

Abstract
In this study, we investigated the role of the nucleotide excision repair (NER) pathway in mycobacterial DNA repair. Mycobacterium smegmatis lacking the NER excinuclease component uvrB or the helicase uvrD1 gene and a double knockout lacking both genes were constructed, and their sensitivities to a series of DNA-damaging agents were analyzed. As anticipated, the mycobacterial NER system was shown to be involved in the processing of bulky DNA adducts and interstrand cross-links. In addition, it could be shown to exert a protective effect against oxidizing and nitrosating agents. Interestingly, inactivation of uvrB and uvrD1 significantly increased marker integration frequencies in gene conversion assays. This implies that in mycobacteria (which lack the postreplicative mismatch repair system) NER, and particularly the UvrD1 helicase, is involved in the processing of a subset of recombination-associated mismatches.
The success of Mycobacterium tuberculosis as a human pathogen lies to some extent in its ability to survive and replicate in macrophages (23). It is currently unknown how it manages to overcome the assault on its DNA by macrophage-generated reactive oxygen intermediates (ROI) and reactive nitrogen intermediates (RNI), which represent an otherwise very effective defense against intracellular pathogens (5, 8, 35). One possibility is that M. tuberculosis effectively detoxifies ROI and RNI. Alternatively, it may possess highly effective DNA repair machineries. In silico analyses of mycobacterial genomes including M. tuberculosis (6, 34), Mycobacterium leprae (7), Mycobacterium bovis (15), Mycobacterium avium, Mycobacterium paratuberculosis, and Mycobacterium smegmatis (The Institute for Genome Research [http://www.tigr.org]) revealed the presence of genes encoding enzymes involved in DNA damage reversal, nucleotide excision repair (NER), base excision repair (BER), recombinational repair, nonhomologous end joining, and SOS repair. Surprisingly, mycobacteria are devoid of the mutLS-based postreplicative mismatch repair (MMR) system (34, 51), which is otherwise highly conserved throughout evolution and contributes to mutation avoidance by correcting replication errors resulting from nucleotide misincorporation and polymerase slippage (27, 46). In addition, MMR inhibits recombination between nonidentical (homeologous) sequences and thus helps to control the fidelity of recombination (32, 41, 56).
The finding that mycobacteria exhibit a general mutation rate comparable to that of MMR-proficient species suggests that they possess alternative or compensating strategies for mismatch recognition and MMR (51). It is conceivable, for example, that their replication and recombination fidelity is higher than in other prokaryotes. Alternatively, it is possible that the fidelity of these processes is controlled by one of the other DNA metabolic pathways, such as NER.
Prokaryotic NER has been extensively studied in Escherichia coli. It is mediated by the UvrABC excinuclease enzyme complex and the helicase UvrD (43), a system capable of dealing with a broad range of bulky and/or helix-distorting lesions, such as UV-induced photodimers and oxidized thymines, intra- and interstrand cross-links, and other large base modifications formed through exposure to DNA-modifying agents (2). Analysis of NER in Bacillus subtilis (43), Streptococcus pneumoniae (47), Mycoplasma genitalium (43), and Deinococcus radiodurans (33) indicated that the repair mechanism originally characterized in E. coli is highly conserved in all prokaryotes. Interestingly, the mycobacterial UvrD1 protein has recently been shown to possess an additional role in DNA metabolism outside of NER; it was shown to physically and functionally interact with Ku, a protein participating in the nonhomologous-end-joining pathway of DNA double-strand break repair. A second UvrD homologue, UvrD2, has a Ku-independent helicase activity (49), but its role in DNA repair has yet to be identified.
To study the role of the mycobacterial NER pathway in the maintenance of genomic integrity, we generated mutants deficient in UvrD1 and UvrB, as well as a mutant lacking both proteins, and studied their responses to a variety of DNA-damaging agents. Moreover, using a plasmid-based assay, we also studied the possible involvement of mycobacterial NER in DNA mismatch recognition. Our data demonstrate that NER is functional in mycobacteria and that UvrB and UvrD1 play important roles in DNA repair and mutation prevention. Our data also suggest that UvrD1 may play a role in the control of homologous recombination.
MATERIALS AND METHODS
Bacterial strains, media, and culture conditions.
E. coli strain XL1 blue (Stratagene) was used for cloning and propagation of plasmids. Bacterial cultures were grown in Luria-Bertani (LB) medium at 37°C. Cultures of M. smegmatis SMR5 rrnB (45), a derivative of M. smegmatis mc2 155, were grown in 7H9 medium at 37°C. For determination of CFU, serial dilutions of three cultures of each strain were plated on solid medium and grown at 37°C for 3 days. Generation times were calculated by optical-density (OD) measurements of cultures in broth medium. When appropriate, antibiotics were added at the following concentrations: ampicillin, 100 μg/ml; hygromycin, 100 μg/ml; kanamycin, 50 μg/ml; streptomycin, 100 μg/ml; paromomycin, 20 μg/ml; and clarithromycin, 50 μg/ml.
Generation of mycobacterial NER mutant strains.
Allelic-replacement techniques were used to generate M. smegmatis knockout mutants (44). Using genomic DNA, 1- to 1.5-kbp fragments upstream (5′) and downstream (3′) of the target gene were amplified by PCR and cloned into pMCS5-rpsL-hyg for generation of unmarked knockouts. The following primers were used: uvrD-1/uvrD-2 for amplification of the uvrD1 upstream region, uvrD-3/uvrD-4 for amplification of the uvrD1 downstream region, uvrB-1/uvrB-2 for amplification of the uvrB upstream region, and uvrB-3/uvrB-4 for amplification of the uvrB downstream region (primer sequences are given in Table 1). The resulting deletion alleles lacked base pairs 219 to 2040 of the 2,352-base-pair uvrD1 (MSMEG5534) open reading frame and base pairs 262 to 1890 of the 2,160-base-pair uvrB (MSMEG3816) open reading frame. Cloning was verified by sequencing. Vectors were transformed into M. smegmatis SMR5 rrnB, a derivative of M. smegmatis mc2 155 carrying a nonrestrictive rpsL mutation conferring streptomycin resistance (45). Transformants were plated on medium containing hygromycin to select for integration of the knockout vector by homologous recombination into the genome. Clones that had undergone a single-crossover event were detected by Southern blot analyses and subjected to counterselection. In brief, single-crossover clones were grown for 3 days in liquid broth and subsequently plated on selective agar containing streptomycin. Putative knockout mutants were colony purified and investigated by Southern blot analysis for disruption of uvrD1 and uvrB. For generation of the M. smegmatis uvrB uvrD1 double mutant, the uvrB knockout vector was transformed into the M. smegmatis uvrD1 mutant and selection procedures were applied as detailed above. Gene disruption was confirmed by Southern blot analysis.
