

Abstract
Respiratory syncytial virus (RSV) is a common cause of infection that is associated with a range of respiratory illnesses, from common cold-like symptoms to serious lower respiratory tract illnesses such as pneumonia and bronchiolitis. RSV is the single most important cause of serious lower respiratory tract illness in children <1 year of age. Host innate and acquired immune responses activated following RSV infection have been suspected to contribute to RSV disease. Toll-like receptors (TLRs) activate innate and acquired immunity and are candidates for playing key roles in the host immune response to RSV. Leukocytes express TLRs, including TLR2, TLR6, TLR3, TLR4, and TLR7, that can interact with RSV and promote immune responses following infection. Using knockout mice, we have demonstrated that TLR2 and TLR6 signaling in leukocytes can activate innate immunity against RSV by promoting tumor necrosis factor alpha, interleukin-6, CCL2 (monocyte chemoattractant protein 1), and CCL5 (RANTES). As previously noted, TLR4 also contributes to cytokine activation (L. M. Haynes, D. D. Moore, E. A. Kurt-Jones, R. W. Finberg, L. J. Anderson, and R. A. Tripp, J. Virol. 75:10730-10737, 2001, and E. A. Kurt-Jones, L. Popova, L. Kwinn, L. M. Haynes, L. P. Jones, R. A. Tripp, E. E. Walsh, M. W. Freeman, D. T. Golenbock, L. J. Anderson, and R. W. Finberg, Nat. Immunol. 1:398-401, 2000). Furthermore, we demonstrated that signals generated following TLR2 and TLR6 activation were important for controlling viral replication in vivo. Additionally, TLR2 interactions with RSV promoted neutrophil migration and dendritic cell activation within the lung. Collectively, these studies indicate that TLR2 is involved in RSV recognition and subsequent innate immune activation.
Respiratory syncytial virus (RSV), a member of the Paramyxoviridae family, is a common cause of serious lower respiratory tract illness in infants and young children and may cause repeated infections throughout life. Both primary and repeat infections are associated with a range of illnesses, from common cold-like symptoms to serious lower respiratory tract diseases such as bronchiolitis and pneumonia. The pathogenesis of human RSV disease is not well understood, and the underlying mechanisms responsible for the broad range of illness associated with infection have not been elucidated (35, 37). Both environmental and genetic factors have been hypothesized to play a significant role in disease pathogenesis, possibly by leading to differences in innate inflammatory responses to infection in the lung (6). RSV is a high priority for vaccine development, but to date attempts to develop a vaccine have been unsuccessful. It is likely that understanding the pathogenesis of RSV disease, including the innate immune response to infection, will help in designing a safe and effective vaccine (7, 36). Therefore, much of the current RSV research is focused on understanding the immunopathogenesis of RSV disease.
Toll-like receptors (TLRs) expressed on leukocytes and within tissues play an important role in activating innate immunity by recognizing invading pathogens, including viruses such as RSV, and initiating signals that promote components of inflammation (25). TLR signaling occurs through a common Toll-interleukin-1 (IL-1) receptor domain. All TLRs recruit MyD88 to the Toll-IL-1 domain with the exception of TLR3, which recruits Toll-IL-1R domain-containing adaptor inducing beta interferon (IFN-β). MyD88 signaling activates transcriptional gene regulators including mitogen-activated protein kinase, NF-κB, and IFN regulatory factors. These transcriptional regulators promote cytokine, chemokine, and IFN production (25). Resident leukocytes, including dendritic cells (DCs), macrophages, and lung epithelial cells, play key roles in mediating inflammatory responses to RSV by expressing TLRs that recognize RSV motifs (2, 44). TLRs are thought to be directly involved in activating innate immunity against many viruses, including RSV, following the recognition of certain conserved viral motifs (28, 40, 41). Early inflammatory signals generated via virus-TLR interactions can contribute to the recruitment of additional inflammatory mediators, including neutrophils and NK cells, into the lung that are thought to be important for clearing RSV-infected cells (5, 33, 35, 44).
Innate inflammatory components generated in part through TLR signaling can influence the type (i.e., Th1 or Th2) of immune response generated against RSV. Genetically predisposed individuals exhibit aberrant immune activation after RSV infection, which can promote bronchiolitis, persistent wheezing, and overactive mucous production (42, 49). Our laboratory previously reported that RSV F protein interacts with TLR4, which can signal through MyD88 to activate an innate immune response (21, 28). These results have been confirmed and extended by others, further supporting the hypothesis that TLR signaling is involved in the innate immune response to RSV (7, 19). Recently, Awomoyi et al. suggested that defects in TLR signaling, particularly in TLR4, are linked to RSV-induced pathology in preterm high-risk infants (3), although the overall effect of TLR4 on RSV pathogenesis remains unclear. Supporting these findings, Holt et al. demonstrated that peripheral blood mononuclear cells isolated from children with TLR4 mutations exhibited decreased NF-κB activation and subsequently displayed decreased cytokine production in response to RSV, suggesting that weakened immune responses contribute to RSV-induced disease (50). Taken together, these studies provide evidence that TLR-dependent signaling is important for activating early inflammatory responses to RSV and that aberrant TLR signaling promotes RSV-induced disease.
In the present study, we further explored the role of TLRs in activating innate immune responses against RSV. We examined whether RSV could promote the activation of innate immunity through TLR2 signaling. TLR2 is expressed on the surface of immune cells and tissues as a heterodimer complex with either TLR1 or TLR6 (1). TLR2 and TLR1 or TLR2 and TLR6 complexes recognize bacterial motifs as well as a diverse range of viruses, including hepatitis C virus, herpes simplex virus, lymphocytic choriomeningitis virus, and human cytomegalovirus, and strongly promote early innate inflammatory responses after viral recognition (8, 9, 27, 56). Genetic analysis and vaccine studies of BALB/c mice indirectly suggest that TLR2 signaling is involved in RSV recognition; however, these studies did not investigate whether TLR2 directly interacts with RSV (11, 20, 23). We wished to further examine the inferred relationship between RSV and TLR2, because identifying novel pathways by which RSV activates innate immunity could aid the development of safe and effective RSV vaccines. In this study, we demonstrate that RSV interacts with TLR2 and TLR6 but not TLR1 and that this interaction activates innate immunity.
