

Abstract
The RNA helicases encoded by melanoma differentiation-associated gene 5 (mda-5) and retinoic acid-inducible gene I (RIG-I) detect foreign cytoplasmic RNA molecules generated during the course of a virus infection, and their activation leads to induction of type I interferon synthesis. Paramyxoviruses limit the amount of interferon produced by infected cells through the action of their V protein, which binds to and inhibits mda-5. Here we show that activation of both mda-5 and RIG-I by double-stranded RNA (dsRNA) leads to the formation of homo-oligomers through self-association of the helicase domains. We identify a region within the helicase domain of mda-5 that is targeted by all paramyxovirus V proteins and demonstrate that they inhibit activation of mda-5 by blocking dsRNA binding and consequent self-association. In addition to this commonly targeted domain, some paramyxovirus V proteins target additional regions of mda-5. In contrast, V proteins cannot bind to RIG-I and consequently have no effect on the ability of RIG-I to bind dsRNA or to form oligomers.
Mammalian cells contain a variety of pattern recognition receptors that recognize foreign macromolecules termed pathogen-associated molecular patterns (PAMPs). Viral PAMPs generated in the cytosol during replication are recognized by the DExD/H-box RNA helicases coded for by melanoma differentiation-associated gene 5 (mda-5) and retinoic acid-inducible gene I (RIG-I) (reviewed in reference 30) and stimulate the production of type I interferon (IFN), which constitutes a major component of the innate immune response to virus infection (reviewed in reference 21). It is becoming clear that viruses generate a variety of different PAMPs and that, rather than being redundant, mda-5 and RIG-I show ligand specificity and are therefore differentially sensitive to activation by different viruses. For example, RIG-I seems to be more important for IFN induction in response to hepatitis C virus (HCV) (6, 24) and influenza A virus (12, 19), while mda-5 is necessary for responses to picornaviruses (7, 12). Both mda-5 and RIG-I can be activated by the synthetic double-stranded RNA (dsRNA) poly(I-C), but a recent study suggests that the length of the dsRNA influences whether IFN induction is dependent on mda-5 or RIG-I, with mda-5 being more important for induction by long dsRNA and RIG-I more important for induction by short dsRNA (11). In addition to length, other structural features of viral RNAs can also determine receptor activation. For example, single-stranded RNA (ssRNA) and dsRNA molecules bearing a 5′ triphosphate induce IFN via RIG-I and not mda-5 (10, 19). This motif is recognized as nonself, since most cellular RNAs are either capped or have a 5′ monophosphate. IFN induction by RNA purified from influenza A virus, vesicular stomatitis virus, and rabies virus requires RIG-I and is dependent on the presence of a 5′ triphosphate, underlining the importance of this motif as a genuine viral PAMP (8, 10, 19).
mda-5 and RIG-I share a common domain structure with two tandem CARD motifs at the N terminus which are responsible for downstream signaling, a central DECH RNA helicase domain with ATPase activity and a C-terminal domain (CTD). Recent structural studies have shown that the binding site for the 5′ triphosphate motif is located on a basic cleft within the CTD of RIG-I (3, 29). Short dsRNAs (∼25 bp) are also recognized through this site, whereas poly(I-C) binding requires both the CTD and the helicase domain (3, 29). A model has been proposed for RIG-I in which the helicase domain and CTD prevent the CARD domains from signaling in the inactive state. Binding of viral RNA stimulates the ATPase activity and triggers a major conformational change which allows dimerization and reveals the CARD domains to interact with the downstream adapter protein IPS-1/VISA/CARDIF/MAVS (hereafter referred to as IPS-1) (23). This leads to activation of the transcription factors IFN regulatory factor 3 (IRF-3) and NF-κB which are required for transcriptional induction of the IFN-β promoter. Consistent with this model, dimers of RIG-I have been detected in cells infected with Sendai virus (SeV) (23), and gel filtration analysis has demonstrated RIG-I dimers in the presence of 5′-triphosphate RNA (3). It would be expected that mda-5 is activated in a similar manner; however, dimerization of mda-5 has not been previously demonstrated.
The Paramyxoviridae are a family of single-stranded, negative-sense RNA viruses which are divided into two subfamilies: (i) the Paramyxovirinae comprising the respiroviruses (e.g., SeV), the rubulaviruses (e.g., parainfluenza virus 5 [PIV5; formerly known as SV5]), the morbilliviruses (e.g., measles virus [MeV]), and the more recently described henipaviruses (e.g., Hendra virus [HeV]) and (ii) the Pneumovirinae (reviewed in reference 14). Members of the Paramyxovirinae use a variety of methods to evade the IFN response, including limiting the amount of IFN produced by infected cells and blocking IFN signaling. These functions are encoded by the P/V/C gene, which makes several products due to the existence of specific RNA editing mechanisms and the use of alternate open reading frames (ORFs). In a mechanism that seems to be conserved among all family members, we found that the V protein is able to antagonize the induction of IFN-β (9, 20). We subsequently identified mda-5 as a cellular binding partner for the PIV5 V protein (PIV5-V) and demonstrated that the consequence of this interaction was inhibition of IFN induction by mda-5 (1). The V proteins from 13 different paramyxoviruses were tested, all were able to interact with mda-5 and inhibit its function, and this was dependent on the highly conserved C terminus of V (1, 2). In contrast, none of the V proteins was able to bind to or inhibit IFN induction by RIG-I (2). Here we investigate the molecular mechanism of inhibition of mda-5 by paramyxovirus V proteins. We show that in a manner similar to activation of RIG-I by 5′-triphosphate RNA, activation of mda-5 by dsRNA involves the formation of homo-oligomers. The V protein binds to the helicase domain of mda-5 and blocks its activation by inhibiting dsRNA binding and consequent self-association.
MATERIALS AND METHODS
Plasmids.
The IFN-β promoter reporter plasmid pIFΔ(−116)lucter (13), the constitutive β-galactosidase reporter plasmid pJatLacZ (18), pEF.plink2 (17), pEF.PIV5-V (4), pdl.HCV.NS3/4a (2), pEF.mda-5, and pEF.mda-5CARD (1) have been described previously. pEF.PKR(1-207) is a pEFplink2-derived mammalian expression vector containing an ORF for amino acids (aa) 1 to 207 of protein kinase R (PKR), and pEF.M2(1-207) contains the same region of PKR but lacks aa 58 to 69. pCMVSPORT6.IPS-1 was obtained from the I.M.A.G.E Consortium (clone identification no. 5751684) (15). For expression of epitope-tagged proteins in mammalian cells, cDNAs were cloned into pEF.myc.plink2 (2), pEF.V5.plink2, or pEF.Flag.plink2. pEF.V5.plink2 and pEF.Flag.plink2 were constructed by inserting the Pk epitope sequence from PIV5-V (26) or the Flag tag sequence upstream of the NcoI site in the multiple-cloning site of pEF.plink2 such that cDNAs cloned into the NcoI site are tagged at the N terminus. cDNAs or partial cDNA fragments encoding mda-5, mda-5 (aa 1 to 935), RIG-I, the mda-5 CARD (mda-5C) domain (aa 1 to 287), the mda-5 helicase (mda-5H) domain (aa 287 to 1025), the RIG-I CARD (RIG-IC) domain (aa 1 to 225), or the RIG-I helicase (RIG-IH) domain (aa 225 to 925) were cloned into these vectors by standard methods.