TABLE 1.
Primers used for gene cloning
| Primer name | Sequence |
|---|---|
| uvrD-1 | 5′-GGAATTCCATATGGTGAGGACGCCTACGAC-3′ |
| uvrD-2 | 5′-GGAAGATCTGGTGAACGTGATGGCCAG-3′ |
| uvrD-3 | 5′-GGAAGATCTGCAGGAACTCATCGACTGGCG-3′ |
| uvrD-4 | 5′-TGCATGCATGATCGCGTCGGGCACCTTC-3′ |
| uvrB-1 | 5′-GGAATTCCATATGGGCCGAGTACGGCCAGTC-3′ |
| uvrB-2 | 5′-GGAAGATCTGCGCCATCACGAGCGTG-3′ |
| uvrB-3 | 5′-GGAAGATCTGAGTCGGTCGAGATCGGTGG-3′ |
| uvrB-4 | 5′-TGCATGCATCGTCATGTGCGCCAGCCGC-3′ |
Complementation was not done, as the results obtained (see Results) and their agreement in general with data reported in the literature, where available, practically excluded polar effects and spontaneous second-site mutations elsewhere in the chromosome.
Determination of spontaneous-mutation frequencies.
At least six parallel cultures of each strain were grown until late log phase in 7H9 medium, with a viable cell number around 109/ml. Subsequently, the cultures were diluted to 2 × 103 cells/ml and incubated for 2 days at 37°C. One hundred microliters of each culture was plated on freshly prepared 7H10 agar plates containing rifampin, and serial dilutions were plated on nonselective medium. After 3 to 6 days of incubation at 37°C, the numbers of CFU were determined. The number of CFU obtained on agar plates containing rifampin was divided by the number of CFU obtained on nonselective medium. For calculation of mutation frequencies, the median of the ratio of cells that gained resistance against rifampin was determined, as previously described (51).
Determination of MIC and MBC.
MICs were determined by Etest (AB Biodisk) according to the manufacturer's instructions. Determination of minimal bactericidal concentrations (MBCs) was performed in a microtiter plate format as described previously (39). In brief, freshly grown cultures were diluted to an absorbance (A600) of 0.01 in 7H9 medium and incubated for 72 h at 37°C in the presence of twofold serial dilutions of rifampin. The rifampin stock solution (2 mg/ml) was made in dimethyl sulfoxide. Aliquots from the wells that showed growth inhibition were plated on drug-free solid agar and incubated at 37°C for a further 72 h. The MBC was defined as the minimal drug concentration that killed >99.9% of the inoculum.
Survival after exposure to UV light.
The UV sensitivities of strains were determined by plating dilutions (in triplicate) on solid medium, followed by irradiation of open plates in a Stratalinker 2400 (Stratagene; 254 nm) at 0 to 160 mJ/cm2 (approximately 0 to 5 s). The plates were incubated in the dark immediately after UV exposure to exclude possible interference with photolyase-mediated light repair. CFU numbers were determined after 3 to 4 days of incubation at 37°C and were compared to those of the untreated control.
Survival after treatment with DNA-damaging agents.
Twenty-five-milliliter cultures of each strain were grown in 7H9 medium until early mid-log phase, and serial dilutions were plated on LB agar for CFU determination. For determination of sensitivity to acidified sodium nitrite (NaNO2), cultures were centrifuged and resuspended in 7H9 medium acidified with HCl (pH 5.4). Subsequently, the cultures were split into six 3-ml aliquots. Freshly prepared compounds (tert-butylhydroperoxide [TBH], 250 μM; NaNO2, 3 mM; mitomycin C (MMC), 0.02 μg/ml) were added to three aliquots, and the remaining aliquots served as untreated controls. After incubation for 24 h at 37°C, serial dilutions were plated onto LB agar. CFU were counted after 3 days growth at 37°C. Survival was calculated from the ratio of CFU of the treated cultures to the CFU of the untreated controls.
Determination of gene conversion frequencies.
M. smegmatis strains were transformed with an integrative vector carrying various versions of mutated rrnA gene fragments. The approximately 1.0-kb partial 16S rRNA gene fragment with a mutated 1491 position covered 16S rRNA position 907 to internal transcribed spacer 1 position 2367, and the partial 23S rRNA gene fragment with a mutated 2058 or 2059 position covered 23S rRNA positions 1426 to 2624. Upon recombination with the chromosomal wild-type rrnA gene, the vector-borne mutated rrnA gene fragment confers drug resistance (paromomycin for the 16S rRNA 1491 mutation and clarithromycin for the 23S rRNA 2058/2059 mutation) (the vectors are listed in Table 2). For each construct and strain, at least 10 independent transformants were picked and grown. Aliquots of these precultures were taken to inoculate 4 ml 7H9 medium with approximately 5 × 105 cells. After 2 days of growth, serial dilutions were plated on permissive medium and on selective medium containing paromomycin or clarithromycin. The relative marker integration frequency was determined by the ratio of the number of cells that gained antibiotic resistance to the number of cells obtained on permissive medium.
TABLE 2.
Recombination substrates containing a mutated rrnA fragmenta
| Vector | rrnA positionb | Antibiotic | Reference |
|---|---|---|---|
| pMV361ΔKan-Gm-rRNA | A2059C | Clarithromycin | 40 |
| pMV361ΔKan-Gm-rRNA | A2059G | Clarithromycin | 40 |
| pMV361ΔKan-Gm-rRNA | A2058C | Clarithromycin | 40 |
| pMV361ΔKan-Gm-rRNA | A2058G | Clarithromycin | 40 |
| pMV361ΔKan-Gm-rRNA | G1491A | Paromomycin | 39 |
| pMV361ΔKan-Gm-rRNA | G1491C | Paromomycin | 39 |
| pMV361ΔKan-Gm-rRNA | G1491T | Paromomycin | 39 |
All vectors harbor a mutated rrnA fragment, which confers antibiotic resistance (the antibiotic is shown in the middle column) upon recombination with the chromosomal wild-type rrnA gene.
The rrnA position of the mutated nucleotide.
RESULTS
To study the role of the NER pathway in mycobacteria, we generated M. smegmatis mutants deficient in uvrB and uvrD1. In addition, a double mutant deficient in both genes was constructed. The correctness of the gene disruptions was verified by Southern blot analysis (Fig. 1).
FIG. 1.

