

Abstract
To enter target cells, human immunodeficiency virus (HIV) first attaches to the cells and fuses with the cell membrane. Attachment and fusion involve envelope glycoprotein trimers on the surface of the virion and the CD4 receptor and chemokine coreceptors on the surface of the target cell. The stoichiometry of entry, that is, the number of bonds between such trimers and CD4 that are required for infection, is unknown. Pseudotyped virions that express mixed trimers consisting of functional and nonfunctional envelope proteins have been used to study how many trimer-receptor interactions are required for virus entry. However, to extract information on the stoichiometry of entry from data generated in in vitro infectivity assays with such viruses, mathematical models are required. Here, we describe mathematical models that can be used to infer the stoichiometry of entry. By fitting our simplest model to previously published data (X. Yang, S. Kurteva, X. Ren, S. Lee, and J. Sodroski, J. Virol. 79: 12132-12147, 2005), we estimated that the number of trimer-receptor interactions required for HIV to infect a target cell is approximately eight, which is higher than previous estimates. We also consider model extensions that explain some systematic deviations of the data from the prediction of the simplest model. However, these extended models yield very different estimates of the stoichiometry of entry ranging from 2 to 19. These results strongly suggest that, based on our present knowledge of HIV entry, the stoichiometry of this process cannot be reliably estimated. Our study identifies parameters that need to be defined to render the estimation of the stoichiometry of HIV entry possible.
Human immunodeficiency virus (HIV) encodes envelope proteins gp120 and gp41, which are expressed on the virion surface as trimers (16). These trimers are involved in virus attachment to and entry into host cells and are targets for neutralizing antibodies and antiretroviral drugs. It has been shown that antibody binding to the envelope protein trimer of HIV is both necessary and sufficient for virus neutralization (10, 13, 20).
Molecular techniques allow the production of pseudotyped viruses with mixed envelope protein (Env) trimers expressed on their surface. By using such mixed-Env viruses, it is possible to study the molecular requirements of virus entry and neutralization quantitatively (14, 17, 18, 19). In particular, it can be investigated how many trimer-receptor interactions are needed for the virus to infect target cells—commonly referred to as the stoichiometry of (virus) entry—on which we focus in this paper. An estimate of the stoichiometry of entry informs us about the molecular characteristics of infectious virions and is the basis for the estimation of additional stoichiometric parameters involved in antibody neutralization. In the context of antibody neutralization, one can ask how many antibodies are needed to neutralize a single trimer—referred to as the stoichiometry of (trimer) neutralization—and how many trimers have to be neutralized to render the entire virion incapable of infection—referred to as the stoichiometry of virion neutralization. The study of these quantitative molecular aspects is important for a rational design of drug treatment and vaccines.
Inferring the stoichiometry of entry, however, requires mathematical models because infectivity assays performed with mixed-Env pseudotyped viruses do not allow direct measurement of the stoichiometry of entry. Rather, the measured infectivity of a virus stock is the result of all of the combinatorial complexity that is involved in the generation of mixed-Env pseudotyped viruses. In this article, we describe mathematical models that can be used to infer the stoichiometry of entry from data generated in in vitro infection experiments with mixed-Env pseudotyped viruses. We finally use our models to reanalyze data published previously (18). Unlike the mathematical treatment in previous studies (4, 18), our models take into consideration the variation in the number of trimers different virions express on their surface. Furthermore, we consider variation in transfection rates across cells, potential segregation of envelope proteins within the transfected cells, and potential proximity requirements of trimers. We find that the stoichiometric parameter obtained depends strongly on the model assumptions.
MATERIALS AND METHODS
Data.
The experimental data we analyzed in this study have been published previously by Yang et al. (18). In this study, pseudotyped virions expressing different types of envelope proteins on their surface were produced as follows. Cells (293T) were transfected with plasmids that encode all of the viral proteins required to form virions, replication-incompetent HIV RNA, and a reporter gene (luciferase). Translation of the plasmids coding for viral proteins and transcription of the plasmids carrying viral RNA result in the production of virions which can infect new cells but not replicate in them. The luciferase allows easy quantification of the infectivity of a given virus stock. Figure 1 is a schematic representation of the transfection system.
FIG. 1.

Generation of mixed-Env pseudotyped virions as described by Yang et al. (18). Cells are transfected with plasmids that give rise to the production of virions, which are able to infect cells but not to replicate. Arbitrary ratios of the plasmids coding for wild-type (wt) and mutant (mut) envelope proteins can be mixed and transfected into cells. The generated virions express four types of trimers: wild-type Env homotrimers, mutant Env homotrimers, and Env heterotrimers with one or two mutant envelope proteins. Infection of cells with these viruses leads to the expression of luciferase, which allows the assessment of infectivity via luminescence.
In the transfection system used by Yang et al. (18), the plasmids coding for the envelope protein of HIV are a mixture of plasmids encoding wild-type and mutant envelope proteins (Fig. 1). The envelope genes on the plasmids are first transcribed and translated, and then the resulting Env proteins are assembled into trimers and expressed on the virion surface. In previous mathematical models (4, 18), it was assumed that the wild-type and mutant envelope proteins are recruited into trimers randomly and independently, according to the frequency of the respective plasmids, giving rise to homo- and heterotrimers on the virion surface. (In the segregation model described below, we relax this assumption.)
The mutant envelope proteins that have been used to determine the stoichiometry of entry by Yang et al. (18) are glycoproteins which either cannot be cleaved into gp120 and gp41 or do not fold properly. When built into a trimer, such mutant envelope proteins prevent the conformational changes in the trimer necessary for the infection of a target cell. One mutated envelope protein is thought to be enough to render a trimer nonfunctional. (These mutants are therefore often called dominant negative.)