For yeast two-hybrid assays, cDNAs and fragments thereof were cloned into pGBKT7 or pGADT7 (Clontech) for expression of proteins as GAL4 DNA-binding domain (DBD) or GAL4 activation domain (AD) fusions, respectively. The amino acids encoded by mda-5C, mda-5H, RIG-IC, and RIG-IH fragments are the same as for the pEF.Tag equivalents. pGADT7 constructs expressing various paramyxovirus V proteins have been described previously (1, 2). pHON7, a vector designed to permit nuclear expression of a non-fusion protein in yeast (the “third” protein) was constructed by deleting the Asp718I/NcoI fragment of the GAL4 activation domain from pGADT7 and replacing the LEU2 gene of the resultant deletion with the URA3 gene from pHisI (Clontech). pHON7.PKR(1-207) and pHON7.PKR.M2(1-207) are pHON7 derivatives containing an ORF for aa 1 to 207 of PKR and an ORF for aa 1 to 207 of PKR lacking aa 58 to 69, respectively. cDNAs for PIV5-V, SeV-V, MeV-V, HeV-V, and PIV5-VΔN104 were cloned into pHON7 from their pGADT7 equivalents (2).
Cells and transfections.
Vero (ATCC CCL-81), HEK-293 (ATCC CRL-1573), and HEp-2 (ATCC CCL-23) cells were maintained in Dulbecco's modified Eagle's medium plus 10% fetal bovine serum and penicillin-streptomycin. Transfections were carried out using linear polyethyleneimine (molecular weight, ∼25,000; Polysciences Inc., Warrington, PA) or Lipofectamine (InVitrogen) under standard conditions. Where indicated, cells were treated with recombinant human IFN-α (Roferon; Roche) at 1,000 IU/ml. Induction of cells with poly(I-C) (2) and measurements of luciferase and β-galactosidase activity were carried out as described elsewhere (13).
Co-IP assays, poly(I-C)-agarose binding assays, and immunoblotting.
To make cell extracts, 6-cm dishes of transfected cells were washed twice in cold phosphate-buffered saline and then lysed in 500 μl coimmunoprecipitation (Co-IP) buffer (50 mM Tris-HCl [pH 7.5], 150 mM NaCl, 1 mM EDTA, 1% NP-40). One hundred microliters of extract was used for Co-IP assays with a monoclonal antibody to the Flag tag (Sigma), the V5 tag (anti-Pk) (22), or the myc tag (clone 4A6; Upstate). Immune complexes were collected on protein A-Sepharose beads (GE Healthcare), which were then washed three times with Co-IP buffer and resuspended in sodium dodecyl sulfate-polyacrylamide gel electrophoresis (SDS-PAGE) loading buffer. For poly(I-C)-agarose binding assays, 150 to 300 μl extract was mixed with an equal volume of a 10% slurry of poly(I-C)-agarose beads (GE Healthcare) in Co-IP buffer. After incubation at 4°C for 2 h, beads were washed three times with Co-IP buffer and resuspended in SDS-PAGE loading buffer. After separation by SDS-PAGE, tagged proteins were detected by immunoblotting using antibodies against the Flag, V5 or myc tags. Western blots for mda-5 and STAT-1 were carried out using an anti-mda-5 antibody (Alexis Biochemicals) or an anti-STAT-1 antibody (Santa Cruz Biotechnology).
Yeast two-hybrid assays.
Combinations of GAL4 DBD and GAL4 AD fusion plasmids were introduced into Saccharomyces cerevisiae strain PJ69-4α using polyethylene glycol-lithium acetate-mediated transformation. Double transformants were selected on synthetic dropout (SD) medium lacking leucine and tryptophan (SD −L −W). Individual colonies were subsequently streaked onto SD −L −W medium also lacking histidine (SD −L −W −H) and containing 5 to 20 mM 3-aminotriazole (3-AT). When the pHON7-derived third plasmid was used, triple transformants were selected on SD medium lacking leucine, tryptophan, and uracil (SD −L −W −U), and individual colonies were subsequently streaked onto SD −L −W −U medium also lacking histidine (SD −L −W −U −H) and containing 5 mM 3-AT. Growth was monitored for 4 to 10 days at 30°C.
RESULTS
Mapping the binding site(s) for paramyxovirus V proteins on mda-5.
We have previously shown that the V proteins from a wide range of paramyxoviruses specifically inhibit the ability of mda-5, but not RIG-I, to induce IFN-β production (1, 2). We demonstrated a direct physical interaction between V and mda-5, but did not determine the mechanism of inhibition by the V protein.
In order to begin to address this question, we first set out to define the region of mda-5 that contains the binding site for paramyxovirus V proteins. We showed previously that PIV5-V interacts with aa 287 to 1025 of mda-5, a sequence which encompasses the entire helicase domain and C-terminal domain but lacks the N-terminal CARDs (2). To define the binding site in more detail, a series of plasmids were constructed that express various fragments of the mda-5 helicase domain, and the ability of these fragments to bind to PIV5-V was assessed using the yeast two-hybrid assay (Fig. 1A). Interestingly, we found that several nonoverlapping fragments of the helicase domain were capable of binding to PIV5-V. The C-terminal domain of mda-5 with most of the helicase domain removed, mda-5(816-1025), retained the ability to bind to PIV5-V; however, neither a fragment containing aa 816 to 935 nor one containing residues 934 to 1025 was sufficient. In addition, mda-5(287-458), mda-5(500-676), and mda-5(676-816), were also capable of interacting with PIV5-V.
FIG. 1.

Mapping the binding sites for paramyxovirus V proteins on mda-5. Interactions between different fragments of mda-5 and various paramyxovirus V proteins were assessed using the yeast two-hybrid assay. Yeast cells were transformed with a plasmid expressing one of the indicated fragments of mda-5 fused to the GAL4 DBD and a plasmid expressing the indicated paramyxovirus V protein fused to the GAL4 AD. Positive transformants were selected on SD −L −W and then streaked onto SD −L −W −H plus 3-AT as indicated to assay for an interaction. All mda-5 fragments used in these experiments failed to promote growth on SD −L −W −H plus 3-AT in the absence of an interacting partner (data not shown). (A) Interactions of mda-5 fragments with PIV5-V. The line diagram shows the domain structure of mda-5 including the two N-terminal CARDs, the RNA helicase domain defined by seven conserved motifs (shaded boxes), and the CTD. The mda-5 deletion constructs used in this work are shown beneath this line. Interactions with PIV5-V are shown to the right of the panel. +, positive interaction; −, no interaction. (B) Interactions of mda-5 fragments with the V proteins of SeV, HeV, Menangle virus, Mapuera virus, and Salem virus were assessed under selection conditions incorporating 5 mM 3-AT, a competitive inhibitor of HIS3. (C) Interactions of mda-5 fragments with the V proteins of PIV5, Menangle virus, and Salem virus under increasingly stringent conditions; the concentration of 3-AT is indicated.