Generation of M. smegmatis NER mutants. (A) Disruption of M. smegmatis uvrD1. (Left) Southern blot analysis. Genomic DNA from wild-type M. smegmatis (lane 1), the uvrD1 mutant (lane 2), and the uvrD1 single-crossover mutant (lane 3) was digested with BstXI and probed with a 653-bp MluI/BamHI DNA fragment containing 5′ flanking sequences of the uvrD1 gene. The presence of a single 10.7-kbp fragment instead of a 2.0-kbp fragment as seen in the parental strain demonstrated successful deletion of uvrD1 coding sequences. (Right) Schematic illustration of the uvrD1 locus and Southern blot analysis. Shown are the genomic organization of the wild type (wt), the knockout vector that contains the uvrD1 deletion allele (vector), the single-crossover genotype (sco), and the mutated genomic uvrD1 region in the knockout mutant (ko). Probe locations and fragments detected by the probe specific for the 5′ flanking region are indicated. (B) Disruption of M. smegmatis uvrB. (Left) Southern blot analysis. Genomic DNA from the M. smegmatis uvrB mutant (lane 1), the uvrB single-crossover mutant (lane 2), and the wild type (lane 3) was digested with NcoI and probed with a 600-bp NdeI/BsmI uvrB gene fragment. The presence of a single 3.9-kbp fragment instead of a 5.6-kbp fragment as seen in the parental strain demonstrated successful deletion of uvrB coding sequences. (Right) Schematic illustration of the uvrB locus and Southern blot analysis. Shown are the genomic organization of the wild type (wt), the knockout vector that contains the uvrB deletion allele (vector), the single-crossover genotype (sco), and the mutated genomic uvrB region in the knockout mutant (ko). Probe locations and fragments detected by the probe specific for the uvrB gene are indicated.
Growth characteristics.
The in vitro growth characteristics of the uvrB and uvrD1 mutant strains in Middlebrook 7H9 medium were determined by OD measurements and CFU counts and were found to be indistinguishable from those of the parental strain (Fig. 2A). The colony morphologies of the single mutants did not show any differences from the wild type. The growth of the uvrB uvrD1 double mutant was considerably impaired (Fig. 2A): the mutant formed visible colonies approximately 20 h later than the wild-type strain when dilutions of broth-grown cultures were plated on solid media, and the colonies were significantly smaller than those of the wild type.
FIG. 2.

In vitro growth analysis and survival of M. smegmatis strains following DNA damage. (A) In vitro growth analysis of M. smegmatis strains. The strains were grown in 7H9 broth in shaking cultures for 2 days at 37°C, and CFU counts were assessed after 0, 9, 15, and 33 h. OD values are shown in a line graph, corresponding to the left y axis; CFU counts are shown as bars, corresponding to the right y axis. Doubling times were as follows: wild type, 3.7 ± 0.1 h; uvrD1 mutant, 3.5 ± 0.2 h; uvrB mutant, 3.4 ± 0.2 h; uvrB uvrD1 mutant, 7.6 ± 0.4 h. (B) Survival following irradiation with UV-C. Survival was calculated from the ratio of cells that survived treatment with UV-C to the untreated-control cell numbers. Shown are the mean values of one representative experiment performed with three independent cultures each. Standard deviations of the means are given by the error bars. The ratio of treated to untreated cells is plotted in logarithmic scale. The uvrB uvrD1 mutant was unable to survive doses higher than 80 mJ/cm2. (C to E) Survival after treatment with different DNA-damaging agents: TBH (250 μM) (C), acidified NaNO2 (pH 5.4; 3 mM) (D), and MMC (0.02 μg/ml) (E). Survival was calculated from the ratio of cells that survived treatment with the DNA-damaging agent to the untreated-control cell numbers. Shown are the mean values of one representative experiment performed with three independent cultures each. The strains were incubated with the DNA-damaging agent for 24 h. Standard deviations from the mean are given by the error bars. *, significant increase in susceptibility to the DNA-damaging agent compared to the wild type (Student's t test; P < 0.05). wt, M. smegmatis wild-type strain; uvrB, uvrD1, and uvrB uvrD1, M. smegmatis NER mutant strains; recA, M. smegmatis recA mutant strain.
Survival after treatment with DNA-damaging agents.
To test whether the absence of NER in M. smegmatis affects DNA repair proficiency, the mutants were challenged with a variety of DNA-damaging agents. The M. smegmatis recA mutant (14) was included as a control.
As NER is primarily responsible for the repair of bulky adducts resulting from exposure to UV light, we irradiated the strains with short-wavelength UV radiation (UV-C). The M. smegmatis uvrB and uvrD1 mutants, as well as the recA mutant, were considerably more sensitive to UV than the wild-type strain. Combined deletion of uvrB and uvrD1 had an additive effect on survival following UV treatment; the double mutant was unable to survive doses in excess of 80 mJ/cm2 (Fig. 2B).
We then studied the cytotoxicity of TBH, which generates ROI that attack the bases and the sugar-phosphate backbone of DNA, leading to strand breaks and base modifications (17). Treatment with TBH may also result in the formation of alkoxy radicals that cause alkylation damage (18). The M. smegmatis NER mutants were significantly more sensitive to TBH than the wild-type strain (Fig. 2C) (P < 0.05; Student's t test), and the sensitivities of all three mutants were similar (Student's t test; P > 0.1). In contrast, the TBH sensitivity of the M. smegmatis recA mutant was only slightly increased compared to that of the wild type (42% surviving recA cells compared to 77% surviving wild-type cells; P < 0.05; Student's t test). These findings indicate that in defense against ROI and alkylation damage in mycobacteria, NER plays a more important role than homologous recombination.
Acidified sodium nitrite generates RNI, which give rise to oxidative damage in DNA, as well as to nitration, nitrosation, and deamination reactions (3). The M. smegmatis recA mutant displayed sensitivity to acidified sodium nitrite similar to that of the wild type (12% and 15% surviving cells; P > 0.08; Student's t test) (Fig. 2D). The NER mutants were substantially more sensitive (P < 0.05; Student's t test), particularly the uvrD1 and uvrB uvrD1 strains (Fig. 2D).
Exposure to MMC results in alkylation and interstrand cross-links (20, 26), which are mainly repaired by NER in E. coli (52). The M. smegmatis NER mutants differed in their sensitivities to MMC. Compared to the recA and the uvrD1 mutants, which showed significantly increased sensitivities, the uvrB and uvrB uvrD1 mutants were even more sensitive to MMC (Fig. 2E).
Spontaneous mutation frequencies.