To study the stoichiometry of entry, infectivity assays were performed. For these assays, mixed-Env pseudotyped viruses were produced with different ratios of mutant to wild-type Env-encoding plasmids. The infectivity of these viruses was then determined and normalized by the infectivity of viruses produced with only wild-type Env-encoding plasmids. The output from such infectivity assays is the relative infectivity (RI) as a function of the fraction of mutant Env-encoding plasmids with which the cells have been transfected.
Mathematical models.
The mathematical models we developed account for the combinatorial aspects involved in the generation of pseudotyped virions and predict the RI of a virus stock. The models have two integral components. First, wild-type and mutant envelope proteins are assumed to be sampled from the pool of envelope proteins within the transfected cells and assembled into trimers. In the basic model, mutant envelope proteins are randomly recruited into a trimer with a probability equal to their frequency in the envelope protein pool and independent of what kind of envelope proteins have already been recruited into the trimer. The frequency of trimers with zero, one, two, or three mutant envelope proteins is then described by the binomial distribution. In the segregation model (see the description of the segregation model below), we relax the assumption of random sampling. Secondly, the number of trimers expressed on each virion varies. We incorporate this variation into our models. We will refer to the distribution which describes the variation in the number of trimers across virions as the distribution of trimer numbers. Note that this distribution describes only the numerical, and not the spatial, distribution of trimers on the surface of a virion. Combining the binomial assembly of wild-type and mutant envelope trimers with the variation of the number of trimers across virions, we obtain an expression for the RI of virus stocks.
Basic model.
Let T be the stoichiometry of entry, i.e., the number of trimer-receptor interactions which the virus needs to infect a cell. To estimate T, infectivity assays have been conducted to determine the RI of virus stocks with different fractions of dominant negative mutant envelope proteins (18). Let fM be the fraction of mutant envelope-encoding plasmids with which the cells are transfected. In the basic model, we assume that after transfection and expression the fraction of mutant envelope proteins in the transfected cells will be equal to fM. (This assumption will be relaxed below in the imperfect-transfection model.)
Assuming that trimers are assembled by randomly and independently sampling envelope proteins from the pool within the transfected cell, the probability that a trimer is functional is given by the equation
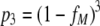 |
(1) |
Mathematically, this corresponds to the probability of three successes in three Bernoulli trials with a probability of success equal to 1 − fM.
If each virtual virion has a constant number of trimers, s, then the probability that a virion has g “good”/functional trimers is:
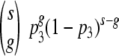 |
(2) |
The expression above is the basic binomial probability of g successes in s trials with success probability p3.
To calculate the RI of virions with exactly s trimers, we simply add the probabilities that a virion has T good trimers more than or equal to:
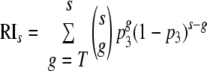 |
(3) |
However, the number of trimers on the virion surface varies (21). Let ηs, where s = 0, …, smax, be the probability that a virion has s trimers. To calculate the RI, we now have to calculate the probability that a virion with s = 0, …, smax trimers has more than T good trimers and then sum the probabilities, weighting them with ηs:
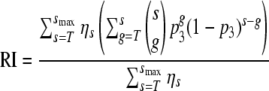 |
(4) |
The expression in the denominator,  ηs, is the fraction of wild-type virions that have more than T good trimers on their surface (which is smaller than 1 if some virions have fewer than T trimers).
ηs, is the fraction of wild-type virions that have more than T good trimers on their surface (which is smaller than 1 if some virions have fewer than T trimers).
Using equation 4, we can infer the stoichiometry of entry from the observed RI for various values of fM and knowing the distribution of trimer numbers given by ηs.
Imperfect transfection model.
In the basic model, we have assumed that the fraction of mutant envelope proteins in the envelope protein pool within each transfected cell is the same as the fraction of mutant envelope protein-encoding plasmids with which the cells have been transfected. The latter is designated fM. This assumption is justified if a high number of plasmids can enter the transfected cell and the relative expression rate of wild-type and mutant envelope-encoding plasmids is the same in each cell. If one of these conditions is violated, the fraction of mutant envelope proteins in the Env pool will vary across the transfected cells.
To incorporate this potential variation into the basic model, we describe the fraction of mutant envelope proteins in the Env pool as a B-distributed random variable with mean fM and variance v. We designate the probability density function of this random variable βfM,v. The variance, v, should be higher the closer the fraction of Env-encoding plasmids comes to 0.5 and is defined as:
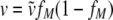 |
(5) |
where 0 < ṽ < 1 denotes the coefficient of variation. For a coefficient of variation close to 0, the fraction of mutant Env proteins in the Env pool of each transfected cell is approximately equal to the fraction of mutant Env-encoding plasmids, fM. This corresponds to the basic model. As the coefficient of variation increases, the variance of the B-distributed fraction of mutant envelope proteins in the Env pool across the transfected cells also increases. A coefficient of variation close to 1 corresponds to a scenario in which the envelope pool of a transfected cell consists of either almost exclusively mutant or almost exclusively wild-type envelope proteins. In this case, the fraction of cells that express only mutant Env is approximately equal to fM.
Because in the imperfect transfection model the fraction of mutant envelope proteins may vary across cells, the probability that a functional trimer is formed may also differ across cells. Let x be the fraction of mutant envelope proteins in a given cell. Then the probability of forming a functional trimer in this cell is (1 − x)3. To calculate the RI, we need to average the probabilities of forming functional trimers over all transfected cells. The average probability of forming functional trimers, 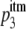 , across all transfected cells is then given as:
, across all transfected cells is then given as:
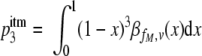 |
(6) |
To obtain the predictions for the RI, we simply replace the probability of forming a functional trimer, p3, in the RI function of the basic model (equation 4) with the probability of forming a functional trimer in the imperfect transfection model, 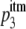 (equation 6):
(equation 6):
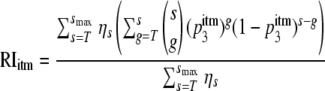 |
(7) |
By fitting this model to the data of Yang et al. (18), we estimate ṽ in addition to the stoichiometry of entry, T.