MATERIALS AND METHODS
Animals.
Knockout (KO) mice were extensively back-bred onto the C57BL/6 background. Backcrossing was confirmed by satellite marker analysis (Charles River Laboratories), and mice were genotyped by the PCR amplification of tail DNA. Eight- to 10-week-old female TLR1 KO (F9), TLR2 KO (F11), TLR4 KO (F11), TLR6 KO (F10), and MyD88 KO (F12) mice (back-bred from KO mice originally provided as a gift from Shizuo Akira) were housed under specific-pathogen-free conditions at the University of Massachusetts, Worcester campus. Age- and sex-matched C57BL/6, BALB/c, and B6.129F2/J wild-type mice were purchased from The Jackson Laboratories.
Growing RSV.
Human RSV strain A2 was grown in Vero cells. Briefly, 85 to 90% confluent T175 flasks of Vero cells were infected with RSV at a multiplicity of infection (MOI) of 1 in 5 ml of Dulbecco's modified Eagle's medium (DMEM). Cells were infected for 2 h at 37°C and 5% CO2. After infection, 7 ml of DMEM with 10% fetal bovine serum (Gibco), 0.1% penicillin-streptomycin (Pen/Strep) (Cellgro), and 0.001% ciprofloxacin (Bayer) was added to the flask. Flasks were incubated for approximately 4 days or until extensive syncytium formation was observed. On the day of harvest, all except 3 ml of supernatant was removed and placed on ice in 50-ml conical tubes, approximately 3 ml per tube. Cells were scraped from the flask, added in equal distribution to conical tubes, and sonicated three times, 5 s per time, at 25 W on ice. Cell debris was removed by centrifugation at 600 × g for 7 min at 4°C. Virus supernatant was stored in 30% sucrose at −80°C. Uninfected flasks were treated identically to generate Vero cell lysate control.
Harvesting peritoneal macrophages.
Mice were intraperitoneally (i.p.) injected with 4% thioglycolate (Sigma). Four days after injection, mice were euthanized and the peritoneum was flushed with normal saline. Contaminating red cells were lysed with red blood cell lysing buffer (Sigma) according to manufacturer specifications. The resulting peritoneal exudate cell suspension was analyzed by flow cytometry and consisted of >90% macrophages based on CD11b+ expression (data not shown). Cells were resuspended in DMEM containing 10% fetal bovine serum (FBS) (HyClone) and 0.1% Pen/Strep, quantified using a hemocytometer, and added to tissue culture plates (Costar) for further analysis.
Macrophage stimulation assays.
Peritoneal macrophages were seeded in 96-well round-bottom tissue culture-treated plates (Costar) at 1.0 × 106 cells/ml. Cells were stimulated with live RSV strain A2 (MOI of 0.3), UV-inactivated RSV strain A2 (MOI equivalent of 0.3), TLR4 ligand LPS (100 ng/ml) (Sigma), TLR2 ligand Pam2CSK4 (100 ng/ml) (Sigma), and Vero cell lysate control (50 μl/well) as indicated. The UV inactivation of RSV was performed using a UV Stratalinker 2400 (Stratagene) at a voltage of 1 × 106 mJ/cm2. Inactivation was confirmed by immunoplaque assay.
Macrophages were stimulated for 24 h at 37°C and 5% CO2. After stimulation, cell supernatants were removed and stored at −20°C for enzyme-linked immunosorbent assay (ELISA) analysis. Intracellular tumor necrosis factor alpha (TNF-α) production was determined using fluorescein isothiocyanate-conjugated anti-TNF-α antibody (BD Pharmingen), and macrophages were gated using phycoerythrin (PE)-conjugated anti-CD11b antibody (BD Pharmingen).
In vivo infection.
Mice were lightly anesthetized with isoflurane and intranasally (i.n.) challenged with 2.4 × 106 PFU of RSV strain A2 or an equal volume of Vero cell lysate control.
Collection and analysis of BAL cells and lung tissue.
Mice were anesthetized with isoflurane and exsanguinated by severing the right caudal artery. Bronchoalveolar lavage (BAL) cells were harvested by lavaging the lung four consecutive times with 1 ml phosphate-buffered saline (PBS) containing 0.1% bovine serum albumin (BSA). BAL cells were pooled and pelleted by centrifugation at 600 × g for 5 min. Cells were surface stained with fluorescein isothiocyanate-conjugated anti-mouse CD11b (clone M1/70; BD Pharmingen), PE-conjugated anti-mouse CD86 (clone GL-1; BD Pharmingen), PE-conjugated anti-mouse F4/80 (clone BM8; eBioscience), allophycocyanin-conjugated anti-mouse CD11c (clone HL3; BD Pharmingen), and/or Alexa-647 conjugated anti-mouse neutrophils (clone 7/4; Serotec).
Lung tissue was removed following BAL harvest. One-half of the total lung tissue was stored in 30% sucrose for plaque assay. The remaining half of the lung tissue was stored in protease inhibitor (Roche) for ELISA analysis. Tissues were stored at −80°C until analysis. For analysis, lungs were thawed on ice and weighed. Lungs were homogenized using a pellet pestle and centrifuged at 2,600 × g for 10 min at 4°C to clarify supernatant.
RSV immunoplaque assay.