Rifampin has a single molecular target, the β subunit of RNA polymerase encoded by rpoB. Various point mutations in rpoB confer high-level drug resistance. The frequency at which bacteria generate rifampin-resistant mutants is widely used to assess the general spontaneous mutation frequency. For selection of spontaneous drug-resistant mutants, we used a concentration of 175 μg/ml rifampin. The wild-type strain displayed a spontaneous mutation frequency of 3.8 × 10−8. In comparison, deletion of uvrD1 resulted in an approximately threefold-higher mutation frequency (9.1 × 10−8; P < 0.05; n = 6; Student's t test), and deletion of uvrB resulted in a fivefold increase (1.9 × 10−7; P < 0.01; n = 6; Student's t test). Inactivation of recA did not affect the mutation frequency (1.7 × 10−8; P > 0.05; n = 6; Student's t test). The mutation frequency of the double mutant could not be assessed by this approach, as the uvrB uvrD1 mutant strain was unable to grow on plates containing 175 μg/ml rifampin. Considering the general growth defect of this strain, we hypothesized that the uvrB uvrD1 mutant might be incapable of acquiring high-level resistance against rifampin. MIC assays revealed that the uvrB uvrD1 mutant displayed a MIC of 4 μg/ml rifampin compared to 32 μg/ml for the wild-type and the uvrB strains and 16 μg/ml for the uvrD1 mutant. The MBC for the uvrB uvrD1 double mutant was 130 μg/ml compared to 500 μg/ml for the wild-type, uvrB, and uvrD1 strains. Given the decreased MBC for the drug, we reassessed the spontaneous-mutation frequencies on plates containing 100 μg/ml rifampin. In view of the prolonged generation time of the uvrB uvrD1 double mutant, CFU on rifampin were determined after 6 days, while the CFU of the wild-type and the single-mutant strains were assessed after 3 days. The wild-type strain displayed a spontaneous mutation frequency of 5.7 × 10−8 (Fig. 3). Deletion of uvrD1 resulted in an approximately twofold increase (1.2 × 10−7; P < 0.05; n = 6; Student's t test), and deletion of uvrB resulted in a fivefold-increased mutation frequency (2.9 × 10−7; P < 0.01; n = 6; Student's t test). Combined deletion of uvrB and uvrD1 resulted in a fourfold increase (2.2 × 10−7; P < 0.01; n = 6; Student's t test), while inactivation of recA did not affect the mutation frequency (6.3 × 10−8; P > 0.05; n = 6; Student's t test).
FIG. 3.
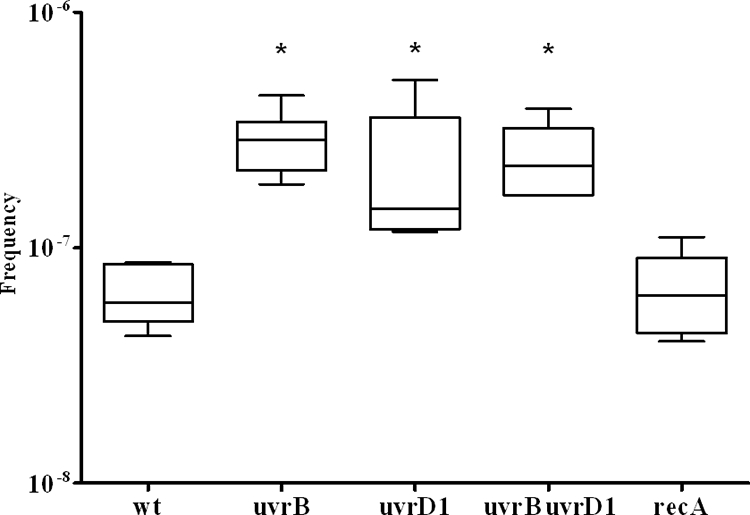
Frequencies of mutation to rifampin resistance. Shown are the median values (horizontal lines), 95% confidence intervals (rectangular boxes), and outliers (vertical lines) of mutation frequencies calculated from the ratio of cells that gained rifampin resistance in six independent cultures of each strain. *, significant increase of mutation frequency compared to the wild type (Student's t test; P < 0.05). wt, M. smegmatis wild-type strain; uvrB, uvrD1, and uvrB uvrD1, M. smegmatis NER mutant strains; recA, M. smegmatis recA mutant strain.
Involvement of NER in mismatch recognition.
Base-base mispairs occur in duplex DNA in the form of purine-purine (G/G, A/A, and G/A), purine-pyrimidine (G/T and A/C), or pyrimidine-pyrimidine (C/C, T/T, and T/C) mismatches. With the exception of C/C, all base-base mispairs are subject to correction by the MMR system (31). As mycobacteria are devoid of this highly conserved repair pathway, we studied the effects of MMR deficiency and the possible compensating role of NER in recombination repair. We deployed a plasmid transformation assay (40) in which intramolecular recombination between the chromosomal M. smegmatis rrnA gene and a plasmid-borne rrnA gene fragment carrying a specific mutation leads to antibiotic resistance. Because the recombination process gives rise to a mismatch-containing heteroduplex, MMR-proficient cells correct the mispair or reject the heteroduplex, which results in low gene conversion frequencies. In MMR-deficient cells, the intermediate is not processed, which gives rise to a mutation of the chromosomal rrnA operon after replication and postmitotic segregation in 50% of the progeny, with a consequent gain of resistance to the antibiotic. This assay provided an opportunity to study the recognition of different mismatches in M. smegmatis. However, because M. smegmatis harbors a second rrn operon (rrnB), which precludes proper measurement of gene conversion frequencies using the above-described assay, the NER mutants were constructed in a ΔrrnB background (45).
The rrnB NER mutant strains were transformed with integrating vectors carrying rrnA gene fragments with different point mutations. For each rrnA fragment, specific gene conversion frequencies were obtained. Comparison of marker integration frequencies in the wild type revealed that the highest gene conversion frequencies (in the range of 10−4 to 10−5) were obtained with A→C mutations that gave rise to C/T and A/G mismatches in 23S rRNA position 2058 or 2059 (Fig. 4A). While gene conversion frequencies in the uvrB mutant were similar to those in the wild type (Student's t test; n = 10; P > 0.05), deletion of uvrD1 resulted in approximately 10-fold-higher gene conversion frequencies. Interestingly, the combined deletion of uvrB and uvrD1 resulted in significantly elevated marker integration frequencies (∼60-fold for the A→C mutation located at position 2058 and 240-fold for the A→C mutation at position 2059). Marker integration frequencies with an rrnA fragment carrying an A→G mutation resulting in G/T and A/C mismatches in 23S rRNA position 2058 or 2059 (Fig. 4A) were lower than with the A→C mutation, yielding frequencies in the range of 10−5 and 10−6 in wild-type M. smegmatis. Gene conversion frequencies increased about 10-fold in the uvrD1 mutant and less (3- to 4-fold) in the uvrB mutant. However, the double mutant displayed marker integration frequencies that were about 200-fold higher than those of the wild type (Fig. 4A).