Segregation model.
Let us again assume perfect transfection as in the basic model. In the basic model, we also assume that envelope proteins are recruited perfectly randomly into trimers, i.e., that they are recruited with a probability equal to their frequency in the envelope protein pool and independent of what kind of envelope proteins have already been recruited into the trimer.
To allow for the possibility of nonrandom or nonindependent formation of trimers from wild-type and mutant envelope proteins, we extend the model by one parameter, ξ, that describes the potential spatial or temporal segregation of envelope proteins within the transfected cells. ξ ranges from 0 to 1. ξ = 0 corresponds to no segregation, i.e., a scenario in which wild-type and mutant envelope proteins are recruited into trimers as in the basic model, whereas ξ = 1 corresponds to full segregation, i.e., a scenario in which heterotrimers are not formed.
Mathematically, we can implement segregation simply by changing the definition of the probability of the formation of wild-type homotrimers, p3, from that in equation 1 to:
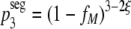 |
(8) |
For ξ = 0, 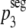 = p3. Substituting
= p3. Substituting 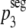 into equation 4, we obtain a prediction for the segregation model, i.e.,
into equation 4, we obtain a prediction for the segregation model, i.e.,
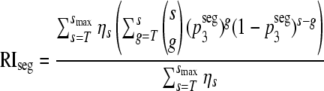 |
(9) |
By fitting this model to the data of Yang et al. (18), we estimate ξ in addition to the stoichiometry of entry, T.
Proximity model.
In the basic model, we assume that functional trimers can engage successfully with the receptor on the cell regardless of their position on the virion. It is conceivable, however, that trimers need to be sufficiently close to each other to cooperate during the infection of the cell. To incorporate this possibility, we extended our basic model by one parameter, a, which describes the critical great circle distance below which trimers can cooperate (Fig. 2).
FIG. 2.

Illustration of the proximity requirements in the proximity model. Three virions are shown with three functional trimers each. Distances between trimers greater than the critical distance a are shown as red lines, and distances smaller than a are green. (A) A virion with trimers that are farther apart from each other than critical distance a. This virion is not infectious in the proximity model. (B) A virion on which two of the three trimers are less than a apart. If the stoichiometry of entry were 2, this virion would be infectious; for T = 3, it would not be infectious. (C) A virion on which the three trimers have pairwise distances that are smaller than critical distance a. This virion is infectious for T = 2 and T = 3.
The RI for this model cannot be derived analytically. We therefore resorted to simulations. To derive the RI, we generate virtual virions in silico. The simulations are done as follows. In step 1, draw a random number, s, of trimers from the distribution specified by ηs. In step 2, generate a random number of functional trimers, g, from a binomial distribution with sample size s and probability of success p3 = (1 − fM)3. In step 3, check if g is larger than the stoichiometry of entry, T; if it is not, then score this virion as not infectious and proceed to the next virion; if it is, then (i) distribute these g trimers randomly across the virion and (ii) check if there is a cluster with a size at least equal to T in which none of the pairwise distances between functional trimers is larger than a. In step 4, repeat this for many virions (we used 104 virions).
The number of infectious virions obtained by this algorithm has to be normalized by the number of infectious virions obtained from an analogous simulation with fM = 0, i.e., wild-type virions. The normalization is required because not every wild-type virion is infectious. (There are two reasons why not every wild-type virion is infectious. First, in general there is a fraction, 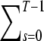 ηs, of virions that have fewer than T trimers and are therefore not infectious. Second, the proximity requirement will render an even larger fraction of wild-type virions not infectious.) This algorithm yields an estimate for the RI of our proximity model.
ηs, of virions that have fewer than T trimers and are therefore not infectious. Second, the proximity requirement will render an even larger fraction of wild-type virions not infectious.) This algorithm yields an estimate for the RI of our proximity model.
By fitting this model to the data of Yang et al. (18), we estimate the critical distance a along with T.
Soft threshold model.
As a last model extension, we relax the assumption of the basic model that there is a strict threshold number of trimers (T) below which virions are not infectious and above which virions have equal infectiousness (Fig. 3).
FIG. 3.

Comparison of the strict threshold assumption of the basic model to the soft threshold assumption. (A) The infectivity of a virion as a function of the number of functional trimers on its surface according to the strict threshold assumption. In this example, we assume that T = 8, i.e., that a virion with eight or more trimers is fully infectious, whereas a virion with fewer than eight trimers is not infectious. (B) The infectivity of a virion as a function of the number of functional trimers on its surface according to the soft threshold model. Two curves with differing Hill coefficients, h, are shown.
To do this, we introduce function γ that describes the relative infectiousness of virions as a function of the number of functional trimers they express on their surface. γ(s) = sh/(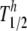 + sh) is a Hill function with parameters T1/2 and h. T1/2 is the number of trimers at which γ = 0.5 and does not have to be integer. h is the Hill coefficient, which is a measure of the sharpness of the threshold.
+ sh) is a Hill function with parameters T1/2 and h. T1/2 is the number of trimers at which γ = 0.5 and does not have to be integer. h is the Hill coefficient, which is a measure of the sharpness of the threshold.
The RI predicted from this model is given as:
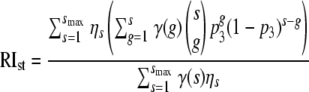 |
(10) |
By fitting this model to the data of Yang et al. (18), we estimate the parameters T1/2 and h.
The expressions in equations 4, 7, 9, and 10 were implemented as functions in the R language for statistical computing (9). Because we did not have an analytical solution for the RI in the proximity model, we approximated it by simulating virtual virions. To reduce run time, the algorithm for this was implemented in C and loaded into R as a shared library. The program is available upon request.
RESULTS
Mean and variation in the number of Env trimers influence RI.