Twenty-four-well tissue culture plates (Costar) were seeded with 1.5 × 105 Vero cells/well in DMEM containing 10% FBS (Gibco), 0.1% Pen/Strep (Cellgro), and 0.001% ciprofloxacin (Bayer). Cells were incubated overnight at 37°C and 5% CO2. Medium was removed from confluent monolayers, and serial dilutions of RSV stock or lung tissue-clarified supernatants were absorbed to monolayers. All samples were run in duplicate wells. Plates were incubated at 37°C and 5% CO2 for 2 h for optimum infection. After incubation, supernatant was removed, and 1 ml of M199 medium (Gibco) containing 0.01% Pen/Strep mixed 1:1 with 2% methylcellulose (Sigma) was overlaid on monolayers. After 6 days of incubation at 37°C and 5% CO2 or when extensive syncytia developed, the overlay was removed and monolayers were fixed with 1 ml of ice-cold acetone:methanol (60:40). Fixative was removed after 10 min, and plates were air dried. Plates were blocked in 5% nonfat dry milk (Bio-Rad) for 10 min at room temperature. Primary RSV anti-F and anti-G antibodies (clones 131-2A and 131-2G; Chemicon) were added to wells for 2 h, followed by secondary horseradish peroxidase anti-mouse immunoglobulin antibody (clone 187.1; BD Pharmingen) for 1 h. Antibodies were diluted in 5% milk, and plates were incubated at 37°C and 5% CO2. Plates were hand washed twice with PBS containing 0.5% Tween 20 (Sigma) after each antibody incubation step. Individual plaques were developed using a DAB substrate kit (Vector) per the manufacture's specifications. The limit of detection for our immunoplaque assay is approximately 1.4 log10 PFU/g.
ELISA analysis.
Clarified lung homogenates and cell supernatants were analyzed for IL-6 (BD Pharmingen), TNF-α (BD Pharmingen), CCL2 (monocyte chemoattractant protein [MCP-1]) (BD Pharmingen), and CCL5 (RANTES) (R&D Systems) using a sandwich ELISA. ELISAs were performed per the manufacturer's specifications.
Type I IFN bioassay.
Macrophages were stimulated with RSV strain A2 (MOI of 2), TLR3 ligand poly(I:C) (Amersham) (50 μg/ml), TLR4 ligand lipopolysaccharide (LPS; 100 ng/ml), or medium alone for 24 h. Supernatants were removed and UV treated to inactivate the infectivity of the virus. Serial twofold dilutions of UV-inactivated supernatant were added to fresh NTCT929 cells at 1 × 105 cells/ml in 96-well flat-bottom tissue culture plates (Costar). Cells were incubated for 24 h at 37°C and 5% CO2. After 24 h, supernatants were removed and the NTCT929 cells were infected with vesicular stomatitis virus at 2.5 × 103 PFU/ml. The highest dilution of supernatant that resulted in a 50% reduction of vesicular stomatitis virus-induced cytopathic effect was defined as 1 U/ml of type I IFN.
Flow cytometric analysis.
Harvested cells were washed twice with cold PBS containing 2.0% BSA, enumerated using a hemocytometer, and transferred to 96-well round-bottom tissue culture plates (Costar). Cells were incubated in anti-mouse CD16/CD32 Fc block (BD Pharmingen) for 15 min at room temperature. Cells were washed twice with wash buffer and surface stained with the indicated antibodies and appropriate isotype controls per the manufacturer's specifications. Cells were washed twice with wash buffer and resuspended in wash buffer for analysis. For intracellular cytokine staining, cells were cultured overnight in the presence of GolgiStop (BD Pharmingen). Following surface staining, cells were treated with a BD Cytofix/Cytoperm Plus kit (BD Pharmingen) per the manufacturer's specifications. Cells were stained with the indicated intracellular antibodies and appropriate isotype controls for 30 min at 4°C in the presence of permeabilization and wash buffer. After incubation, cells were washed twice with permeabilization and wash buffer and resuspended in wash buffer for analysis. Cells were analyzed using a BD LSR II flow cytometer and FlowJo 8.4.2 software (TreeStar).
Statistical analysis.
Statistical significance was determined using an unpaired, two-tailed Student's t test. Values of P < 0.05 were considered significant. Error bars are ± standard deviations or ± standard errors of the means (SEM), as indicated in the figure legends. Statistics were generated using GraphPad software v4.0c (Prism).
RESULTS
TLR2 and TLR6 recognize RSV and mediate proinflammatory responses.
The role of TLRs in response to RSV was examined using TLR-deficient mice. Thioglycolate-elicited peritoneal macrophages from C57BL/6, TLR2 KO, and TLR4 KO mice were stimulated with RSV in the presence of brefeldin A, and intracellular cytokine levels were measured by flow cytometry. Macrophages from both TLR2 KO and TLR4 KO mice produced lower levels of intracellular TNF-α than wild-type cells following RSV infection (Fig. 1a). Interestingly, macrophages from TLR2 KO mice produced the lowest levels of TNF-α, i.e., less TNF-α than TLR4 KO mice and wild-type mice (4, 33, and 51%, respectively), suggesting a major role for TLR2 in the induction of proinflammatory cytokines following RSV stimulation. Controls indicated that TLR2 KO cells responded normally to LPS (TLR4 ligand) stimulation, and TLR4 KO cells responded normally to Pam2CSK4 (TLR2 ligand). To examine whether RSV replication was required to elicit TNF-α production, wild-type macrophages were stimulated with live RSV or UV-inactivated RSV (confirmed by immunoplaque assay; data not shown), and intracellular TNF-α was measured. The response to RSV and UV-inactivated RSV was equivalent, indicating that RSV replication was not required to stimulate intracellular TNF-α production (Fig. 1c). To exclude the possibility that contaminating products in the Vero cell-propagated RSV activated TLR2 signaling, sucrose-purified RSV and UV-inactivated purified RSV (a generous gift from Trudy Morrison) were used to stimulate wild-type macrophages. RSV was purified using methods previously described (17, 26). Purified RSV contained approximately fivefold higher viral protein concentrations than unpurified RSV based on Western blot analysis (data not shown). Purified RSV stimulated wild-type macrophages equivalently to Vero cell-propagated RSV, indicating that RSV itself and not contaminating products in the medium activated TLR2 signaling (Fig. 1d).
FIG. 1.