FIG. 4.

Relative gene conversion frequencies obtained with rrnA fragments carrying a resistance-conferring mutation at 23S rRNA position 2058 or 2059 (A) and rrnA fragments carrying a resistance-conferring mutation at 16S rRNA position 1491 (B). Shown are the median values (horizontal lines), 95% confidence intervals (rectangular boxes), and outliers (vertical lines) of gene conversion frequencies calculated from the numbers of cells that gained paromomycin or clarithromycin resistance in 10 independent cultures of each strain. *, significant increase of gene conversion frequencies compared to the wild type (Student's t test; P < 0.05). wt, M. smegmatis wild-type strain; uvrB, uvrD1, and uvrB uvrD1, M. smegmatis NER mutant strains.
Metabolism of A/C and G/T or C/C and G/G mismatches was determined with rrnA fragments carrying a G→A or a G→C mutation at position 1491 of 16S rRNA. The gene conversion frequencies of the wild type were in the 1 × 10−6 range, and those of the uvrB, uvrD1, and uvrB uvrD1 mutants were only marginally (about fourfold) increased (Fig. 4B).
Analysis of marker integration frequencies using an rrnA fragment carrying a G→T mutation provided insights into the processing of T/C and G/A mismatches. The uvrD1 mutant strain and the uvrD1 uvrB mutant strain displayed more than 10-fold-higher gene conversion frequencies, while the frequency in the uvrB mutant was increased 4-fold (Fig. 4B).
DISCUSSION
NER is a highly versatile pathway of DNA metabolism, which can efficiently remove DNA adducts that bring about gross distortions of the helical structure. In the prototypic organism, E. coli, the NER system protects against UV radiation by removing cyclobutane pyrimidine dimers and six to four photoproducts but also deals with thymine glycols and both inter- and intrastrand cross-linking agents, such as cisplatin and MMC, respectively (2, 13). In this study, we showed that M. smegmatis uvrD1 and uvrB mutants are hypersensitive to UV and MMC. Moreover, we also show that the mutants are sensitive to ROI- and RNI-generating substances, such as TBH and sodium nitrite. These reagents generate DNA modifications that give rise primarily to single-strand breaks through the processing of modified bases by the BER system. In this respect, M. smegmatis appears to differ from E. coli, as uvrA, uvrB, and uvrC mutants of the latter organism show wild-type-like sensitivity to the ROI-generating substance, hydrogen peroxide (19); indeed, oxidative damage in E. coli is primarily repaired by homologous recombination and BER (24). Interestingly, the M. smegmatis uvrD1 mutant has been reported to sensitize cells to ionizing radiation (49), apparently through association with Ku. The latter factor is involved in the processing of double-strand DNA breaks, which can arise by the direct action of ionizing radiation, but also in cases where single-strand breaks are in close proximity, such as in DNA containing extensive amounts of DNA base damage. Thus, this link requires further study, especially as TBH also gives rise to alkylation damage (18). Although this type of modification is mainly repaired by alkyl transferases and BER (36) rather than NER (42, 52), at least in E. coli, the tert-butyl group is rather bulky, and the possibility that it is this type of damage rather than oxidized bases that is addressed by the M. smegmatis NER system cannot be disregarded at this time.
The M. smegmatis NER mutants were more sensitive to TBH than the wild-type strain and the recA mutant, and a similar situation was observed for acidified sodium nitrite. This differs from E. coli, in which repair of RNI-induced DNA damage (like that induced by ROI) is mainly accomplished by BER and homologous recombination (50), although a role for UvrA in resistance to acidified sodium nitrite has been reported (48). Thus, the mild RNI/ROI phenotype of the M. smegmatis recA mutant, coupled with the hypersensitivity of the NER mutants to the tested agents, suggested that homologous recombination plays only a minor part in dealing with this type of damage.
The above-mentioned experiments indicated that mycobacterial NER is involved in the processing of induced oxidative damage. As this type of damage is believed to be the most important contributor to spontaneous mutagenesis, we measured the spontaneous-mutation frequencies of M. smegmatis uvrD1, uvrB, and uvrB uvrD1 mutants to rifampin resistance. As shown in Fig. 3, all three strains displayed elevated spontaneous mutation frequencies, indicating that disruption of NER impacts genomic integrity. The observed increases were similar for the uvrB, uvrD1, and double mutants, which indicates that the two proteins are involved in the same pathway that is responsible for dealing with spontaneous DNA damage.
Combined deletion of uvrB and uvrD1 has not been analyzed in prokaryotes so far. Loss of both proteins rendered cells more sensitive to UV than single deletion of uvrB and uvrD1, implying a specific role for one of the proteins in addition to NER. This assumption is corroborated by the observation that the in vitro growth rate of the double mutant was significantly affected, while the growth of the uvrB and uvrD1 single mutants was similar to that of the wild type. A statistically significant additive effect of combined lack of UvrB and UvrD1 was not observed when the strains were assayed for sensitivity to MMC, TBH, and acidified sodium nitrite. Other repair pathways, like BER and recombinational repair, may have overlapping repair specificities for DNA damage generated by these compounds and thus may partially compensate for the lack of UvrB and UvrD1 under these conditions. In addition, a second UvrD homologue, UvrD2, has recently been described in M. smegmatis (49). However, UvrD2 function—in particular, a possible involvement in NER—remains to be elucidated. Taken together, the above data provide evidence that NER in mycobacteria is functional and that, as anticipated, UvrB and UvrD1 play important roles in the repair of bulky lesions. Moreover, our findings also suggest that the substrate spectrum of mycobacterial NER differs from that of E. coli, inasmuch as mycobacterial NER appears to be involved in the detoxification of oxidation damage, both induced and spontaneous.