We also analyzed binding of several other paramyxovirus V proteins to these fragments of mda-5 to determine whether they all share the ability to bind to multiple sites. The V proteins of Sendai, Hendra, Menangle, Mapuera, and Salem viruses all bound to the intact helicase domain lacking the CARDs, mda-5(287-1025) (Fig. 1B). Interestingly, the only smaller fragment of mda-5 that could bind to all of the V proteins examined was mda-5(676-816). In addition, both Salem and Menangle V proteins could interact with mda-5(500-676), and the Salem virus V protein could also interact with mda-5(287-458). Interaction with mda-5(816-1025) was restricted to PIV5-V. These results show that the conserved binding site for the paramyxovirus V protein lies between aa 676 and 816 of mda-5 and that some V proteins also make additional contacts with other parts of mda-5.
For those V proteins that were able to bind to more than one fragment of mda-5, the relative affinities of these interactions were assessed by plating the yeast onto selective media containing increasing concentrations of 3-AT (Fig. 1C). 3-AT is a competitive inhibitor of the HIS3 gene product, and therefore only the strongest interactions which give the highest levels of reporter gene activity will be detected in the presence of high concentrations of 3-AT. On 10 mM 3-AT, yeast cells expressing mda-5(816-1025) and PIV5-V showed impaired growth relative to growth on 5 mM 3-AT (compare Fig. 1B with C), but all other interacting pairs grew equally well. For PIV5-V and Menangle virus V, the only interaction [apart from the intact helicase domain, mda-5(287-1025)] that could support growth of yeast on 20 mM 3-AT was with mda-5(676-816), indicating that this fragment contains the highest-affinity binding site for these V proteins. The interaction between Salem V and mda-5(287-458) was also strong enough to support yeast growth on 20 mM 3-AT, but the interaction with mda-5(500-676) was not.
For those V proteins that can interact with multiple sites on mda-5, there are three possibilities: first that several molecules of V can bind to a single molecule of mda-5 at different sites along the length of the helicase domain; second, that a single molecule of V binds to a single mda-5 but makes contacts with multiple sites that are juxtaposed in the context of the folded protein; and third, that individual molecules of V interact with distinct molecules of mda-5. We reasoned that if a single molecule of V can bind to several sites on mda-5, it might be possible to see recruitment of one mda-5 fragment to another in the presence of V. None of the three fragments mda-5(287-458), mda-5(500-816), or mda-5(816-1025) could interact with themselves or each other in the yeast two-hybrid assay, and when we coexpressed PIV5-V, we failed to observe any bridging between different fragments (data not shown). Conversely, we also expressed mda-5 to see if it could bridge two V proteins, but again no interaction was observed (data not shown).
PIV5-V does not target mda-5 for degradation.
We then considered possible mechanisms through which the V protein might inhibit mda-5. In addition to their role in limiting IFN production, paramyxovirus V proteins can also block IFN signaling. In the case of PIV5, the V protein targets STAT1 for proteasome-mediated degradation (4), so we therefore investigated whether PIV5-V could also target mda-5 for degradation. We analyzed the levels of mda-5 in cells that had been infected with the W3 strain of PIV5 (Fig. 2) and found that the amount of mda-5 protein in the infected cells was comparable to that found in uninfected cells. As expected, mda-5 was highly inducible by IFN pretreatment, and the induced levels were similarly unaffected by PIV5 infection of the IFN-pretreated cells. For comparison, we also looked at STAT1 levels in the same cell extracts and observed that in the infected cells all the STAT1 had been completely degraded. We therefore conclude that PIV5-V does not target mda-5 for proteasome-mediated degradation.
FIG. 2.
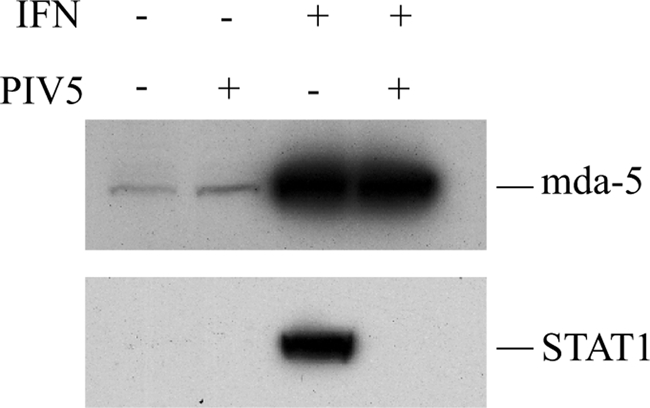
PIV5 infection does not cause degradation of mda-5. HEp-2 cells were or were not pretreated for 12 h with type I IFN before mock infection or infection with the W3A strain of PIV5 at a multiplicity of infection of 50 for 12 h. Levels of mda-5 and STAT1 were analyzed by Western blotting.
PIV5-V prevents activation of mda-5.
Since mda-5 levels are unaffected by PIV5 infection, V proteins must either prevent the activation of mda-5 or block its downstream signaling to IPS-1. The location of the binding site for the V proteins being within the mda-5 helicase domain, rather than the CARD domain, which interacts with IPS-1, makes it seem more likely that they would prevent activation. Indeed, in contrast to its effect on full-length mda-5, PIV5-V is unable to block induction by the isolated CARD domains or by overexpression of IPS-1, while HCV NS3/4a, which cleaves IPS-1, is able to block all three (Fig. 3). Also, our previous data have shown that a chimera consisting of the CARD domain of RIG-I and the helicase domain of mda-5 is sensitive to inhibition by V, whereas an equivalent chimera containing the CARD domain of mda-5 and the helicase domain of RIG-I is insensitive to V (2). These results indicate that the V protein blocks mda-5 function by targeting the helicase domain and preventing its activation and not by inhibiting downstream signaling.
FIG. 3.