RSV activates murine macrophages through TLR2 and TLR6. Macrophages were harvested 4 days after thioglycolate treatment from wild-type, TLR2 KO, and TLR4 KO mice. (a to c) Macrophages were stimulated with RSV (MOI of 0.3), UV-inactivated RSV (MOI equivalent of 0.3 MOI), LPS (TLR4 ligand; 100 ng/ml), Pam2CSK4 (TLR2 ligand; 100 ng/ml), Vero cell lysate (50 μl), or UV-inactivated Vero cell lysate (50 μl) for 24 h in the presence of brefeldin A as indicated. (d) Wild-type macrophages were stimulated with sucrose-purified RSV (MOI of 0.02) or UV-inactivated sucrose purified RSV (MOI equivalent of 0.02). Cells were stained for CDllb, permeabilized, and stained for intracellular TNF-α. Values indicate the percentages of CDllb+ macrophages producing TNF-α. TNF-α production (black line) was compared to that of the isotype control (gray line).
Since TLR2 can partner with either TLR1 or TLR6 on the cell surface to propagate downstream signaling, we examined whether TLR1 or TLR6 in conjunction with TLR2 was involved in RSV signaling. Wild-type, TLR2 KO, TLR1 KO, and TLR6 KO macrophages were stimulated with RSV, and TNF-α levels were measured. A role for TLR6, but not TLR1, was observed in RSV-induced, TLR-mediated cytokine production (Fig. 1b). As expected, MyD88 KO cells, which lack signaling via TLR2, TLR1, and TLR6, did not produce TNF-α in response to RSV (data not shown). We further examined the role of TLRs in cytokine and chemokine secretion following RSV challenge by measuring cytokine protein levels from in vitro-cultured macrophages using ELISA. Macrophages from TLR2 KO and TLR6 KO mice produced significantly less IL-6 and CCL2 (MCP-1) than wild-type cells following RSV infection (Fig. 2a and b). Additionally, a role for TLR4 in the production of inflammatory cytokines IL-6 and CCL2 was observed in the TLR4 KO cells. CCL5 (RANTES) production also was decreased in both TLR2 KO and TLR6 KO cells compared to that of wild-type macrophages after RSV challenge (Fig. 2c). CCL5 production in TLR4 KO was lower compared to that of wild-type cells but was higher compared to that of TLR2 KO or TLR6 KO macrophages. Furthermore, MyD88 KO mice exhibited the decreased production of each cytokine and chemokine assayed in response to RSV. Since MyD88 is essential for TLR2, TLR6, and, partially, TLR4 signaling, these data support the hypothesis that TLR-driven MyD88-dependent signaling was important for early cytokine and chemokine production in response to RSV.
FIG. 2.
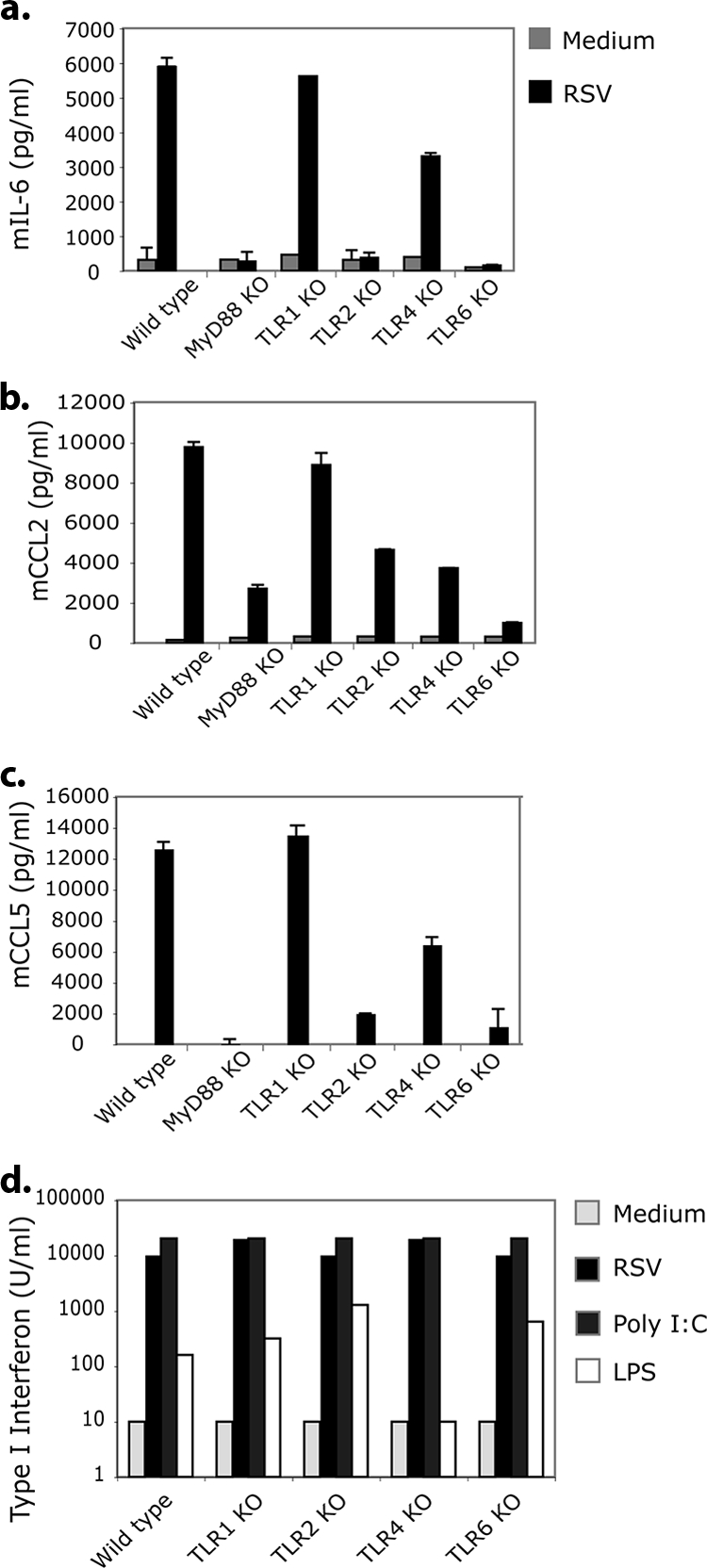
RSV induces inflammatory mediators through TLR2 and TLR6. Macrophages were stimulated with RSV (MOI of 0.3) or medium control for 24 h. Culture supernatants were harvested and tested for IL-6 (a), CCL2 (b), and CCL5 (c) production using ELISA. Error bars indicate ± standard deviations. Macrophages were stimulated with RSV (MOI of 2), poly(I:C) (50 μg/ml), LPS (100 ng/ml), or medium alone for 24 h. (d) Supernatants were tested for type I IFN production.