As mentioned in the introduction, mycobacteria lack the MutLS MMR system. Interestingly, their spontaneous mutation frequencies appear to be similar to those of MMR-proficient bacteria. One possible explanation for this phenomenon is that mycobacterial replication fidelity is higher than that of other organisms. However, there is currently no evidence to support this notion. The second possibility is that another repair pathway compensates for the lack of MMR. This is a difficult task, because successful MMR must not only ensure that the mismatch is recognized, but also target the excision to the newly synthesized strand. In most organisms, this task is accomplished by the MutS homologues, which undergo an ATP-driven conformational change upon mismatch binding that allows them to diffuse away from the mismatch. When they encounter a strand discontinuity (such as one end of an Okazaki fragment during replication or the terminus of the invading strand during recombination), they load an exonuclease, which then degrades the error-containing strand. Such a scheme is difficult to imagine without the MutS function. However, it is conceivable that another mismatch recognition factor might be able to activate a DNA helicase that could load at the mismatch and unwind the DNA helix. Should this process reach a strand discontinuity, the flap could be degraded by a single-strand-specific endonuclease.
We decided to test whether UvrB and UvrD1 might possess the above-described functionalities, primarily because NER was shown to be involved in mismatch processing in MMR-deficient Schizosaccharomyces pombe (10, 28) and because UvrD deficiency was reported to give rise to a hyperrecombinogenic phenotype in E. coli (55) and in Helicobacter pylori (22). These observations were explained by two different models. The first proposed that replication forks would be arrested at persistent nicks in the DNA that arose as a result of incomplete NER or MMR. This would give rise to double-strand breaks at the collapsed forks, which would be repaired by homologous recombination (1). The second model proposed that UvrD participates in the degradation of toxic recombination intermediates by actively removing RecA proteins from DNA (4, 30). This model is supported by observations made in E. coli and Saccharomyces cerevisiae, in which UvrD (4, 11, 12, 38, 53) and the Srs2 helicase (54) were found to act as antirecombinases that prevent potentially deleterious recombination events.
We made use of gene conversion assays, which allow M. smegmatis cells to acquire antibiotic resistance upon recombination between the chromosomal rrnA operon and a plasmid-borne gene fragment carrying a specific resistance mutation. This system allowed us to test several different mutations in two different sequence contexts, which allowed us to discover whether marker integration frequencies depended on the specific mismatch, as in other systems (16, 21, 29). In MMR-proficient organisms, repair efficiency was shown to depend on the type of mismatch and its sequence context (9, 21, 31). In E. coli, for example, the correction of G/T, A/C, G/G, and A/A mismatches by the MMR system is highly effective, while C/T, T/T, and A/G mismatches are repaired less efficiently and C/C mispairs are generally not corrected (9, 25, 37). Our present findings show similar fluctuations in gene conversion frequencies in M. smegmatis, and the fact that the frequencies increase in the mutants indicates that mycobacterial uvrB and uvrD1 may control the fidelity of homologous recombination, as gene conversion is the result of recombination in the absence of MMR.
Interestingly, inactivation of M. smegmatis uvrD1 had a more profound effect on marker integration frequencies than uvrB loss, and the uvrB uvrD1 double mutant displayed higher gene conversion frequencies than either single mutant. This implies that the two polypeptides either act, at least in part, in different processes of DNA metabolism or that they address different substrates. Our experiments showing that the additive effect of UvrB and UvrD1 deletions on gene conversion was not observed in the G→A or G→C mutations in 16S rrnA position 1491 may be taken as support for the latter hypothesis, but the reason for this phenomenon is difficult to identify, given the large number of variables that contribute to gene conversion efficiency. However, the dominant role of the UvrD1 helicase can be understood if it is assumed that in all cases the rejection of the nascent heteroduplex requires the helix-unwinding function but that, due to the varying helix-destabilizing effects of different mispairs, the UvrB function is required only in a subset of cases.
In conclusion, our observations provide evidence that the mycobacterial NER is capable of repairing a wider range of DNA damage than the E. coli system. Furthermore, our data suggest that the NER system of M. smegmatis may play a role in controlling the fidelity of DNA recombination even in the absence of an MMR pathway. We therefore propose that the resolution of DNA heteroduplexes arising during recombination is accomplished differently in mycobacteria than in model species possessing an MMR system. Further investigations aimed at elucidating the molecular mechanisms governing genome stability in mycobacteria are currently in progress in our laboratory.
Acknowledgments
This work was supported in part by grants from the University of Zurich (to B.S.), the Swiss National Science Foundation (BO-3200-68488 to P.S. and E.C.B.), and the European Community (CSI_LTB and LSHP-CT-2007-037235 to B.S., E.C.B., and E.O.D.). E.O.D. was supported by the Medical Research Council (United Kingdom).
Footnotes
Published ahead of print on 14 November 2008.
REFERENCES
- 1.Arthur, H. M., and R. G. Lloyd. 1980. Hyper-recombination in uvrD mutants of Escherichia coli K-12. Mol. Gen. Genet. 180185-191. [DOI] [PubMed] [Google Scholar]
- 2.Batty, D. P., and R. D. Wood. 2000. Damage recognition in nucleotide excision repair of DNA. Gene 241193-204. [DOI] [PubMed] [Google Scholar]
- 3.Burney, S., J. L. Caulfield, J. C. Niles, J. S. Wishnok, and S. R. Tannenbaum. 1999. The chemistry of DNA damage from nitric oxide and peroxynitrite. Mutat. Res. 42437-49. [DOI] [PubMed] [Google Scholar]