The stoichiometry of entry, T, can be estimated from data generated in infectivity assays performed as described above. For these infectivity assays, mixed-Env pseudotyped viruses are produced with different fractions of plasmids encoding mutant envelope proteins, fM, and their RI is determined. The stoichiometry of entry can then be obtained by fitting a mathematical model to the RI measurements.
In Fig. 4A, we show how the predicted RI depends on the stoichiometry of entry, T. The higher the value of T, the sooner the RI drops as a function of fM. This is intuitive because if many trimer-receptor interactions are needed for virus entry, a virion will be able to tolerate only few mutant envelope proteins that render trimers nonfunctional. For example, if a virion has 10 trimers and requires 7 for infection, the RI drops to 0.5 for fM = 0.14, whereas it drops to 0.5 for fM = 0.62 if T = 1. These relationships among RI, fM, and T are consistent with earlier findings (4). This dependence of the predicted RI on the stoichiometry of entry forms the basis for the inference of this parameter.
FIG. 4.

The RI as a function of the fraction of mutant envelope proteins, fM, predicted by equation 4. (A) Dependence of the RI on stoichiometry parameter T. For this plot, we assumed that every virion has exactly 10 trimers. (B) Dependence of the RI on the mean and variance of the distribution of trimer numbers, ηs. For the plot, we assumed T = 1 and distributions with means of 10 (solid lines) and 36 (dashed lines) with either no variance (black lines) or high variance (red lines). The high variances derive from uniform distributed trimer numbers between 2 and 18 (which results in a mean of 10 trimers) or 0 and 72 (which results in a mean of 36 trimers).
However, the RI predicted by the mathematical model also depends on additional parameters. An important such parameter, which we consider here, is the distribution of trimer numbers, ηs. Because the distribution of trimer numbers affects the predicted RI, it will also affect the estimates of the stoichiometric parameter that one infers from data by fitting the model. Figure 4B shows how the predicted RI depends on the mean number of trimers and the variance of the distribution of trimer numbers.
The higher the mean number of trimers, the larger fM has to be for the RI to drop. For example, if we assume that there are 36 trimers on each virion and the stoichiometry of entry is 1, the predicted RI is 0.5 when fM = 0.43, whereas if we assume that each virion has 10 trimers, the RI is 0.5 for fM = 0.14.
If we keep the mean number of trimers constant and change the variance of the distribution of trimer numbers, we find that the RIs become flatter functions of fM. Experimentally determined RIs tend to drop more smoothly for increasing fM than models with a constant number of trimers on virions predict. Thus, it is important to incorporate as much information about the distribution of trimer numbers as possible into the model when inferring stoichiometric parameters. We therefore base our statistical inferences in the following on a “realistic” distribution of trimer numbers with a mean of 14 and a standard deviation of 7. These numbers were recently defined experimentally by cryoelectron microscopy (21). Figure 5 shows a histogram of this “realistic” distribution. By using this distribution for our mathematical models, we assume that the trimers identified by electron microscopy are functional.
FIG. 5.

Histogram of the “realistic” distribution of trimer numbers. We defined this distribution as a discretized B-distribution with a mean of 14 and a standard deviation of 7, as found in reference 21. This distribution is also in agreement with the range of 4 to 35 trimers found in a sample of 40 virions by Zhu et al. (21).
Is T equal to 1?
It has been suggested that the stoichiometry of entry is 1 (18), i.e., that a single trimer-receptor interaction is sufficient to mediate viral entry into a target cell. Reanalyzing the data of Yang et al. (18) by using a mathematical model in which all virions were assumed to have exactly nine trimers, Klasse (4) found that the stoichiometry of entry is 5. Using the same data set (18) and our basic model that incorporates a “realistic” distribution of trimer numbers defined above (Fig. 5), we came to different conclusions.
The analyzed data are described in detail in reference 18. In short, two mutant envelope proteins, V513E and R508S/R511S, have been used to generate pseudotyped viruses, and RIs were measured for fM = 0, 0.2, 0.4, 0.6, 0.8, and 1.0. Both mutant envelope proteins are thought to have a dominant negative effect on trimer functionality; i.e., one mutant envelope will render a trimer unable to engage in an interaction with the CD4 receptor that results in the infection of the target cell.
We fitted our model (equation 4) to the RIs for both mutant envelope proteins V513E and R508S/R511S, assuming that there is no difference in the stoichiometry of entry between the two mutants. This resulted in an estimated stoichiometry of entry (T) of 8. A bootstrap procedure resulted in estimates of T ranging from 7 to 9 in 99.3% of the cases. This estimate is consistent with empirical evidence (15). Figure 6A shows the best fit of our model, and Fig. 6B shows the residual sum of squares as a function of the stoichiometry of entry, T. If we fit our model (equation 4) by allowing for differences in the stoichiometry of entry between V513E and R508S/R511S, we obtain T = 7 and T = 9, respectively. However, this extension of the model by allowing for mutant-specific stoichiometric parameters does not improve the fit significantly (F-test on the residual sum of squares, P = 0.50) and the difference in T for the two mutants is not significant (data not shown).
FIG. 6.

Reanalysis of the data from reference 18 with our basic model (equation 4). (A) Best fit of our model, which results in an estimate of T = 8. (B) Histogram of bootstrap estimates of T. The bootstrap sample size was 1,000, and 99.3% of the estimates lie between 7 and 9. (C) Residual sum of squares for different T values. From this plot, it is clear that T = 8 is the best fit and that there are no other local minima.
Fitting the imperfect transfection model.
In the basic model, we assume that the fraction of mutant envelope proteins within each transfected cell is identical to the fraction of mutant envelope-encoding plasmids with which the cells have been transfected. However, this may not be the case because of variable transfection efficiency or expression rates across cells. Such differences in transfection and expression will result in fractions of mutant envelope proteins that vary across the transfected cells. To allow for this possibility, we developed the imperfect transfection model.