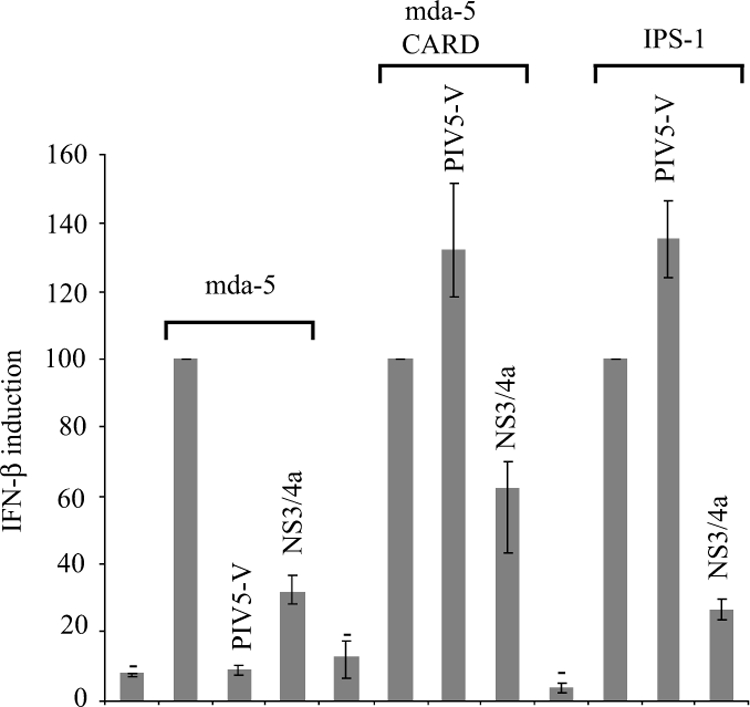
PIV5-V prevents activation of mda-5. Vero cells were transfected with the IFN-β reporter plasmid pIFΔ(−116)lucter, the β-galactosidase expression vector pJatlacZ, a plasmid expressing mda-5 (pEF.mda-5), the CARD domains of mda-5 (pEF.mda-5C) and IPS-1 (pCMVSPORT6.IPS-1), and either pEF.PIV5-V, pdl.NS3/4a, or the empty vector pEFpl2. Luciferase and β-galactosidase assays were carried out 48 h after transfection, and relative expression levels were calculated. Results shown represent the average from three independent experiments, with the highest and lowest values shown as error bars. A reference value of 100% has been assigned to the level of induction seen with mda-5, mda-5C, and IPS-1.
mda-5 and RIG-I form homo-oligomers in response to dsRNA.
Current models of RIG-I activation suggest that following binding of the helicase domain to an RNA ligand, a conformational change occurs which allows RIG-I to dimerize and reveals the CARD domains for signaling. In support of this, dimers of RIG-I have been detected in the presence of 5′-triphosphate RNA and in SeV-infected cells (3, 23). It seemed likely that activation of mda-5 would proceed in a similar manner, so we initially sought to determine whether oligomers of mda-5 and RIG-I could be detected in cells treated with the synthetic dsRNA poly(I-C), a ligand to which both helicases are known to respond. 293 cells were cotransfected with myc- and Flag-tagged mda-5 or myc- and Flag-tagged RIG-I and then were either left untreated or treated with poly(I-C) for 2 to 16 h. Extracts from these cells were then subjected to immunoprecipitation with an antibody against the Flag tag, and the presence of the myc-tagged protein in the immunoprecipitate was determined to assess the formation of oligomers. In unstimulated cells, no myc-tagged mda-5 is coimmunoprecipitated with Flag-tagged mda-5 (Fig. 4A, lane 1), and similarly, no myc-tagged RIG-I is coimmunoprecipitated with Flag-tagged RIG-I (Fig. 4B, lane 1), indicating that in untreated cells both mda-5 and RIG-I exist as monomers. Significantly, in cells treated with poly(I-C), myc-mda-5 is coprecipitated with Flag-mda-5 (Fig. 4A), and also myc-RIG-I is coprecipitated with Flag-RIG-I (Fig. 4B), thus demonstrating that poly(I-C) induces the formation of homo-oligomers of both mda-5 and RIG-I. In cells transfected with myc-tagged RIG-I and Flag-tagged mda-5, we were not able to observe any coprecipitation after treatment with poly(I-C) (Fig. 4C), demonstrating that hetero-oligomerization of these helicases is not observed.
FIG. 4.

mda-5 and RIG-I self-associate in response to poly(I-C). 293 cells were transfected with plasmids expressing (A) myc- and Flag-tagged forms of mda-5, (B) myc- and Flag-tagged forms of RIG-I, or (C) myc-tagged RIG-I and Flag-tagged mda-5. Twenty-four hours after transfection, cells were transfected with poly(I-C) and left for a further 2 to 16 h before harvesting. Cell extracts were subjected to immunoprecipitation (IP) with an antibody against the Flag tag, and proteins present in the immunoprecipitate were analyzed by Western blotting (WB). (D) Extracts from cells transfected with myc- and Flag-tagged mda-5 (top panel) or myc- and Flag-tagged RIG-I (lower panel) were incubated with increasing concentrations of poly(I-C) in the presence of 1 mM ATP for 1 h at 4°C. Samples were then subjected to immunoprecipitation with an antibody against the Flag tag, and the presence of myc-tagged protein in the immunoprecipitate was determined by Western blotting for the myc tag. (E) Western blots were carried out on the extracts used in panel D to determine expression levels of tagged proteins.
Poly(I-C) could also induce oligomerization of mda-5 and RIG-I in vitro. Extracts from untreated cells transfected with tagged versions of mda-5 or RIG-I were incubated with increasing concentrations of poly(I-C) for 1 h prior to coimmunoprecipitation. Oligomerization of mda-5 was observed in the presence of 0.1 μg/ml poly(I-C), was maximal in the presence of 1 μg/ml poly(I-C), and declined thereafter (Fig. 4D). Interestingly, oligomerization of RIG-I did not occur in the presence of 0.1 μg/ml poly(I-C), but required a higher concentration, with oligomers only being formed in the presence of 1 μg/ml and 10 μg/ml poly(I-C) (Fig. 4D). This was not due to differences in protein concentrations since both mda-5 and RIG-I were expressed to similar levels (Fig. 4E), suggesting that mda-5 is more sensitive to activation by poly(I-C) than RIG-I, although it is possible that the difference may be due to the relative concentrations of poly(I-C) of different lengths in the preparation.
The helicase domains of mda-5 and RIG-I contain a dsRNA-dependent oligomerization domain.
We next determined which domains of mda-5 and RIG-I are required for oligomerization. To do this, we constructed plasmids that express V5- and Flag-tagged versions of the CARD or the helicase domains alone and assayed by Co-IP whether they were able to form oligomers in the presence or absence of poly(I-C). Figure 5A shows that the CARD domains of mda-5 self-associate in the absence of an inducer (lane 1) and that this is unaffected by treatment of the cells with poly(I-C) (lane 2). In contrast, oligomerization of the RIG-I CARD domain was not observed under either condition (lanes 5 and 6). Interestingly, the helicase domain of mda-5 was also found to self-associate in uninduced cells, but the amount of Flag-tagged mda-5H being coimmunoprecipitated with the V5-tagged mda-5H was reproducibly increased in the induced cells, suggesting that this interaction may be regulated by poly(I-C) (lanes 3 and 4). In contrast to mda-5, the helicase domain of RIG-I does not constitutively oligomerize, but self-associates only in the presence of exogenously added poly(I-C) (lanes 7 and 8). As expected, no interaction was detected between mda-5H and RIG-IH in the absence or presence of poly(I-C) (lanes 9 and 10).
FIG. 5.