Type I IFN responses to RSV in TLR KO mice were examined next. RSV induced levels of type I IFN in each TLR KO group that were similar to those of the wild-type mice (Fig. 2d). Poly(I:C) (TLR3 ligand) has been shown to stimulate type I IFN production independently of TLR1, TLR2, TLR4, and TLR6 (55). RSV-induced type I IFN production was similar to that of poly(I:C)-induced type one IFN production in wild-type and TLR KO mice. LPS has also been shown to induce type one IFN production albeit to a lesser extent than poly(I:C). As expected, wild-type and TLR1, TLR2, and TLR6 KO cells produced equivalent amounts of type I IFN in response to LPS, while TLR4 KO cells did not respond to LPS. These data suggest that RSV induces type I IFN production independently of TLR2 and TLR6 signaling.
RSV replication in TLR KO mice.
To examine the role of TLRs in RSV replication after in vivo infection, C57BL/6, TLR2 KO, TLR4 KO, and TLR6 KO mice were infected by an i.n. instillation of 2.4 × 106 PFU/mouse of RSV. Kinetic studies examining the day of peak virus titer in RSV-resistant C57BL/6 mice as well as RSV-permissive BALB/c and semipermissive B6.129F2/J mice indicated that the virus titer peaked at day 4 in all strains tested (Table 1). These findings were in agreement with previous studies that demonstrated that peak viral titer in mouse models occurs approximately 4 days after infection (43). Importantly, data summarized in Table 1 and previous studies have shown that wild-type C57BL/6 mice readily cleared RSV, resulting in significantly diminished virus recovery compared to that of more susceptible mouse strains, such as BALB/c, even at the day of peak viral load, day 4 postinfection (43). Although the TLR KO mice were back-bred onto the resistant C57BL/6 background, RSV was readily recovered from the lungs of TLR2, TLR4, and TLR6 KO mice on day 4. A significantly higher peak viral load was observed in TLR2 KO and TLR6 KO mice compared to that of wild-type mice on the C57BL/6 background (Fig. 3). These data also indicated a significant difference in peak viral titer between TLR4 KO mice and C57BL/6 wild-type mice, with higher viral loads in TLR4 KO mice. These findings confirm our original observations using mice with deficiencies in TLR4 on the C3H and the C57BL/10 genetic backgrounds, which indicated that TLR4 and CD14 were involved in RSV clearance (28). Significantly higher virus titers in TLR2 KO and TLR6 KO lungs suggested an important role for TLR2 and TLR6 in controlling RSV replication in vivo.
TABLE 1.
Kinetics of RSV infection in various inbred mouse strainsa
| Mouse strain | RSV titer on day:
|
||||||||
|---|---|---|---|---|---|---|---|---|---|
| 2 | 3 | 4 | 5 | 7 | 8 | 10 | 13 | 14 | |
| C57BL/6 | <LLD (12) | <LLD (3) | 0.82 ± 0.07 (22) | <LLD (3) | <LLD (3) | <LLD (3) | ND | <LLD (3) | <LLD (3) |
| BALB/c | 0.93 ± 0.11 (9) | ND | 2.54 ± 0.16 (9) | ND | 1.98 ± -.23 (9) | ND | <LLD (6) | ND | <LLD (3) |
| B6.129F2/J | 1.51 ± 0.19 (6) | ND | 2.32 ± -.07 (9) | ND | 1.8 ± -.28 (3) | ND | ND | ND | ND |
Wild type C57BL/6, BALB/c, and B6.129F2/J mice were infected i.n. with RSV strain A2 (2.4 × 106 PFU/mouse). Lung samples were collected postinfection at the time point indicated and enumerated for PFU using an immunoplaque assay. The mean titer is represented as log10 PFU/gram of lung tissue ± SEM. LLD indicates below the lower limit of detection. Numbers in parentheses indicate the number of animals tested. ND indicates not determined.
FIG. 3.
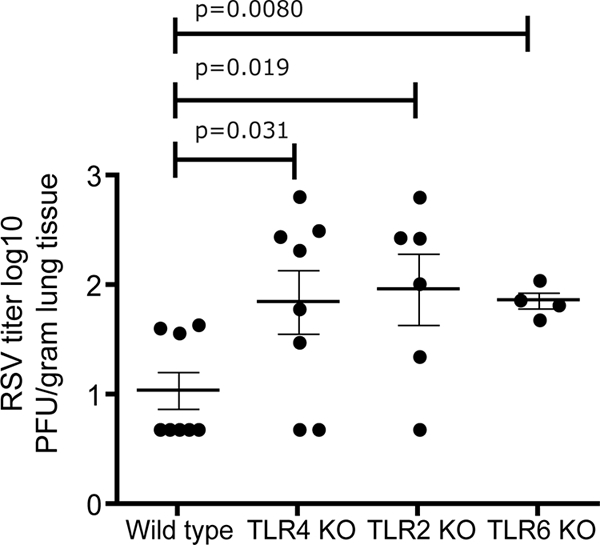
TLR2 and TLR6 are required for controlling RSV replication in vivo. Wild-type (n = 8), TLR4 KO (n = 8), TLR2 KO (n = 6), and TLR6 KO (n = 4) mice were infected by i.n. inoculation with RSV strain A2 (2.4 × 106 PFU/mouse). Lungs were harvested 4 days after infection, and plaques were enumerated using a Vero cell immunoplaque assay. Titers are represented as log10 PFU/gram of lung tissue. Solid lines represent the mean 1og10 value for each group. Error bars indicate ± SEM. Mice that were negative for RSV by plaque assay were scored at 0.5 times the lower limit of detection.