The imperfect transfection model is an extension of the basic model by parameter ṽ that measures the variation of the fraction of mutant envelope proteins in the envelope protein pool across the transfected cells. More precisely, we assume that the fraction of mutated envelope proteins in the envelope protein pool is a B-distributed random variable with mean fM and coefficient of variation ṽ. According to this model, the predictions for the RI (equation 7) are dependent on the stoichiometry of entry and the coefficient of variation, ṽ (see Materials and Methods). As in all of our models, we assume that the number of trimers on the virion surface follows the “realistic” distribution described in Fig. 5.
Fitting the imperfect transfection model to the data in reference 18 improves the fit significantly (F-test, P = 3 × 10−11). The stoichiometry parameter T is estimated to be 19, and the coefficient of variation (ṽ) is estimated to be 0.99992. Hence, according to this model, 19 trimer-receptor interactions are needed for cell entry and the envelope protein pool is enormously heterogeneous between cells, consisting mainly of either wild-type or mutant envelope proteins. A bootstrap routine with 1,000 replicates results in 95% of the bootstrap estimates lying in a region with high stoichiometry (17 ≤ T ≤ 19) and a high coefficient of variation (0.9563 ≤ ṽ ≤ 0.99995).
The better fit of the imperfect transfection model, however, should not be interpreted as proof that transfection or subsequent expression of envelope proteins is actually variable across the transfected cells. Rather, this result shows that the estimate of the stoichiometry of entry sensitively depends on the assumptions of the mathematical model on which its estimation is based. The main lesson from the fit of the imperfect transfection model is that the transfection and expression efficacy should be determined independently to obtain a better estimate of the stoichiometry of entry.
Fitting the segregation model.
In the basic model, we assume that wild-type and mutant envelope proteins are recruited into trimers perfectly randomly, i.e., with a probability equal to their frequency in the envelope protein pool and independent of what kind of envelope proteins have already been recruited into the trimer. However, the expression of envelope proteins may be spatially or temporally segregated. We developed a model that allows for the potential segregation of envelope proteins (equation 9). In addition to the stoichiometry of entry, this model features a parameter, ξ, describing the segregation of envelope proteins within the transfected cell. ξ ranges from 0 to 1, corresponding to no segregation and full segregation, respectively.
Including potential segregation in the model improves the fit to the data reported by Yang et al. (18) significantly (F-test, P = 4 × 10−11). The fit of the segregation model yields an estimate of the stoichiometry of entry of 19 and a segregation coefficient (ξ) of 1.0, i.e., full segregation. A bootstrap routine with 1,000 replicates results in 98% of the bootstrap estimates lying in a region characterized by a high stoichiometry of entry (18 ≤ T ≤ 20) and high segregation (ξ > 0.98).
By the same logic that we applied in the case of the imperfect transfection model, the better fit of the segregation model should not be interpreted as definitive evidence in favor of a high segregation coefficient. This result rather suggests that for reliable estimation of stoichiometry, the segregation coefficient should be determined independently. Figure 7 shows the fraction of virions that carry heterotrimers as a function of the fraction of mutant envelopes, fM, and the segregation coefficient, ξ. For intermediate values of fM, our basic model (ξ = 0) predicts that almost all virions carry heterotrimers, whereas the best fit of the segregation model predicts that almost no virion should carry heterotrimers. An analysis such as the one presented in Fig. 7 could be used to estimate the segregation coefficient independently from data on the fraction of virions carrying heterotrimers.
FIG. 7.

Fraction of virions with heterotrimers according to the segregation model (seg) for different segregation parameters, ξ.
Fitting the proximity model.
Does the estimate of the stoichiometry of entry we obtain from the basic model (T = 8) mean that eight trimer-receptor interactions are required for HIV entry? No, not necessarily. If trimers have to be close to each other in order to mediate entry, fewer than eight trimers may have to engage with the CD4 receptor.
We have developed a mathematical model (the proximity model) that predicts the RI for the case in which T trimers which are less than a critical distance, a, apart are necessary for infection (see Materials and Methods). A comparison of the proximity model and the basic model is shown in Fig. 8.
FIG. 8.

Comparison of the proximity model (pc) with the basic model (bm, without proximity correction). Predictions of the proximity model are based on simulating 10,000 virtual virions.
Fitting the proximity model to the data by Yang improves the fit of the basic model significantly (F-test, P = 5 × 10−10). The fit of the proximity model yields estimates of the stoichiometry of entry of T = 2 and a = 12 nm (assuming a virion diameter of 100 nm). A bootstrap routine with 700 replicates results in 93% of the bootstrap estimates being in the ranges 2 < T < 5 and 10 nm < a < 100 nm.
By cryoelectron microscopy Sougrat et al. (15) recently succeeded in visualizing virions in the act of infecting CD4+ T cells. The authors established from this analysis that virions form an “entry claw” with a diameter of approximately 40 nm with five to seven trimers between the virion and the cell. Although the estimates of the critical distance, a, which we obtain by fitting our proximity model is quite consistent with the observations in reference 15, we have to be cautious not to overinterpret this consistency. In the Discussion, we elaborate on the commonalities and discrepancies between the observations of Sougrat et al. (15) and our proximity model.
Fitting the soft threshold model.
In our models so far, we assumed that there is a strict threshold in the number of functional trimers above which a virion is infectious. It is conceivable that the infectivity of a virion increases with the number of functional trimers on its surface. To allow for this possibility, we have developed a soft threshold model (equation 10). This model has two parameters: T1/2, describing the trimer number at which the RI reaches 50%, and h, describing the softness of the threshold (the Hill coefficient). We limited the possible values of T1/2 to 0 to 100 because our “realistic” distribution is cut off at 100 trimers too; i.e., according to the realistic trimer distribution, there are no virions with more than 100 trimers.