- 4.Centore, R. C., and S. J. Sandler. 2007. UvrD limits the number and intensities of RecA-green fluorescent protein structures in Escherichia coli K-12. J. Bacteriol. 1892915-2920. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Chan, J., Y. Xing, R. S. Magliozzo, and B. R. Bloom. 1992. Killing of virulent Mycobacterium tuberculosis by reactive nitrogen intermediates produced by activated murine macrophages. J. Exp. Med. 1751111-1122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Cole, S. T., R. Brosch, J. Parkhill, T. Garnier, C. Churcher, D. Harris, S. V. Gordon, K. Eiglmeier, S. Gas, C. E. Barry III, F. Tekaia, K. Badcock, D. Basham, D. Brown, T. Chillingworth, R. Connor, R. Davies, K. Devlin, T. Feltwell, S. Gentles, N. Hamlin, S. Holroyd, T. Hornsby, K. Jagels, A. Krogh, J. McLean, S. Moule, L. Murphy, K. Oliver, J. Osborne, M. A. Quail, M. A. Rajandream, J. Rogers, S. Rutter, K. Seeger, J. Skelton, R. Squares, S. Squares, J. E. Sulston, K. Taylor, S. Whitehead, and B. G. Barrell. 1998. Deciphering the biology of Mycobacterium tuberculosis from the complete genome sequence. Nature 393537-544. [DOI] [PubMed] [Google Scholar]
- 7.Cole, S. T., K. Eiglmeier, J. Parkhill, K. D. James, N. R. Thomson, P. R. Wheeler, N. Honore, T. Garnier, C. Churcher, D. Harris, K. Mungall, D. Basham, D. Brown, T. Chillingworth, R. Connor, R. M. Davies, K. Devlin, S. Duthoy, T. Feltwell, A. Fraser, N. Hamlin, S. Holroyd, T. Hornsby, K. Jagels, C. Lacroix, J. Maclean, S. Moule, L. Murphy, K. Oliver, M. A. Quail, M.-A. Rajandream, K. M. Rutherford, S. Rutter, K. Seeger, S. Simon, M. Simmonds, J. Skelton, R. Squares, S. Squares, K. Stevens, K. Taylor, S. Whitehead, J. R. Woodward, and B. G. Barrell. 2001. Massive gene decay in the leprosy bacillus. Nature 4091007-1011. [DOI] [PubMed] [Google Scholar]
- 8.Ding, A., C. Nathan, and D. Stuehr. 1988. Release of reactive nitrogen intermediates and reactive oxygen intermediates from mouse peritoneal macrophages. Comparison of activating cytokines and evidence for independent production. J. Immunol. 1412407-2412. [PubMed] [Google Scholar]
- 9.Dohet, C., R. Wagner, and M. Radman. 1985. Repair of defined single base-pair mismatches in Escherichia coli. Proc. Natl. Acad. Sci. USA 82503-505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Fleck, O., E. Lehmann, P. Schar, and J. Kohli. 1999. Involvement of nucleotide-excision repair in msh2 pms1-independent mismatch repair. Nat. Genet. 21314-317. [DOI] [PubMed] [Google Scholar]
- 11.Flores, M. J., V. Bidnenko, and B. Michel. 2004. The DNA repair helicase UvrD is essential for replication fork reversal in replication mutants. EMBO Rep. 5983-988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Flores, M. J., N. Sanchez, and B. Michel. 2005. A fork-clearing role for UvrD. Mol. Microbiol. 571664-1675. [DOI] [PubMed] [Google Scholar]
- 13.Friedberg, E. C., Graham C. Walker, Wolfram Siede, Richard D. Wood, Roger A. Schultz, and Tom Ellenberger. 2005. DNA repair and mutagenesis, 2nd ed. ASM Press, Washington, DC.
- 14.Frischkorn, K., P. Sander, M. Scholz, K. Teschner, T. Prammananan, and E. C. Böttger. 1998. Investigation of mycobacterial recA function: protein introns in the RecA of pathogenic mycobacteria do not affect competency for homologous recombination. Mol. Microbiol. 291203-1214. [DOI] [PubMed] [Google Scholar]
- 15.Garnier, T., K. Eiglmeier, J.-C. Camus, N. Medina, H. Mansoor, M. Pryor, S. Duthoy, S. Grondin, C. Lacroix, C. Monsempe, S. Simon, B. Harris, R. Atkin, J. Doggett, R. Mayes, L. Keating, P. R. Wheeler, J. Parkhill, B. G. Barrell, S. T. Cole, S. V. Gordon, and R. G. Hewinson. 2003. The complete genome sequence of Mycobacterium bovis. Proc. Natl. Acad. Sci. USA 1007877-7882. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Gasc, A. M., A. M. Sicard, and J. P. Claverys. 1989. Repair of single- and multiple-substitution mismatches during recombination in Streptococcus pneumoniae. Genetics 12129-36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Halliwell, B., and O. I. Aruoma. 1991. DNA damage by oxygen-derived species. Its mechanism and measurement in mammalian systems. FEBS Lett. 2819-19. [DOI] [PubMed] [Google Scholar]
- 18.Hix, S., S. Morais Mda, and O. Augusto. 1995. DNA methylation by tert-butyl hydroperoxide-iron(II). Free Radic Biol. Med. 19293-301. [DOI] [PubMed] [Google Scholar]
- 19.Imlay, J. A., and S. Linn. 1987. Mutagenesis and stress responses induced in Escherichia coli by hydrogen peroxide. J. Bacteriol. 1692967-2976. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Iyer, V. N., and W. Szybalski. 1963. A molecular mechanism of mitomycin action: linking of complementary DNA strands. Proc. Natl. Acad. Sci. USA 50355-362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Jones, M., R. Wagner, and M. Radman. 1987. Repair of a mismatch is influenced by the base composition of the surrounding nucleotide sequence. Genetics 115605-610. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Kang, J., and M. J. Blaser. 2006. UvrD helicase suppresses recombination and DNA damage-induced deletions. J. Bacteriol. 1885450-5459. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Kaufmann, S. H. 2001. How can immunology contribute to the control of tuberculosis? Nat. Rev. Immunol. 120-30. [DOI] [PubMed] [Google Scholar]
- 24.Konola, J. T., K. E. Sargent, and J. B. Gow. 2000. Efficient repair of hydrogen peroxide-induced DNA damage by Escherichia coli requires SOS induction of RecA and RuvA proteins. Mutat. Res. 459187-194. [DOI] [PubMed] [Google Scholar]
- 25.Kramer, B., W. Kramer, and H. J. Fritz. 1984. Different base/base mismatches are corrected with different efficiencies by the methyl-directed DNA mismatch-repair system of E. coli. Cell 38879-887. [DOI] [PubMed] [Google Scholar]
- 26.Kumar, S., R. Lipman, and M. Tomasz. 1992. Recognition of specific DNA sequences by mitomycin C for alkylation. Biochemistry 311399-1407. [DOI] [PubMed] [Google Scholar]
- 27.Kunkel, T. A., and D. A. Erie. 2005. DNA mismatch repair. Annu. Rev. Biochem. 74681-710. [DOI] [PubMed] [Google Scholar]
- 28.Kunz, C., and O. Fleck. 2001. Role of the DNA repair nucleases Rad13, Rad2 and Uve1 of Schizosaccharomyces pombe in mismatch correction. J. Mol. Biol. 313241-253. [DOI] [PubMed] [Google Scholar]