The helicase domains of mda-5 and RIG-I contain a dsRNA-dependent oligomerization domain. (A). 293 cells were transfected with plasmids expressing V5- and Flag-tagged forms of the mda-5C domains (lanes 1 and 2), V5- and Flag-tagged forms of the mda-5H domain (lanes 3 and 4), V5- and Flag-tagged forms of the RIG-IC domains (lanes 5 and 6), V5- and Flag-tagged forms of the RIG-IH domain (lanes 7 and 8), or V5-tagged mda-5H and Flag-tagged RIG-IH (lanes 9 and 10). Where indicated (+), one of each pair was transfected the following day with poly(I-C) for 8 h prior to harvesting. Cell extracts were subjected to immunoprecipitation (IP) with an antibody against the V5 tag, and then the presence of the Flag-tagged protein in the immunoprecipitate was analyzed by Western blotting (WB). Expression of all tagged proteins was confirmed by Western blotting on the cell extracts (data not shown). The experiment was performed three times, images were scanned using a Molecular Dynamics Storm 840 PhosphorImager and quantitated using ImageQuant software, and the increase in the amount of Flag-tagged mda-5H in lane 4 compared to lane 3 was determined to be statistically significant by Student's t test with a P value of <0.05. (B). Extracts were prepared from 293 cells transfected with plasmids expressing either Flag-mda-5H, V5-mda-5H, the dsRNA-binding domains of PKR [PKR(1-207)], a mutant form of PKR that is unable to bind dsRNA [M2(1-207)], or the PIV5-V protein. In vitro oligomerization assays were performed by mixing together 50 μl extract from cells expressing Flag-mda-5H with 50 μl extract from cells expressing V5-mda-5H in the presence of 300 μl buffer (lane 1), 300 μl extract from cells expressing PKR(1-207) (lane 2), 300 μl extract from cells expressing M2(1-207) (lane 3), or 300 μl extract from cells expressing PIV5-V (lane 4). Following incubation for 1 h at 4°C to allow complexes to form, immunoprecipitation assays were carried out using an antibody against the Flag tag and the presence of the V5-tagged protein in the immunoprecipitate was determined by Western blotting. The experiment was performed three times, and the reduction in V5-tagged protein in the immunoprecipitate in the presence of PKR(1-207) and PIV5-V was determined to be statistically significant according to Student's t test with P values of <0.01 and <0.05, respectively. (C) 293 cells were transfected with pIFΔ(−116)lucter, pJatlacZ, 80 ng of a plasmid expressing mda-5 and 160 ng of either the empty vector pEFpl2, a plasmid expressing the dsRNA-binding domains of PKR [PKR(1-207)], or a plasmid expressing a mutant form of PKR that is unable to bind dsRNA [M2(1-207)]. Luciferase and β-galactosidase assays were carried out 48 h after transfection, and relative expression levels were calculated. The results shown represent the average from three independent experiments, with the highest and lowest values shown as error bars. A reference value of 100% has been assigned to the level of induction seen in the presence of full-length mda-5. (D) Extracts from 293 cells expressing Flag-tagged full-length mda-5(1-1025) or Flag-tagged mda-5(1-935) were incubated with poly(I-C)-agarose beads for 1 h at 4°C. After washing the beads, bound proteins were analyzed by Western blotting for the Flag tag (upper panel). Expression of both proteins was confirmed by Western blotting of the cell extracts (lower panel). (E) Vero cells were transfected with pIFΔ(−116)lucter, pJatlacZ, and either the empty vector pEFpl2, a plasmid expressing full-length mda-5, or a plasmid expressing mda-5(1-935). After 24 h, the cells were either left untreated or were transfected with poly(I-C). Luciferase and β-galactosidase assays were carried out 48 h after transfection, and relative expression levels were calculated. The results shown represent the average from three independent experiments, with the highest and lowest values shown as error bars. A reference value of 100% has been assigned to the level of induction seen in the presence of full-length mda-5 and poly(I-C). (F) Interactions between the CARD and helicase domains of mda-5 and RIG-I were assessed in the yeast two-hybrid assay. Yeast cells were transformed with plasmids expressing the indicated fragments of mda-5 and RIG-I as GAL4 DBD fusions (given first) and GAL4 AD fusions (given second). Positive transformants were selected on SD −L −W, and growth on this medium demonstrates that the yeast cells have been transformed by both plasmids. They were then streaked onto SD −L −W −H plus 5 mM 3-AT, and growth on this medium demonstrates an interaction between the two proteins. (G) The dependence of the helicase domain interactions on the presence of yeast dsRNA was assessed. Yeast cells were transformed with a plasmid expressing a GAL4 DBD fusion protein (given first), a plasmid expressing a GAL4 AD fusion protein (given third), and either the empty vector pHON7, pHON7 expressing the dsRNA-binding domains of PKR [PKR(1-207)], or pHON7 expressing a mutant form of PKR that is unable to bind dsRNA [M2(1-207)] (given second). Positive transformants were selected on SD −L −W −U, and growth on this medium indicates that the yeast cells have been transformed by all three plasmids. They were then streaked onto SD −L −W −U −H plus 5 mM 3-AT, and growth on this medium demonstrates an interaction between the GAL4 DBD fusion protein and the GAL4 AD fusion protein.
We speculated that the self-association of the mda-5 helicase domain observed in uninduced cells (Fig. 5A, lane 3) is caused by endogenous dsRNA, perhaps generated during the transfection process, and we tested this by coexpressing an N-terminal fragment of PKR(1-207) which contains the dsRNA-binding domains of PKR without the catalytic domain and should function to sequester cellular dsRNA. Figure 5B (lanes 1-3) demonstrates that the self-association of the mda-5 helicase domain is indeed significantly reduced by coexpression of PKR(1-207), but not a mutant form of PKR that is unable to bind dsRNA [M2(1-207)] (5), and, as expected, self-association of the mda-5 helicase domain was also disrupted by PIV5-V (Fig. 5B, lane 4). Expression of PKR(1-207) but not M2(1-207) also inhibited the activation of the IFN-β promoter by mda-5 (Fig. 5C), indicating that the constitutive activation associated with the overexpression of mda-5 is a function of a response to endogenous dsRNA molecules. The fact that mda-5H but not RIG-IH is constitutively oligomerized in uninduced cells may reflect the fact that mda-5 is more sensitive to dsRNA than RIG-I. We also generated a form of mda-5 that lacks the C-terminal 90 aa [mda-5 (1-935)]; in contrast to full-length mda-5 [mda-5(1-1025)], this truncated form is unable to bind to poly(I-C) (Fig. 5D), and is unable to stimulate transcription of the IFN-β promoter in uninduced cells or to stimulate a response above vector alone in cells treated with poly(I-C) (Fig. 5E). As discussed below the loss of the C-terminal 90 aa abolished the ability of the mda-5 helicase domain to self-associate (Fig. 5F).