Including the possibility of a soft threshold again improves the fit to the data of Yang et al. (18) significantly (F-test, P = 5 × 10−15). Fitting the soft threshold model, we estimate T1/2 = 100 and h = 1.26. A bootstrap routine with 1,000 bootstrap replicates yields 95% confidence intervals for T1/2 ranging from 22 to 100 (hereby we set the maximum allowed estimate of T1/2 at 100) and for h ranging from 1.14 to 1.67. The low confidence in the estimate of T1/2 is due to the fact that—according to the “realistic” distribution we assume in our analysis—virions with high numbers of trimers are rare. This limits the accuracy with which T1/2 can be estimated. Figure 9C shows the RIs of virions with various numbers of functional trimers on their surface according to the best fit of the soft threshold model. The fit results in an almost linear increase in RIs with the number of trimers, rather than in a soft threshold. Thus, the assumptions of a strict threshold in the models above may not even hold approximately. However, what we show here is only that infectivities which increase linearly with the number of trimers explain some of the deviation from the fit of our basic model (as did the assumption of segregation and proximity requirements). We cannot conclude from this analysis that there is no threshold (see Discussion).
FIG. 9.

Fits of the four extended models to the RI data. Shown are the fits obtained with the imperfect transfection model (A), the segregation model (B), the proximity model (C), and the soft threshold model (D). All four extended models improve the fit of the basic model significantly.
DISCUSSION
In this study, we developed a framework for the estimation of the stoichiometry of HIV entry. Our basic model incorporates the variation in the number of trimers virions express on their surface and yields an estimate of the stoichiometry of entry of 8. However, as we show by investigating four extensions of the basic model, this estimate is not robust. If trimers need to be close to each other in order to cooperate in the infection process, the estimate of the stoichiometry of entry may be as low as 2. If the transfection efficiency varies across cells or envelope proteins segregate within the transfected cell, giving rise to virions that express only wild-type or mutant Env homotrimers, the stoichiometry of entry is estimated to be 19. Our analysis illustrates the need for further experimental studies that shed light on the factors that influence the estimate of the stoichiometry of entry. The data generated so far do not allow reliable estimation of the stoichiometry of entry.
Our basic model goes beyond the mathematical model of Yang et al. (18) by accounting for the fact that virions have multiple trimers on their surface. In the model of Yang et al., RI = (1 − fM)3T, the predicted RI of a virus stock depends only on the fraction of mutant envelope proteins, fM, and the stoichiometry of entry, T. It is independent of the number of trimers on the virions.
In a recent study, Klasse (4) reanalyzed the data of Yang et al. (18). Klasse's model assumes a constant number (n = 9) of trimers on each virion. He estimates a stoichiometry of entry of 5. The difference between Klasse's estimate of the stoichiometry of entry and the one we derived with our basic model is due to different assumptions about the distribution of trimer numbers. Klasse assumes that each virion has 9 trimers (i.e., a singular distribution), whereas we assume a mean number of trimers of 14 with a standard deviation of 7, in agreement with the latest estimates by Zhu et al. (21).
In addition to the different estimates of the stoichiometry of entry we obtained, there are two other consequences of taking into account the variation in the number of trimers across virions. First, unlike in models that assume that virions have a constant number of trimers, there is no fixed number of trimers which have to be neutralized to neutralize a virion. For example, in reference 4, every virion has nine trimers and needs five to be infectious. Therefore, neutralizing five trimers renders the virion noninfectious. In our basic model, virions are estimated to require eight trimers to be infectious. However, how many trimers have to be neutralized to render a given virion noninfectious depends on the total number of trimers on the virion in question. Second, whereas in reference 4 every virion is infectious, in our models not every virion is predicted to be infectious. The fraction of infectious virions depends on the distribution of trimer numbers. According to our basic model with the “realistic” distribution, 18% of the virions have fewer than eight trimers and are therefore not infectious. The remaining 82% can potentially infect, at least as judged by the number of trimers they express on their surface. In the other extended models, the fraction of potentially infectious virions is of the same order of magnitude. This fraction of infectious virions is still far larger than the fraction of virions that is observed to infect cells in culture. Estimates of this fraction range from 1 in 100,000 (8) to 1 in 5,000 (11). However, the high fraction of potentially infectious virions predicted by our basic model should not be misinterpreted as an inconsistency with observations. It is very likely that there are many other factors—factors not related to the number of trimers on virions—which may influence the infection success of a virion.
The dependence of the estimate of the stoichiometry of entry on the distribution of trimer numbers is particularly relevant for attempts to study stoichiometric parameters by using hybrid viruses (6, 7, 18). For example, the stoichiometry of entry of influenza virus has been studied by generating HIV virions which display trimers of the influenza virus hemagglutinin protein (18). Such approaches may lead to false estimates of the stoichiometry of entry if they are not based on the appropriate distribution of trimer numbers. The appropriate distribution to use in the above example is the one describing the hemagglutinin trimers on the hybrid pseudotyped virions. (Yang et al. [18] did not factor the distribution of trimer numbers into their estimation of the stoichiometry of entry and therefore did not encounter this problem.) By the same logic, even for the nonhybrid HIV system, one should base the estimation of the stoichiometry of entry on the distribution of trimer numbers found on the pseudotyped virions that are used in the infectivity assays rather than on the distribution of clinical isolates. Since HIV pseudotyped virions have been suggested to have fewer trimers, on average, than normal virions (5), a definitive estimate of the stoichiometry of entry should be based on the distribution of trimer numbers of pseudotyped virions. However, the estimates of the stoichiometry of entry we present here will be unaffected if pseudotyped virions simply have a larger fraction of virions lacking any trimers.