TLR2 signals promote neutrophil recruitment and DC activation in response to RSV.
To examine a possible role for TLR2 in the production of early inflammatory mediators in wild-type and TLR2 KO mice in response to RSV, wild-type C57BL/6, TLR2 KO, and MyD88 KO mice were infected i.n. with 2.4 × 106 PFU/mouse of RSV. Uninfected groups of each type of mouse were used to control for lung cytokine production and leukocyte infiltrate. Whole lungs and BAL were collected at 24, 48, and 96 h postinfection. Total cellular CCL2 levels were measured from lung homogenates. A significant decrease in CCL2 was observed in TLR2 KO and MyD88 KO mouse lungs compared to that in wild-type lungs at 24 h postinfection (Fig. 4a). CCL2 was undetectable at 48 and 96 h postinfection in all groups (data not shown). MyD88 KO mice did not produce detectable levels of CCL2 compared to that of the wild type. Although uninfected mice produced detectable basal levels of TNF-α, IL-6, and CCL5, we were unable to observe differences in lung homogenates between the groups (data not shown). These data suggested that TLR2 signaling is involved in early CCL2 production within the lungs of RSV-infected mice.
FIG. 4.

TLR2 signaling influences early innate cellular response against RSV in vivo. Wild-type, TLR2 KO, and MyD88 KO mice were infected by i.n. inoculation with RSV strain A2 (2.4 × 106 PFU/mouse). Uninfected mice (n = 3) were used to control for lung cytokine production and BAL infiltrate for each group. At 24, 48, and 96 h after infection, BAL and lung tissue were harvested from mice. (a) Lung tissue was homogenized and tested for CCL2 production by ELISA (wild-type and TLR2 KO mice, n = 7; MyD88 KO mice, n = 6). Data are represented as picograms/gram of lung tissue. Solid lines represent the means for each group. Error bars are ± SEM. BAL from each group (n = 3 per group) was pooled, and neutrophils (b) and activated DCs (c) were enumerated by flow cytometry. Neutrophil (7.4+ F4/80−) and DC (CD11b+ CD11c+ CD86+) populations are indicated as the percent positive cells of the total number of gated cells.
Since CCL2 is a potent leukocyte chemoattractant, the early recruitment of leukocytes to the lung following RSV infection was studied. A large neutrophil influx was observed in the BAL of wild-type mice at 24 h postinfection. In contrast to wild-type mice, fewer neutrophils were observed in TLR2 KO mice BAL at 24 h postinfection, suggesting that TLR2 signaling induced neutrophil migration into RSV-infected lungs (Fig. 4b). Furthermore, low levels of neutrophils were detected in the BAL of wild-type and TLR2 KO mice at 48 h postinfection and were undetected in BAL at 96 h postinfection, suggesting that neutrophil migration occurs very early during the innate immune response to RSV. Additionally, a reduced number of activated DCs (CD11hi CD11bhi CD86+) was observed in the BAL of TLR2 KO mice compared to that of the wild type at 24 and 48 h postinfection (Fig. 4c). MyD88 KO mice exhibited a reduced number of neutrophils and activated DCs in response to RSV infection, further indicating a role for TLR2-dependent signaling in activating innate immunity against RSV. Additionally, T-cell, NK cell, and eosinophil migration into the BAL of wild-type and TLR2 KO mice was examined. These cell populations were detected in the BAL at very low frequencies, and no differences were observed between the groups (data not shown).
DISCUSSION
Activating host immune responses during RSV infection is dependent on complex signaling events initiated in part by TLRs. These coordinated signaling events promote the production of cytokines, chemokines, and IFN-α, IFN-β, and IFN-γ in the lung. Interestingly, the type of inflammatory environment generated during these early signaling events likely alters subsequent T-cell responses, because susceptible infants exhibit characteristics of Th2 responses (6). Since recent findings have implicated TLR-mediated inflammation in the immunopathogenesis of RSV disease, emphasis has been placed on understanding how TLRs can influence immune responses against RSV (3, 7, 28, 36, 39, 50).
Initial studies conducted by Kurt-Jones et al. used strain C57BL/10ScNCr (a TLR4 mutant with a deletion mutation) to examine the interactions between RSV and TLR4. However, during the final publication review the strain designation was changed to C57BL/10ScCr (TLR4 deficient and IL-12 deficient), an error that was not caught during revision (unpublished data). Nevertheless, the authors demonstrated that the i.n. infection of RSV in the TLR4 mutant mice resulted in delayed viral clearance at 10 days postinfection compared to that of wild-type mice. In a follow-up study published by the same group, Haynes et al. again used the C57BL/10ScNCr mice (TLR4 deficient, IL-12 responsive) to confirm their original findings (21). Importantly, Haynes et al. indicated in the text that “C57BL/10ScNCr strain is homozygous for a null mutation of the TLR4 gene… a related mouse strain, C57BL/10ScCr (not used in these studies), has a reported defect in interleukin 12 (IL-12)-induced production of gamma IFN (IFN-γ)… . The C57BL/10ScNCr mice used in the present study were IL-12 responsive and expressed IFN-γ at levels similar to those of the wild type.” In contrast to these studies, other investigators examined the interaction between TLR4 and paramyxoviruses, including RSV, and found no association. Ehl et al. investigated whether deficiencies in TLR4 or in IL-12R could contribute to RSV susceptibility in the C57BL/10ScCr mouse (13). The authors concluded that increased susceptibility to RSV in this mouse model was due to defects in IL-12R rather than defects in TLR4. Studies by Van der Sluijs et al. explored the possible interaction between Sendai virus and TLR4 using C3H/HeJ (TLR4 mutant) mice (51). The authors were unable to find evidence of an interaction between Sendai virus and TLR4. Faisca et al. examined the interaction between PVM, a member of the Pneumovirus genus, and TLR4 using TLR4 mutant mice (14). Since RSV and PVM share many similarities, the authors suggested that the responses to both pathogens in the mouse should be similar. Additionally, the authors indicated that PVM is a natural mouse pathogen while RSV is not, suggesting that PVM would be a more suitable model for examining TLR4 and pneumovirus interactions in the mouse. In summary, the authors concluded that there was no evidence of an interaction between TLR4 and PVM. Collectively, these studies seemed to suggest some members of the Paramyxoviridae family, including Sendai virus and PVM, do not interact with TLR4 to activate innate immune responses; however, data briefly summarized below suggests that RSV does indeed interact with TLR4 to promote innate immune activation.