Since 293 cells contain endogenous mda-5 and RIG-I and also other proteins such as IPS-1 that may be involved in the formation of high-molecular-weight complexes, it is possible that the interactions observed in Co-IP experiments are formed indirectly through association with these endogenous molecules. Therefore, in order to look at whether oligomerization of mda-5 and RIG-I is mediated through direct protein-protein interactions, we chose to use the yeast two-hybrid system since yeast have no homologues of these proteins. In this assay the CARD domains of mda-5 were unable to interact (Fig. 5F), which suggests that the interaction seen by Co-IP from mammalian cells may require the presence of another protein(s) that is not present in yeast. In contrast, we found that the helicase domains of mda-5 and RIG-I were both able to self-associate in yeast, indicating that these interactions are direct. Deletion of the C-terminal 90 aa of mda-5 (103 aa from RIG-I) or deletion of around 200 aa from the N terminus of either helicase domain resulted in a loss of the ability of mda-5H and RIG-IH to homo-oligomerize, indicating that both the helicase domain and the C-terminal domain are involved in the interaction (Fig. 5F). Again, no heterologous interaction was observed between the helicase domain of mda-5 and the helicase domain of RIG-I.
Oligomerization of mda-5 and RIG-I is dependent upon poly(I-C) in mammalian cells, and yet we see these interactions occurring in yeast without adding exogenous dsRNA. It has been known for some time that most yeast strains are naturally infected with dsRNA viruses (reviewed in references 25 and 31), so it was of interest to determine whether the interactions we observe are dependent on the presence of dsRNA in yeast. To do this, we coexpressed the dsRNA-binding domain of PKR for the purpose of sequestering any dsRNA present in the yeast cells and thus suppressing any interactions that are dsRNA dependent. When this is coexpressed with the helicase domains of mda-5 or RIG-I, it clearly inhibits their oligomerization (Fig. 5G). In contrast, the mutant form of PKR, M2(1-207), which is unable to bind dsRNA, had no effect on oligomerization of mda-5H or RIG-IH.
Paramyxovirus V proteins inhibit dsRNA binding to mda-5 and the consequent oligomerization of the mda-5H domain.
Having shown that activation of mda-5 occurs through dsRNA-dependent oligomerization of the helicase domain, we then wanted to establish how paramyxovirus V proteins are able to interfere with this. The interaction between mda-5 and PIV5-V occurs irrespective of the presence of dsRNA since it is not affected by coexpression of the dsRNA-binding domain of PKR (Fig. 6A). Therefore, we assessed the effect of PIV5-V on the ability of Flag-tagged mda-5H to bind poly(I-C)-agarose beads. Figure 6B shows that mda-5H bound strongly to the beads in the absence of PIV5-V, but binding was substantially reduced when PIV5-V was added to the reaction in a dose-dependent manner. In contrast, PIV5-V had no effect on the ability of RIG-IH to bind poly(I-C).
FIG. 6.

The V protein inhibits poly(I-C) binding and oligomerization of the helicase domain. (A) Yeast cells were transformed with pGBKT7.mda-5, pGADT7.PIV5-V, and either pHON7, pHON7.PKR(1-207), or pHON7.M2(1-207). Positive transformants were selected on SD −L −W −U and streaked onto SD −L −W −U −H plus 5 mM 3-AT to assay for interactions. (B) Extracts from cells expressing Flag-tagged mda-5H (top panel) or Flag-tagged RIG-IH (lower panel) were mixed with buffer (lane 1), an equal volume of an extract from cells expressing PIV5-V (lane 2), or twice the volume of an extract from cells expressing PIV5-V (lane 3) and incubated for 1 h at 4°C to allow V/mda-5 binding. Poly(I-C)-agarose beads were then added to the samples and incubated for a further hour. Proteins that had bound to the beads were analyzed by Western blotting for the Flag tag. (C) Yeast cells were transformed with plasmids expressing either mda-5H or RIG-IH as GAL4 DBD and GAL4 AD fusion proteins (given first and third, respectively) and either the empty vector pHON7 or pHON7 expressing PIV5-V, the C terminus of PIV5-V (PIV5-VΔN104), the V protein from SeV, the V protein from MeV, or the V protein from HeV (given second). Positive transformants were selected on SD −L −W −U and then streaked onto SD −L −W −U −H plus 5 mM 3-AT to assay for interactions.
We then used the yeast assay to determine whether the V protein could also prevent oligomerization of mda-5. As demonstrated previously, self-association of the mda-5H domain can be detected when it is expressed from both the DBD vector and the AD vector (Fig. 6C). Significantly, when PIV5-V is expressed from a third plasmid, no self-association is observed, indicating that V is able to block the interaction between the mda-5H domains. We have previously shown that the unique cysteine-rich region at the C terminus of the V protein is sufficient for binding to mda-5 and inhibiting IFN induction (1). Expression of this C-terminal fragment of PIV5-V (PIV5-VΔN104) is also sufficient to block oligomerization of the helicase domain in yeast (Fig. 6C). In contrast, the expression of PIV5-V had no effect on oligomerization of the RIG-IH domain, which is consistent with our previous data showing that PIV5-V could not interact with RIG-I nor inhibit its function (2).
We have studied the V proteins of 13 paramyxoviruses from several genera, and we have found that they all share the capacity to bind to mda-5 and inhibit IFN-β induction (2). Having shown that PIV5-V is able to block oligomerization of mda-5, we then repeated the experiment with the V proteins from SeV, HeV, and MeV (Fig. 6C). All three V proteins were capable of blocking oligomerization of mda-5, which suggests that this mechanism of inhibition is conserved.
DISCUSSION
The activation of RIG-I by 5′-triphosphate RNA and SeV has been shown to be accompanied by the formation of RIG-I dimers (3, 23). Here we show that this is also true for activation of RIG-I by the synthetic dsRNA, poly(I-C), and provide the first evidence of mda-5 oligomerization in response to dsRNA. Interestingly, although both mda-5 and RIG-I can both respond to poly(I-C), oligomerization was restricted to the formation of homo-oligomeric complexes. In addition, mda-5 was activated by lower concentrations of poly(I-C) than RIG-I in in vitro assays, which suggests that mda-5 is more sensitive than RIG-I to activation by this ligand and may help to explain why under some conditions it has been reported that RIG-I plays a limited role in poly(I-C) responses. It is also possible that the length of the poly(I-C) molecules in this particular preparation makes it a better ligand for mda-5 than RIG-I or that it contains a mixture of molecules of different lengths and that the relative concentrations of those that activate mda-5 and those that activate RIG-I are different (11).
Oligomerization of mda-5 and RIG-I occurred independently of the N-terminal CARD domains but required the entire helicase domain and the CTD and was also dependent on dsRNA binding. Previous studies have shown that the CTD of RIG-I contains the ligand binding site, but while it is sufficient for binding to 5′-triphosphate RNA and short dsRNAs, poly(I-C) binding involves both the CTD and the helicase domain (23, 29). Therefore, the requirement for both domains for oligomerization may reflect the need for both domains to bind dsRNA. Consistent with this, we observed that excess dsRNA could function as a competitive inhibitor of oligomerization in in vitro assays (Fig. 4D). Interestingly, self-association of the helicase domains in the yeast two-hybrid assay could be suppressed by expression of the dsRNA-binding domain of PKR, but not by a mutant which is defective in dsRNA binding. This implies that yeast contain some sort of dsRNA which can act as a ligand for both mda-5 and RIG-I. There are many ssRNA and dsRNA viruses that infect yeast, and in fact, most strains of S. cerevisiae are known to contain at least one type of dsRNA virus from the L-A and L-BC families (reviewed in references 25 and 31). These viruses may therefore provide a source of dsRNA that can facilitate oligomerization of mda-5 and RIG-I. It is important to note that we failed to see an interaction between the mda-5 helicase domain and the RIG-I helicase domain in yeast, indicating that the interactions we observe are not a result of two proteins being indirectly tethered via a dsRNA intermediate.