We extended our basic model in four different ways. In the first extension, we allowed for imperfect transfection of cells by wild-type and mutant Env-encoding plasmids. In the second extension, we incorporated the possibility that envelope proteins segregate within transfected cells. In the third extension, we considered the possibility that trimers might need to be sufficiently close to each other on the virion to cooperate during the infection of a cell. In the last extension, we relaxed the assumption that there is a strict threshold number of trimers above which a virion is infectious but assigned an RI to virions with any number of trimers. All of our model extensions resulted in a significantly better fit to the data. This does not imply, however, that any of the mechanisms we incorporated into the extended models reflect the biological reality. Rather, the factors we consider—such as transfection efficiency, segregation, or proximity requirements—should be quantified independently to allow better estimation of the stoichiometry of entry. Moreover, additional mechanisms which we did not consider here may also lead to an improvement of the fit to the data.
The imperfect transfection model allows for variation of the fraction of mutant envelope proteins in the Env pool across the transfected cells. When this model was fitted to the data of Yang et al. (18), the stoichiometry of entry was estimated to be 19. The coefficient of variation is almost 1, suggesting that there are only two different types of transfected cells: one expressing almost exclusively wild-type envelope proteins and another expressing almost exclusively mutant envelope proteins. If this were indeed the case, the generated pseudotyped virions would carry mostly homotrimers. Whether this model extension is biologically relevant should be tested by studying the envelope pool in more detail. This could be done by linking the wild-type and mutant Env-encoding regions on the plasmids to different luminescent markers to test if cells coexpress both wild-type and mutant envelope proteins. Furthermore, one could quantify mutant and wild-type envelope-encoding plasmids and proteins on a single-cell level to obtain estimates of the variation in transfection and expression. These estimates can then be used to obtain a better estimate of the stoichiometry of entry.
Fitting the segregation model, we estimated the stoichiometry of entry to be 19, as in the imperfect transfection model. Further, we estimated a segregation coefficient of almost 1, which corresponds to practically full segregation. Although the model with almost full segregation fits the data of Yang et al. (18) significantly better than our basic model, full segregation is unlikely since influenza virus hemagglutinin trimers are found to be formed almost perfectly at random (1), and for HIV, some studies have documented the formation of heterotrimers (2, 3, 12, 14). By measuring the fraction of virions that have heterotrimers on their surface, it would be possible to estimate the segregation coefficient independently (Fig. 7). This estimate of the segregation coefficient can then be used to refine estimates of the stoichiometry of entry.
Extending our basic model by potential proximity requirements also significantly increases the quality of the fit to the data of Yang et al. (18). We estimate that the critical distance between trimers ranges from 10 to 100 nm and the stoichiometry of entry ranges from 2 to 5. On the one hand, the estimates of the critical distance are quite consistent with the entry claw size of 40 nm that Sougrat et al. (15) observed. On the other hand, they observed five to seven rods of high density between the virion and the cell, which is higher than our estimate of the stoichiometry of entry. Sougrat et al. (15), however, raise the possibility that first five to seven trimer-receptor interactions form a scaffold (the entry claw) but that fusion of the virion and the cell then requires a lower number of trimers. Moreover, the virions Sougrat et al. (15) observed mostly did not have any trimers outside of the entry claw; whether they could not be detected or whether trimers are either shed or recruited into the contact region between the virion and the cell remains to be determined. If trimers were indeed recruited into the contact region, an essential assumption of the proximity model would be violated, i.e., that trimer positions are fixed on the virion surface. More information on the spatial-temporal dynamics of trimers on the virion surface is needed in this context.
The last model we considered was the soft threshold model. We initially conceived this model to investigate if a softer threshold may be able to explain some of the residual deviation between the prediction of our basic model and the data. The resulting estimates of the parameters of the soft threshold model, however, are in contradiction to a threshold assumption. Rather, the RI of virions is estimated to increase approximately linearly with the number of functional trimers on them.
We find that the estimate of the stoichiometry of entry depends sensitively on the assumptions about the generation of pseudotyped virions. Our estimates range from 2 to 19. We identify key parameters which should be determined independently to allow a better estimation of the stoichiometry of entry. We suggest the following experiments (in order of their importance for the estimation of the stoichiometry of entry). (i) In the first and most important line of experiments, the distribution of trimer numbers (our parameters ηs) has to be determined. This distribution is a crucial input parameter for all models and can be studied by electron tomography (15, 21). The number of trimers on a sample of a few hundred virions should be determined. It would be advisable to use the same pseudotyped virions for this as one is going to use for the generation of virions with mixed trimers. (ii) A second line of experiments should clarify if the fraction of mutant envelope proteins in the Env pool of transfected cells varies or if it is equal to the fraction of mutant Env-encoding plasmids with which the cells have been transfected. (iii) In a third line of experiments, the fraction of heterotrimers on virions should be quantified. From this experiment, the segregation parameter can be estimated. (iv) A last line of experiments should determine whether the trimers have fixed positions on the virion surface or whether they can change their positions. If they can change their positions, the proximity model would be proven invalid because it assumes that the trimers have fixed positions on the virion surface.
The stoichiometry of HIV entry is essential for our understanding of which virions are infectious. Only once we know the molecular characteristics of infectious virions can we proceed to investigate how to best target these virions with vaccines or drugs. The next step in a quantitative understanding of HIV entry will be to find out how many antibodies or drug molecules are required to prevent HIV entry into cells. However, before we can proceed to investigate the quantitative aspects of antibody neutralization or drug action, a reliable estimate of the stoichiometry of entry is needed. In this article, we outlined which parameters have to be determined to allow a reliable estimation of the stoichiometry of entry, i.e., the variation in the number of trimers between virions, the variation in transfection efficiency, the segregation coefficient of envelope proteins within transfected cells, and the spatial-temporal dynamics of trimers on the virion surface.
Acknowledgments
We gratefully acknowledge Xinzhen Yang for generously sharing his data and Martin Ackermann, Rustom Antia, Simon Frost, Frederik Graw, Trevor Hinkley, Michael Huber, and Andrew Yates for helpful discussions. We are also grateful to Robert Doms for constructive criticism of our manuscript.
