PECAM, platelet-endothelial cell adhesion molecule, CD31; ref. 19). For this analysis, we selected tumor samples from each of the groups with different levels of residual PTN. We took care that tumors of equal size were used in the assessment. A significant reduction of the number of microvessels per field by approximately 70% (P < 0.01 vs. control) was found in the medium PTN group and by 85% (P < 0.002 vs. control) in the low PTN group (Fig. 2c). We conclude from this result that PTN acts as a tumor angiogenesis factor in vivo. Furthermore, we propose that the reduced blood supply of the tumors due to the reduced levels of PTN results in the slower rate of tumor growth observed.
Apoptosis in Tumors with Reduced PTN Levels.
A recent study describing the targeting of integrins to inhibit angiogenesis reported that the extent of apoptosis in adjacent tumor tissues was increased and may cause tumor regression in a clinical setting (20). In our study, a significant increase in apoptotic DNA fragmentation [detected as DNA–histone complexes (15)] was found in tumors from the low PTN group (149 ± 25% of control; P < 0.05), and a small but insignificant increase of this parameter was observed in the medium PTN group (115 ± 23% of control). The increase in apoptosis was corroborated by staining of the tumor tissues for apoptotic nuclei using the TUNEL method [TdT-mediated dUTP nick end-labeling (16), data not shown]. This finding suggests to us that the decreased angiogenesis in the tumors due to decreased levels of PTN causes a shift of more tumor cells into apoptosis.
Metastasis and PTN Levels.
The ultimate goal of our studies was to assess the metastatic spread of the 1205LU melanoma cells expressing different levels of PTN. In most of the groups, we resected the subcutaneous tumors when they reached at least 50 mm2 and followed the development of lung metastases for up to 11 weeks (Fig. 2d). One set of subcutaneous tumors growing from a low PTN cell line (pRz66-6) was left to grow for the complete study period to reach (or surpass) the size of tumors in the control group (see Fig. 2a).
Animals carrying tumors from the control group (high PTN) showed metastases to their lungs 7–9 weeks after tumor cell inoculation (Fig. 2d) and the majority of these metastases was visible by macroscopic inspection of the lungs (see Fig. 4a). In contrast with this, none of the tumor cell lines in which PTN was reduced generated macroscopic metastases (see Fig. 4 b and c). Upon further microscopic inspection of the lungs (Fig. 4 e and f), no micrometastases were found in any of the low PTN group even after 11 weeks (incidence of metastasis P < 0.002 vs. control; Fig. 2d). This was independent from the continuous presence (pRz66-6) or surgical resection of the primary tumors (pRz66-10, pRz66-4). In the medium PTN group, we found one or more micrometastases in one-third of the animals at this time point (P < 0.01 vs. control; Figs. 2d and 4e). Obviously, the reduction of PTN mRNA to different levels is reflected in a gradual reduction of the incidence of metastasis. This result supports the thesis that the reduction of angiogenesis in the primary tumors is the cause for this effect on metastasis.
DISCUSSION
Clinical studies support a direct connection between the ability of a tumor to induce new blood vessels (angiogenesis) and its ability to metastasize in the body (see Introduction). In the past decade, more than a dozen distinct proteins have been described that can drive physiological as well as pathological angiogenesis (for review, see ref. 21), and a major question has been which of these factors is significant for tumor development in vivo. This task is complicated by the finding that tumor cells in tissue culture simultaneously express multiple angiogenic factors (18, 22, 23), and the mere presence of a particular angiogenic growth factor thus does not prove a role in metastasis. Furthermore, in tumor tissues in vivo, angiogenic factors can also be found in the stroma, and tumor cells can mobilize and activate these stored growth factors through binding proteins or proteases (see, e.g., refs. 24 and 25). It is also conceivable that tumor cells depend on the combined activity of a number of different angiogenic molecules and to search for an individual significant factor could be futile. Our current paper addresses this issue.
Recent studies have shown that one of the tumor-cell-derived angiogenesis factors, VEGF, is expressed in tumors and blockade of the pathway is rate-limiting for local growth of some tumors (26–28). Furthermore, anti-VEGF antibodies blocked seeding of fibrosarcoma cells into the lungs after intravenous injection (7) and blocked metastasis into the liver of intrasplenically injected colon carcinoma cells (8). We chose to study a tumor cell line that metastasizes spontaneously after subcutaneous growth and expresses high levels of PTN, a growth factor that can serve as an angiogenesis factor (see Introduction). Earlier studies using ribozyme targeting of PTN mRNA showed that this gene product can be rate-limiting for in vitro colony formation and animal tumor growth of WM852 human melanoma cells (14). The development of this molecular targeting approach put us into a position to ask whether PTN plays a role in tumor angiogenesis and metastasis and allowed a direct assessment of the functional redundancy of the various growth factors produced by melanoma cells (18).