Following the Kurt-Jones et al. and Haynes et al. observations, Haeberle et al. used C3H/HeJ mice (with a TLR4 point mutation leading to LPS unresponsiveness) and demonstrated that NF-κB activation by RSV in vivo involves TLR4 (19). Additionally, studies by Boukhvalova et al. in cotton rats establish that the formalin-inactivated vaccine strain of RSV stimulates a Th2-like response, and this effect can be blocked by using TLR4 antagonists (7). Furthermore, studies by Vogel and colleagues demonstrate a clear linkage between RSV disease severity in high-risk infants and particular TLR4 polymorphism alleles, suggesting that TLR4 plays a significant role in RSV pathogenesis in humans (3). Interestingly, Rassa et al. demonstrated that mouse mammary tumor virus, a murine retrovirus, also activated innate immunity via TLR4 (38). Therefore, the literature published by several different groups seems to indicate an important role for TLR4 in the activation of innate immunity in response to RSV infection. Herein we have outlined some important factors that could highly affect study outcomes. First, different inbred mouse strains do express different levels of baseline and induced TLR, including TLR4 and TLR2, which could contribute to differences in innate and acquired immune activation. Importantly, C57BL/10ScNCr, C57BL/10ScCr, and C57BL/6 are distinct mouse strains that may differ in TLR expression. Second, the dose of RSV administered can influence disease outcome, because high inocula of RSV are required to elicit immune activation in mice. Third, frozen RSV stocks have been shown to lose a substantial amount of activity over time (>6 months); therefore, it is critical to use recently propagated stocks for in vivo experiments to ensure optimum immune activation in mice. Fourth, the cell lines that RSV is propagated in can greatly affect infectivity. For example, studies by Taylor et al. have shown that genetically similar mice infected i.n. with equivalent titers of RSV isolated from different cell lines show significant differences in the amount of virus recovered from the lungs (47). Collectively, differences in these parameters could influence experimental results by different investigators.
The present study used C57BL6/J mice lacking the TLR4 gene (TLR4 KO mice), and the data presented here support our original observations, further suggesting a role for TLR4-mediated innate immune activation in response to RSV (i.e., TLR4 deficiency blunted the cytokine response to RSV infection). Additionally, we extended our initial observations by demonstrating that TLR2 signaling also can activate innate immunity in response to RSV.
Studies presented here provide evidence for interactions between RSV and TLR2 and TLR6 but not TLR1. These findings are important, because the lung cytokine and chemokine milieu is thought to directly shape cellular processes within the lung that prevent or promote RSV-induced disease (6). Macrophages from TLR2 and TLR6 KO mice produce less TNF-α than wild-type mice in vitro. TNF-α has been linked to both viral clearance and exacerbated airway disease, suggesting one mechanism by which the characteristics of the immune response dictates disease outcome. In fact, previous work indicates that RSV does indeed modulate TNF-α secretion; i.e., the small hydrophobic protein inhibits TNF-α secretion (16). RSV likely inhibits a major inflammatory component of TLR2 signaling by limiting TNF-α signaling. Since TLR2 KO and TLR6 KO mice were less effective in controlling viral replication, it is possible that the lowered secretion of TNF-α in these mice decreased the host response and subsequently affected viral clearance. Additionally, recent findings suggest that TNF-α can modulate TLR2 expression, therefore it is possible RSV could further alter immune activation by limiting TLR2 expression (46). Signaling through TLR2 and TLR6 controlled the expression of IL-6, an important inflammatory mediator that is linked to RSV pathology (31). In a model of airway allergic inflammation, TLR2 can induce IL-6 production in response to mycoplasma, and the lack of IL-6 production impaired bacterial clearance (54). Therefore, TLR2-dependent IL-6 production in conjunction with TNF-α may play a key role in mediating viral clearance.
TLR2 and TLR6 signaling promoted the production of chemokines CCL5 and CCL2. Both chemokines are linked to RSV pathogenesis (10, 35, 48). CCL5 is produced primarily by Th1-polarizing cells and can influence both innate and acquired leukocyte migration (29). CCL2 functions as a potent chemotaxic molecule for monocytes, including neutrophils, and can influence Th polarization toward Th2 by stimulating IL-4 production in T cells (18). In fact, recent studies associate increased CCL2 production with skewed Th2 responses in RSV G protein-primed BALB/c mice; however, this study did not address whether CCL2 causes or results from a Th2-polarized inflammatory environment (10). Interestingly, studies by Liu et al. demonstrate a role for TLR3 and retinoic acid-inducible gene I in mediating IFN-β production and NF-κB/RelA transcription by lung epithelial cells in response to RSV (30). Furthermore, our studies indicated that TLR2 and TLR6 signaling was important for NF-κB-dependent cytokine and chemokine production but not for type I IFN production, suggesting that the activation of innate immunity occurs via multiple TLRs in response to RSV to provide optimum protection against RSV-induced disease. Our in vitro studies examined the early inflammatory response initiated by peritoneal macrophages in response to RSV. Additionally, Suzuki et al. demonstrated that murine alveolar macrophages and peritoneal macrophages express similar levels of TLR2 and respond equally to TLR2 ligands, suggesting that alveolar macrophages and peritoneal macrophages exhibit similar responses to RSV (45). Lung epithelial cells express TLR2 and may produce additional inflammatory components that further contribute to the early inflammatory environment (2, 44). Collectively, these findings suggested that the control of cytokine and chemokine production by TLR2 is critical for supporting viral clearance while limiting RSV-induced disease.