- 29.Lefevre, J. C., P. Mostachfi, A. M. Gasc, E. Guillot, F. Pasta, and M. Sicard. 1989. Conversion of deletions during recombination in pneumococcal transformation. Genetics 123455-464. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Lestini, R., and B. Michel. 2007. UvrD controls the access of recombination proteins to blocked replication forks. EMBO J. 263804-3814. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Marra, G., and P. Schär. 1999. Recognition of DNA alterations by the mismatch repair system. Biochem. J. 3381-13. [PMC free article] [PubMed] [Google Scholar]
- 32.Matic, I., C. Rayssiguier, and M. Radman. 1995. Interspecies gene exchange in bacteria: the role of SOS and mismatch repair systems in evolution of species. Cell 80507-515. [DOI] [PubMed] [Google Scholar]
- 33.Minton, K. W. 1994. DNA repair in the extremely radioresistant bacterium Deinococcus radiodurans. Mol. Microbiol. 139-15. [DOI] [PubMed] [Google Scholar]
- 34.Mizrahi, V., and S. J. Andersen. 1998. DNA repair in Mycobacterium tuberculosis. What have we learnt from the genome sequence? Mol. Microbiol. 291331-1339. [DOI] [PubMed] [Google Scholar]
- 35.Nathan, C., and M. U. Shiloh. 2000. Reactive oxygen and nitrogen intermediates in the relationship between mammalian hosts and microbial pathogens. Proc. Natl. Acad. Sci. USA 978841-8848. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Nowosielska, A., S. A. Smith, B. P. Engelward, and M. G. Marinus. 2006. Homologous recombination prevents methylation-induced toxicity in Escherichia coli. Nucleic Acids Res. 342258-2268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Parker, B. O., and M. G. Marinus. 1992. Repair of DNA heteroduplexes containing small heterologous sequences in Escherichia coli. Proc. Natl. Acad. Sci. USA 891730-1734. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Petit, M. A., and D. Ehrlich. 2002. Essential bacterial helicases that counteract the toxicity of recombination proteins. EMBO J. 213137-3147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Pfister, P., S. Hobbie, C. Brüll, N. Corti, A. Vasella, E. Westhof, and E. C. Böttger. 2005. Mutagenesis of 16S rRNA C1409-G1491 base-pair differentiates between 6′OH and 6′NH3+ aminoglycosides. J. Mol. Biol. 346467-475. [DOI] [PubMed] [Google Scholar]
- 40.Pfister, P., S. Jenni, J. Poehlsgaard, A. Thomas, S. Douthwaite, N. Ban, and E. C. Böttger. 2004. The structural basis of macrolide-ribosome binding assessed using mutagenesis of 23S rRNA positions 2058 and 2059. J. Mol. Biol. 3421569-1581. [DOI] [PubMed] [Google Scholar]
- 41.Rayssiguier, C., D. S. Thaler, and M. Radman. 1989. The barrier to recombination between Escherichia coli and Salmonella typhimurium is disrupted in mismatch-repair mutants. Nature 342396-401. [DOI] [PubMed] [Google Scholar]
- 42.Samson, L., J. Thomale, and M. F. Rajewsky. 1988. Alternative pathways for the in vivo repair of O6-alkylguanine and O4-alkylthymine in Escherichia coli: the adaptive response and nucleotide excision repair. EMBO J. 72261-2267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Sancar, A. 1996. DNA excision repair. Annu. Rev. Biochem. 6543-81. [DOI] [PubMed] [Google Scholar]
- 44.Sander, P., A. Meier, and E. C. Böttger. 1995. rpsL+: a dominant selectable marker for gene replacement in mycobacteria. Mol. Microbiol. 16991-1000. [DOI] [PubMed] [Google Scholar]
- 45.Sander, P., T. Prammananan, and E. C. Böttger. 1996. Introducing mutations into a chromosomal rRNA gene using a genetically modified eubacterial host with a single rRNA operon. Mol. Microbiol. 22841-848. [DOI] [PubMed] [Google Scholar]
- 46.Schofield, M. J., and P. Hsieh. 2003. DNA mismatch repair: molecular mechanisms and biological function. Annu. Rev. Microbiol. 57579-608. [DOI] [PubMed] [Google Scholar]
- 47.Sicard, N., J. Oreglia, and A. M. Estevenon. 1992. Structure of the gene complementing uvr-402 in Streptococcus pneumoniae: homology with Escherichia coli uvrB and the homologous gene in Micrococcus luteus. J. Bacteriol. 1742412-2415. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Sidorkina, O., M. Saparbaev, and J. Laval. 1997. Effects of nitrous acid treatment on the survival and mutagenesis of Escherichia coli cells lacking base excision repair (hypoxanthine-DNA glycosylase-ALK A protein) and/or nucleotide excision repair. Mutagenesis 1223-28. [DOI] [PubMed] [Google Scholar]
- 49.Sinha, K. M., N. C. Stephanou, F. Gao, M. S. Glickman, and S. Shuman. 2007. Mycobacterial UvrD1 is a Ku-dependent DNA helicase that plays a role in multiple DNA repair events, including double-strand break repair. J. Biol. Chem. 28215114-15125. [DOI] [PubMed] [Google Scholar]
- 50.Spek, E. J., T. L. Wright, M. S. Stitt, N. R. Taghizadeh, S. R. Tannenbaum, M. G. Marinus, and B. P. Engelward. 2001. Recombinational repair is critical for survival of Escherichia coli exposed to nitric oxide. J. Bacteriol. 183131-138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Springer, B., P. Sander, L. Sedlacek, W.-D. Hardt, V. Mizrahi, P. Schar, and E. C. Böttger. 2004. Lack of mismatch correction facilitates genome evolution in mycobacteria. Mol. Microbiol. 531601-1609. [DOI] [PubMed] [Google Scholar]
- 52.Van Houten, B., D. L. Croteau, M. J. DellaVecchia, H. Wang, and C. Kisker. 2005. ‘Close-fitting sleeves’: DNA damage recognition by the UvrABC nuclease system. Mutat. Res. 57792-117. [DOI] [PubMed] [Google Scholar]
- 53.Veaute, X., S. Delmas, M. Selva, J. Jeusset, E. Le Cam, I. Matic, F. Fabre, and M. A. Petit. 2005. UvrD helicase, unlike Rep helicase, dismantles RecA nucleoprotein filaments in Escherichia coli. EMBO J. 24180-189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Veaute, X., J. Jeusset, C. Soustelle, S. C. Kowalczykowski, E. Le Cam, and F. Fabre. 2003. The Srs2 helicase prevents recombination by disrupting Rad51 nucleoprotein filaments. Nature 423309-312. [DOI] [PubMed] [Google Scholar]
- 55.Washburn, B. K., and S. R. Kushner. 1991. Construction and analysis of deletions in the structural gene (uvrD) for DNA helicase II of Escherichia coli. J. Bacteriol. 1732569-2575. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Worth, L., Jr., S. Clark, M. Radman, and P. Modrich. 1994. Mismatch repair proteins MutS and MutL inhibit RecA-catalyzed strand transfer between diverged DNAs. Proc. Natl. Acad. Sci. USA 913238-3241. [DOI] [PMC free article] [PubMed] [Google Scholar]