We demonstrate that reduction of a candidate angiogenesis factor (PTN) can reduce the number of blood vessels in the primary subcutaneous tumor site and, subsequently, the metastatic spread of human melanoma cells. Our results support the thesis that the reduction of angiogenesis in the primary tumors is the cause for this effect. Slower growth or smaller size of the primary tumors can be ruled out as a reason for this finding, since we allowed tumors to reach the same size, thus prolonging the exposure of the animals to the primary tumors and increasing the chances of tumor cells to seed. In contrast, effects of tumor-derived inhibitors of angiogenesis found in some tumor models (29) do not appear to play a role, since no difference in metastasis was seen whether the primary tumors were removed early or not at all.
These in vivo effects on tumor growth, angiogenesis, and metastasis were “gene dose” dependent inasmuch as the extent of the effects followed the extent of the reduction of PTN mRNA in the tumor cells due to ribozyme targeting. Most surprising for us, a relatively small reduction of PTN mRNA by less than one-half already showed a significant effect in vivo. This is supported by the experiment with mixtures of different ratios of tumor cell populations producing high and low levels of PTN. From this data, it is tempting to speculate that therapeutic targeting of PTN using ribozymes would only require partial efficacy to achieve a significant therapeutic effect on tumors. It remains to be seen if this hypothesis can be sustained for angiogenesis factors produced in other tumors.
In conclusion, our results demonstrate that specific and dose-dependent reduction in tumor cells of a single growth factor that can mediate angiogenesis can have a significant inhibitory effect on tumor metastasis. Furthermore, our data suggest that ribozyme targeting has the potential to be of therapeutic significance for metastatic malignancies. It will be up to further studies to evaluate which other candidate gene products are rate-limiting in the chain of events that leads to the growth and metastatic spread of tumors.
Acknowledgments
We thank Drs. Anna Tate Riegel (Georgetown University), Meenhard Herlyn and Ullrich Rodeck (Wistar Institute, Philadelphia, PA) for discussions and helpful suggestions. M. Herlyn also provided the 1205LU melanoma cell line. Furthermore, we thank Andrea Jaetsch for her general help with the experiments. The studies were supported by grants from the National Cancer Institute (Bethesda; Special Program of Research Excellence CA58185) and the U.S. Army Medical Research Materiel Command Breast Cancer Program to A.W.
Footnotes
Abbreviations: PTN, pleiotrophin; VEGF/VPF, vascular endothelial cell growth factor/vascular permeability factor.
References
- 1.Weidner N, Semple J P, Welch W R, Folkman J. N Engl J Med. 1991;324:1–8. doi: 10.1056/NEJM199101033240101. [DOI] [PubMed] [Google Scholar]
- 2.Weidner N. Am J Pathol. 1995;147:9–19. [PMC free article] [PubMed] [Google Scholar]
- 3.Liotta L A, Steeg P S, Stetler-Stevenson W G. Cell. 1991;64:327–336. doi: 10.1016/0092-8674(91)90642-c. [DOI] [PubMed] [Google Scholar]
- 4.Fidler I F, Ellis L M. Cell. 1994;79:185–188. doi: 10.1016/0092-8674(94)90187-2. [DOI] [PubMed] [Google Scholar]
- 5.Dvorak H F, Brown L F, Detmar M, Dvorak A M. Am J Pathol. 1995;146:1029–1039. [PMC free article] [PubMed] [Google Scholar]