We generated an extensive panel of deletion constructs in order to map the V binding site on mda-5, and surprisingly we found that PIV5-V could bind to several nonoverlapping fragments of the helicase domain. Analysis of the amino acid sequences of these fragments did not reveal any regions of internal homology which might account for this, and although there are conserved motifs, all three sections showed relatively low homology (ranging from 34 to 45%) with the equivalent regions in RIG-I. Interestingly, the only site that was conserved in its ability to bind the V proteins of several diverse paramyxoviruses was mda-5(676-816), and indeed this fragment gave the strongest interaction with PIV5-V. It therefore seems likely that this region contains the main, conserved binding site for the paramyxovirus V protein and that different V proteins might make further contacts in a virus-specific manner. We were unable to demonstrate V binding to more than one fragment of mda-5 simultaneously or show that mda-5 could interact with two molecules of V at the same time. It will be of interest to obtain the crystal structure of mda-5 bound to V in order to determine the stoichiometry of the complex and to further define the residues involved in the interaction.
In this report, we have established that paramyxovirus V proteins inhibit mda-5 activation by binding to the helicase domain and preventing it from binding dsRNA, thus inhibiting oligomerization. Viruses from diverse genera within the Paramyxovirinae share what appears to be a universal mechanism, which is contrary to the many different ways in which paramyxoviruses have been shown to block IFN signaling (reviewed in reference 21). Interestingly, the V protein specifically targets mda-5 and not RIG-I. None of the V proteins we have looked at can either bind RIG-I or inhibit IFN induction by RIG-I overexpression, and consistent with this, we also show that PIV5-V has no effect on poly(I-C) binding or oligomerization of RIG-I. It is possible that paramyxovirus infections fail to generate RNAs that are efficient RIG-I inducers or that RIG-I is not generally present in the types of cells that they infect, and as a consequence, they may have no real need to block RIG-I activation. However, several studies have indicated a potential role for RIG-I in IFN induction in response to SeV and Newcastle disease virus (12, 27, 28, 32, 33), implying that paramyxoviruses can induce through RIG-I, at least under some circumstances. This raises the possibility that paramyxoviruses have other mechanisms to block signaling responses through RIG-I. In this context, it has recently been reported that SeV-C can block RIG-I (28) and that some paramyxovirus V proteins, including PIV5-V, can block IRF-3 phosphorylation by interacting with IKKɛ/TBK1 and acting as alternative substrates (16). The latter phenomenon is presumably cell type specific, since we have failed to observe the expected inhibition of RIG-I by PIV5-V (2), and furthermore PIV5-V does not block induction by overexpression of the CARD domains of mda-5 or IPS-1 (Fig. 3). Consistent with this, we have observed that PIV5-V has an inhibitory effect on TBK1 in 293 cells but not in Vero cells (unpublished data). Paramyxoviruses thus appear to interact with the host IFN response on multiple levels, and this may reflect the need to target different pathways depending on the cell type and stage of the replicative cycle.
Acknowledgments
This work was supported by The Wellcome Trust.
We thank Craig Ross for comments on the manuscript.
Footnotes
Published ahead of print on 19 November 2008.
REFERENCES
- 1.Andrejeva, J., K. S. Childs, D. F. Young, T. S. Carlos, N. Stock, S. Goodbourn, and R. E. Randall. 2004. The V proteins of paramyxoviruses bind the IFN-inducible RNA helicase, mda-5, and inhibit its activation of the IFN-beta promoter. Proc. Natl. Acad. Sci. USA 10117264-17269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Childs, K., N. Stock, C. Ross, J. Andrejeva, L. Hilton, M. Skinner, R. Randall, and S. Goodbourn. 2007. mda-5, but not RIG-I, is a common target for paramyxovirus V proteins. Virology 359190-200. [DOI] [PubMed] [Google Scholar]
- 3.Cui, S., K. Eisenacher, A. Kirchhofer, K. Brzozka, A. Lammens, K. Lammens, T. Fujita, K. K. Conzelmann, A. Krug, and K. P. Hopfner. 2008. The C-terminal regulatory domain is the RNA 5′-triphosphate sensor of RIG-I. Mol. Cell 29169-179. [DOI] [PubMed] [Google Scholar]
- 4.Didcock, L., D. F. Young, S. Goodbourn, and R. E. Randall. 1999. The V protein of simian virus 5 inhibits interferon signalling by targeting STAT1 for proteasome-mediated degradation. J. Virol. 739928-9933. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Feng, G. S., K. Chong, A. Kumar, and B. R. Williams. 1992. Identification of double-stranded RNA-binding domains in the interferon-induced double-stranded RNA-activated p68 kinase. Proc. Natl. Acad. Sci. USA 895447-5451. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Foy, E., K. Li, R. Sumpter, Jr., Y. M. Loo, C. L. Johnson, C. Wang, P. M. Fish, M. Yoneyama, T. Fujita, S. M. Lemon, and M. Gale, Jr. 2005. Control of antiviral defenses through hepatitis C virus disruption of retinoic acid-inducible gene-I signaling. Proc. Natl. Acad. Sci. USA 1022986-2991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Gitlin, L., W. Barchet, S. Gilfillan, M. Cella, B. Beutler, R. A. Flavell, M. S. Diamond, and M. Colonna. 2006. Essential role of mda-5 in type I IFN responses to polyriboinosinic:polyribocytidylic acid and encephalomyocarditis picornavirus. Proc. Natl. Acad. Sci. USA 1038459-8464. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Habjan, M., I. Andersson, J. Klingstrom, M. Schumann, A. Martin, P. Zimmermann, V. Wagner, A. Pichlmair, U. Schneider, E. Muhlberger, A. Mirazimi, and F. Weber. 2008. Processing of genome 5′ termini as a strategy of negative-strand RNA viruses to avoid RIG-I-dependent interferon induction. PLoS ONE 3e2032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.He, B., R. G. Paterson, N. Stock, J. E. Durbin, R. K. Durbin, S. Goodbourn, R. E. Randall, and R. A. Lamb. 2002. Recovery of paramyxovirus simian virus 5 with a V protein lacking the conserved cysteine-rich domain: the multifunctional V protein blocks both interferon-beta induction and interferon signaling. Virology 30315-32. [DOI] [PubMed] [Google Scholar]
- 10.Hornung, V., J. Ellegast, S. Kim, K. Brzozka, A. Jung, H. Kato, H. Poeck, S. Akira, K. K. Conzelmann, M. Schlee, S. Endres, and G. Hartmann. 2006. 5′-Triphosphate RNA is the ligand for RIG-I. Science 314994-997. [DOI] [PubMed] [Google Scholar]
- 11.Kato, H., O. Takeuchi, E. Mikamo-Satoh, R. Hirai, T. Kawai, K. Matsushita, A. Hiiragi, T. S. Dermody, T. Fujita, and S. Akira. 2008. Length-dependent recognition of double-stranded ribonucleic acids by retinoic acid-inducible gene-I and melanoma differentiation-associated gene 5. J. Exp. Med. 2051601-1610. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Kato, H., O. Takeuchi, S. Sato, M. Yoneyama, M. Yamamoto, K. Matsui, S. Uematsu, A. Jung, T. Kawai, K. J. Ishii, O. Yamaguchi, K. Otsu, T. Tsujimura, C. S. Koh, C. Reis e Sousa, Y. Matsuura, T. Fujita, and S. Akira. 2006. Differential roles of MDA5 and RIG-I helicases in the recognition of RNA viruses. Nature 441101-105. [DOI] [PubMed] [Google Scholar]
- 13.King, P., and S. Goodbourn. 1994. The beta-interferon promoter responds to priming through multiple independent regulatory elements. J. Biol. Chem. 26930609-30615. [PubMed] [Google Scholar]
- 14.Lamb, R. A., and G. D. Park. 2007. Paramyxoviridae: the viruses and their replication, p. 1449-1496. In D. M. Knipe, P. M. Howley, D. E. Griffin, R. A. Lamb, M. A. Martin, B. Roizman, and S. E. Straus (ed.), Fields virology, 7th ed. Lippincott Williams and Wilkins, Philadelphia, PA.