Support was provided by the Swiss National Science Foundation (315200-114148 to R.R.R. and 310000-120739 to A.T.). A.T. is an Elizabeth Glaser Scientist supported by the Elizabeth Glaser Pediatric AIDS Foundation.
Footnotes
Published ahead of print on 19 November 2008.
REFERENCES
- 1.Boulay, F., R. W. Doms, R. G. Webster, and A. Helenius. 1988. Posttranslational oligomerization and cooperative acid activation of mixed influenza hemagglutinin trimers. J. Cell Biol. 106629-639. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Doms, R. W., P. L. Earl, S. Chakrabarti, and B. Moss. 1990. Human immunodeficiency virus type 1 and 2 and simian immunodeficiency virus env proteins possess a functionally conserved assembly domain. J. Virol. 643537-3540. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Herrera, C., P. J. Klasse, C. W. Kibler, E. Michael, J. P. Moore, and S. Beddows. 2006. Dominant-negative effect of hetero-oligomerization on the function of the human immunodeficiency virus type 1 envelope glycoprotein complex. Virology 351121-132. [DOI] [PubMed] [Google Scholar]
- 4.Klasse, P. J. 2007. Modeling how many envelope glycoprotein trimers per virion participate in human immunodeficiency virus infectivity and its neutralization by antibody. Virology 369245-262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Louder, M. K. K., A. Sambor, E. Chertova, T. Hunte, S. Barrett, F. Ojong, E. Sanders-Buell, S. Zolla-Pazner, F. E. E. McCutchan, J. D. D. Roser, D. Gabuzda, J. D. D. Lifson, and J. R. R. Mascola. 2005. HIV-1 envelope pseudotyped viral vectors and infectious molecular clones expressing the same envelope glycoprotein have a similar neutralization phenotype, but culture in peripheral blood mononuclear cells is associated with decreased neutralization sensitivity. Virology 339226-238. [DOI] [PubMed] [Google Scholar]
- 6.Marzi, A., A. Wegele, and S. Pohlmann. 2006. Modulation of virion incorporation of ebolavirus glycoprotein: effects on attachment, cellular entry and neutralization. Virology 352345-356. [DOI] [PubMed] [Google Scholar]
- 7.Ou, W., and J. Silver. 2006. Stoichiometry of murine leukemia virus envelope protein-mediated fusion and its neutralization. J. Virol. 8011982-11990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Piatak, M., Jr., M. S. Saag, L. C. Yang, S. J. Clark, J. C. Kappes, K. C. Luk, B. H. Hahn, G. M. Shaw, and J. D. Lifson. 1993. High levels of HIV-1 in plasma during all stages of infection determined by competitive PCR. Science 2591749-1754. [DOI] [PubMed] [Google Scholar]
- 9.R Development Core Team. 2008. R: a language and environment for statistical computing. R Foundation for Statistical Computing, Vienna, Austria.
- 10.Ren, X., J. Sodroski, and X. Yang. 2005. An unrelated monoclonal antibody neutralizes human immunodeficiency virus type 1 by binding to an artificial epitope engineered in a functionally neutral region of the viral envelope glycoproteins. J. Virol. 795616-5624. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Rusert, P., M. Fischer, B. Joos, C. Leemann, H. Kuster, M. Flepp, S. Bonhoeffer, H. F. Gunthard, and A. Trkola. 2004. Quantification of infectious HIV-1 plasma viral load using a boosted in vitro infection protocol. Virology 326113-129. [DOI] [PubMed] [Google Scholar]
- 12.Salzwedel, K., and E. A. Berger. 2000. Cooperative subunit interactions within the oligomeric envelope glycoprotein of HIV-1: functional complementation of specific defects in gp120 and gp41. Proc. Natl. Acad. Sci. USA 9712794-12799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Sattentau, Q. J., and J. P. Moore. 1995. Human immunodeficiency virus type 1 neutralization is determined by epitope exposure on the gp120 oligomer. J. Exp. Med. 182185-196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Schønning, K., O. Lund, O. S. Lund, and J.-E. S. Hansen. 1999. Stoichiometry of monoclonal antibody neutralization of T-cell line-adapted human immunodeficiency virus type 1. J. Virol. 738364-8370. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Sougrat, R., A. Bartesaghi, J. D. Lifson, A. E. Bennett, J. W. Bess, D. J. Zabransky, and S. Subramaniam. 2007. Electron tomography of the contact between T cells and SIV/HIV-1: implications for viral entry. PLoS Pathog. 3571-581. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Wyatt, R., and J. Sodroski. 1998. The HIV-1 envelope glycoproteins: fusogens, antigens, and immunogens. Science 2801884-1888. [DOI] [PubMed] [Google Scholar]
- 17.Yang, X., S. Kurteva, S. Lee, and J. Sodroski. 2005. Stoichiometry of antibody neutralization of human immunodeficiency virus type 1. J. Virol. 793500-3508. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Yang, X., S. Kurteva, X. Ren, S. Lee, and J. Sodroski. 2005. Stoichiometry of envelope glycoprotein trimers in the entry of human immunodeficiency virus type 1. J. Virol. 7912132-12147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Yang, X., S. Kurteva, X. Ren, S. Lee, and J. Sodroski. 2006. Subunit stoichiometry of human immunodeficiency virus type 1 envelope glycoprotein trimers during virus entry into host cells. J. Virol. 804388-4395. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Yang, X., I. Lipchina, S. Cocklin, I. Chaiken, and J. Sodroski. 2006. Antibody binding is a dominant determinant of the efficiency of human immunodeficiency virus type 1 neutralization. J. Virol. 8011404-11408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Zhu, P., J. Liu, J. Bess, Jr., E. Chertova, J. D. Lifson, H. Grise, G. A. Ofek, K. A. Taylor, and K. H. Roux. 2006. Distribution and three-dimensional structure of AIDS virus envelope spikes. Nature 441847-852. [DOI] [PubMed] [Google Scholar]