- 6.Thomas K A. J Biol Chem. 1996;271:603–606. doi: 10.1074/jbc.271.2.603. [DOI] [PubMed] [Google Scholar]
- 7.Asano M, Yukita A, Matsumoto T, Kondo S, Suzuki H. Cancer Res. 1995;55:5296–5301. [PubMed] [Google Scholar]
- 8.Warren R S, Yuan H, Matli M R, Gillett N A, Ferrara N. J Clin Invest. 1995;95:1789–1797. doi: 10.1172/JCI117857. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Li Y S, Milner P G, Chauhan A K, Watson M A, Hoffman R M, Kodner C M, Milbrandt J, Deuel T F. Science. 1990;250:1690–1694. doi: 10.1126/science.2270483. [DOI] [PubMed] [Google Scholar]
- 10.Fang W J, Hartmann N, Chow D, Riegel A T, Wellstein A. J Biol Chem. 1992;267:25889–25897. [PubMed] [Google Scholar]
- 11.Laaroubi K, Delbe J, Vacherot F, Desgranges P, Tardieu M, Jaye M, Barritault D, Courty J. Growth Factors. 1994;10:89–98. doi: 10.3109/08977199409010982. [DOI] [PubMed] [Google Scholar]
- 12.Wellstein A, Fang W J, Khatri A, Lu Y, Swain S S, Dickson R B, Sasse J, Riegel A T, Lippman M E. J Biol Chem. 1992;267:2582–2587. [PubMed] [Google Scholar]
- 13.Kojima S, Inui T, Muramatsu H, Kimuara H, Sakakibara S, Muramatsu T. Biochem Biophys Res Commun. 1995;216:574–581. doi: 10.1006/bbrc.1995.2661. [DOI] [PubMed] [Google Scholar]
- 14.Czubayko F, Riegel A T, Wellstein A. J Biol Chem. 1994;269:21358–21363. [PubMed] [Google Scholar]
- 15.Sumantran V N, Ealovega M W, Nunez G, Clarke M F, Wicha M S. Cancer Res. 1995;55:2507–2510. [PubMed] [Google Scholar]
- 16.Gavrieli Y, Sherman Y, Ben-Sasson S A. J Cell Biol. 1992;119:493–501. doi: 10.1083/jcb.119.3.493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Chauhan A K, Li Y S, Deuel T F. Proc Natl Acad Sci USA. 1993;90:679–682. doi: 10.1073/pnas.90.2.679. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Shih I M, Herlyn M. J Invest Dermatol. 1993;100:196S–203S. [PubMed] [Google Scholar]
- 19.Horak E R, Leek R, Klenk N, Lejeunde S, Smith K, Stuart N, Greenall M, Stepniewska K, Harris A L. Lancet. 1992;340:1120–1124. doi: 10.1016/0140-6736(92)93150-l. [DOI] [PubMed] [Google Scholar]
- 20.Brooks P C, Montgomery A M P, Rosenfeld M, Reisfeld R A, Hu T, Klier G, Cheresh D A. Cell. 1994;79:1157–1164. doi: 10.1016/0092-8674(94)90007-8. [DOI] [PubMed] [Google Scholar]
- 21.Folkman J, Shing Y. J Biol Chem. 1992;267:10931–10934. [PubMed] [Google Scholar]
- 22.Cross M, Dexter T M. Cell. 1991;64:271–280. doi: 10.1016/0092-8674(91)90638-f. [DOI] [PubMed] [Google Scholar]
- 23.Denekamp J. Br J Cancer. 1982;45:136–139. doi: 10.1038/bjc.1982.16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Czubayko F, Smith R V, Chung H C, Wellstein A. J Biol Chem. 1994;269:28243–28248. [PubMed] [Google Scholar]
- 25.Vlodavsky I, Fuks Z, Ishai-Michaeli R, Bashkin P, Levi E, Korner G, Bar-Shavit R, Klagsbrun M. J Cell Biochem. 1991;45:167–176. doi: 10.1002/jcb.240450208. [DOI] [PubMed] [Google Scholar]
- 26.Plate K H, Breier G, Weich H A, Risau W. Nature (London) 1992;359:845–848. doi: 10.1038/359845a0. [DOI] [PubMed] [Google Scholar]
- 27.Millauer B, Shawver L K, Plate K H, Risau W, Ullrich A. Nature (London) 1994;367:576–579. doi: 10.1038/367576a0. [DOI] [PubMed] [Google Scholar]
- 28.Kim K J, Li B, Winer J, Armanini M, Gillett N, Phillips H S, Ferrara N. Nature (London) 1993;362:841–844. doi: 10.1038/362841a0. [DOI] [PubMed] [Google Scholar]
- 29.O’Reilly M S, Holmgren L, Shing Y, Chen C, Rosenthal R A, Moses M, Lane W S, Cao Y, Sage E H, Folkman J. Cell. 1994;79:315–328. doi: 10.1016/0092-8674(94)90200-3. [DOI] [PubMed] [Google Scholar]