- 15.Lennon, G., C. Auffray, M. Polymeropoulos, and M. B. Soares. 1996. The I.M.A.G.E. Consortium: an integrated molecular analysis of genomes and their expression. Genomics 33151-152. [DOI] [PubMed] [Google Scholar]
- 16.Lu, L. L., M. Puri, C. M. Horvath, and G. C. Sen. 2008. Select paramyxoviral V proteins inhibit IRF3 activation by acting as alternative substrates for inhibitor of kappaB kinase epsilon (IKKe)/TBK1. J. Biol. Chem. 28314269-14276. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Marais, R., Y. Light, H. F. Paterson, and C. J. Marshall. 1995. Ras recruits Raf-1 to the plasma membrane for activation by tyrosine phosphorylation. EMBO J. 143136-3145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Masson, N., M. Ellis, S. Goodbourn, and K. A. W. Lee. 1992. Cyclic AMP response element-binding protein and the catalytic subunit of protein kinase A are present in F9 embryonal carcinoma cells but are unable to activate the somatostatin promoter. Mol. Cell. Biol. 121096-1106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Pichlmair, A., O. Schulz, C. P. Tan, T. I. Naslund, P. Liljestrom, F. Weber, and C. Reis e Sousa. 2006. RIG-I-mediated antiviral responses to single-stranded RNA bearing 5′-phosphates. Science 314997-1001. [DOI] [PubMed] [Google Scholar]
- 20.Poole, E., B. He, R. A. Lamb, R. E. Randall, and S. Goodbourn. 2002. The V proteins of simian virus 5 and other paramyxoviruses inhibit induction of interferon-beta. Virology 30333-46. [DOI] [PubMed] [Google Scholar]
- 21.Randall, R. E., and S. Goodbourn. 2008. Interferons and viruses: an interplay between induction, signalling, antiviral responses and virus countermeasures. J. Gen. Virol. 891-47. [DOI] [PubMed] [Google Scholar]
- 22.Randall, R. E., D. F. Young, K. K. Goswami, and W. C. Russell. 1987. Isolation and characterization of monoclonal antibodies to simian virus 5 and their use in revealing antigenic differences between human, canine and simian isolates. J. Gen. Virol. 682769-2780. [DOI] [PubMed] [Google Scholar]
- 23.Saito, T., R. Hirai, Y. M. Loo, D. Owen, C. L. Johnson, S. C. Sinha, S. Akira, T. Fujita, and M. Gale, Jr. 2007. Regulation of innate antiviral defenses through a shared repressor domain in RIG-I and LGP2. Proc. Natl. Acad. Sci. USA 104582-587. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Saito, T., D. M. Owen, F. Jiang, J. Marcotrigiano, and M. Gale, Jr. 2008. Innate immunity induced by composition-dependent RIG-I recognition of hepatitis C virus RNA. Nature 454523-527. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Schmitt, M. J., and F. Breinig. 2006. Yeast viral killer toxins: lethality and self-protection. Nat. Rev. Microbiol. 4212-221. [DOI] [PubMed] [Google Scholar]
- 26.Southern, J. A., D. F. Young, F. Heaney, W. K. Baumgartner, and R. E. Randall. 1991. Identification of an epitope on the P and V proteins of simian virus 5 that distinguishes between two isolates with different biological characteristics. J. Gen. Virol. 721551-1557. [DOI] [PubMed] [Google Scholar]
- 27.Strahle, L., D. Garcin, and D. Kolakofsky. 2006. Sendai virus defective-interfering genomes and the activation of interferon-beta. Virology 351101-111. [DOI] [PubMed] [Google Scholar]
- 28.Strähle, L., J.-B. Marq, A. Brini, S. Hausmann, D. Kolakofsky, and D. Garcin. 2007. Activation of the beta interferon promoter by unnatural Sendai virus infection requires RIG-I and is inhibited by viral C proteins. J. Virol. 8112227-12237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Takahasi, K., M. Yoneyama, T. Nishihori, R. Hirai, H. Kumeta, R. Narita, M. Gale, Jr., F. Inagaki, and T. Fujita. 2008. Nonself RNA-sensing mechanism of RIG-I helicase and activation of antiviral immune responses. Mol. Cell 29428-440. [DOI] [PubMed] [Google Scholar]
- 30.Takeuchi, O., and S. Akira. 2008. MDA5/RIG-I and virus recognition. Curr. Opin. Immunol. 2017-22. [DOI] [PubMed] [Google Scholar]
- 31.Wickner, R. B. 1996. Double-stranded RNA viruses of Saccharomyces cerevisiae. Microbiol. Rev. 60250-265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Yoneyama, M., M. Kikuchi, K. Matsumoto, T. Imaizumi, M. Miyagishi, K. Taira, E. Foy, Y. M. Loo, M. Gale, Jr., S. Akira, S. Yonehara, A. Kato, and T. Fujita. 2005. Shared and unique functions of the DExD/H-box helicases RIG-I, MDA5, and LGP2 in antiviral innate immunity. J. Immunol. 1752851-2858. [DOI] [PubMed] [Google Scholar]
- 33.Yoneyama, M., M. Kikuchi, T. Natsukawa, N. Shinobu, T. Imaizumi, M. Miyagishi, K. Taira, S. Akira, and T. Fujita. 2004. The RNA helicase RIG-I has an essential function in double-stranded RNA-induced innate antiviral responses. Nat. Immunol. 5730-737. [DOI] [PubMed] [Google Scholar]