TLR2 signaling was examined in vivo by infecting TLR2 KO mice with RSV. We were unable to detect differences in TNF-α, IL-6, and CCL5 production in vivo at 24 h postinfection, suggesting that the peak production of these factors occurred at an earlier time point during the response. TLR2 signaling promoted CCL2 production in vivo. Interestingly, studies examining acute sepsis peritonitis using mouse models showed that CCL2 production was indirectly involved in neutrophil influx through the production of leukotriene B4, suggesting that CCL2 production influences neutrophil migration (32). Moreover, we observed a decrease in lung neutrophils in the BAL of TLR2 KO mice at 24 h postinfection. These findings indicated that TLR2-mediated CCL2 production is important for early neutrophil migration to the lung in response to RSV infection. Neutrophils are innate leukocytes that traffic to sites of infection via chemokine gradients and greatly aid the clearance of virus-infected cells. The presence of neutrophils correlates with RSV disease severity and, further, these cells secrete inflammatory mediators, including reactive oxygen species and cytokines (i.e., IL-8), that further drive inflammation and indiscriminately kill both infected and uninfected tissues (4, 24, 52, 53). Therefore, heightened TLR2 signaling may mediate neutrophil-associated airway inflammation through CCL2 production.
Early inflammatory components can profoundly influence DCs in their ability to shape innate and acquired immune responses. Therefore, we examined the affect of TLR2 signaling on DC activation. Within the lung, immature DCs sense viruses, including RSV, in part through TLRs and can promote T- and B-cell responses after receiving activation signals from components that make up the inflammatory environment (12, 22). We observed a decrease in activated lung DCs (CD11bhi CD11chi CD86+) in TLR2 KO mice at 24 and 48 h postinfection, suggesting that TLR2-mediated signals generated during RSV infection could influence DC activation. Interestingly, Rudd et al. showed that a lack of MyD88-dependent signaling in DCs can promote Th2 responses after RSV infection (39). Additionally, TLR2 signaling may further contribute to RSV responses by facilitating interactions between DCs and neutrophils that can further shape T- and B-cell polarization (34).
Collectively, these studies indicated that TLR2, in addition to previously discovered TLRs, including TLR3, TLR7, and TLR4 plus CD14, activate innate immune responses upon RSV recognition (28, 40, 41). TLR2 and TLR6 signaling, but not TLR1 signaling, activated inflammatory components that are thought to promote the clearance of RSV and prevent RSV-induced disease. Since the early inflammatory environment greatly shapes subsequent immune responses, these findings provide new insight into how TLR signaling could contribute to a beneficial anti-RSV immune response. Interestingly, TLR4 signaling can influence TLR2 expression following certain stimuli, suggesting that the optimal induction of multiple signaling pathways is required to elicit protective rather than deleterious innate immune responses following RSV infection, consistently with a role for both TLR4 and TLR2 in the response to RSV (15). Finally, data presented here suggested that TLR2 signaling was involved in activating T-cell-like responses; therefore, RSV vaccines incorporating TLR2 adjuvant may help elicit strong Th1 responses. This strategy was evaluated in mouse models of RSV disease using the TLR2 ligand peptidoglycan as an adjuvant and showed effectiveness in limiting RSV-induced disease (20). Alternatively, TLR2 antagonist therapies may provide some benefit to susceptible children during active infection by dampening inflammation and limiting neutrophil-associated airway inflammation. In light of the findings presented here, therapies targeting TLR2 signaling may prove beneficial in limiting RSV infection and preventing RSV-induced disease.
Acknowledgments
This work was supported by Public Health Service grants from the NIH: RO1 AI51405 (to E.K.-J.) and RO1 AI049309, U54 AI057159, P01 AI0577484, RO1 AI64349, and P30 DK032520 (to R.W.F.).
We thank Shenghua Zhou for suggestions and the critical reading of the manuscript. We thank Trudy Morrison for providing purified RSV and UV-inactivated purified RSV for our studies.
Footnotes
Published ahead of print on 19 November 2008.
REFERENCES
- 1.Akira, S., S. Uematsu, and O. Takeuchi. 2006. Pathogen recognition and innate immunity. Cell 124783-801. [DOI] [PubMed] [Google Scholar]
- 2.Applequist, S. E., R. P. Wallin, and H. G. Ljunggren. 2002. Variable expression of Toll-like receptor in murine innate and adaptive immune cell lines. Int. Immunol. 141065-1074. [DOI] [PubMed] [Google Scholar]
- 3.Awomoyi, A. A., P. Rallabhandi, T. I. Pollin, E. Lorenz, M. B. Sztein, M. S. Boukhvalova, V. G. Hemming, J. C. Blanco, and S. N. Vogel. 2007. Association of TLR4 polymorphisms with symptomatic respiratory syncytial virus infection in high-risk infants and young children. J. Immunol. 1793171-3177. [DOI] [PubMed] [Google Scholar]
- 4.Bataki, E. L., G. S. Evans, and M. L. Everard. 2005. Respiratory syncytial virus and neutrophil activation. Clin. Exp. Immunol. 140470-477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Becker, S., J. Quay, and J. Soukup. 1991. Cytokine (tumor necrosis factor, IL-6, and IL-8) production by respiratory syncytial virus-infected human alveolar macrophages. J. Immunol. 1474307-4312. [PubMed] [Google Scholar]
- 6.Becker, Y. 2006. Respiratory syncytial virus (RSV) evades the human adaptive immune system by skewing the Th1/Th2 cytokine balance toward increased levels of Th2 cytokines and IgE, markers of allergy—a review. Virus Genes 33235-252. [DOI] [PubMed] [Google Scholar]
- 7.Boukhvalova, M. S., G. A. Prince, L. Soroush, D. C. Harrigan, S. N. Vogel, and J. C. Blanco. 2006. The TLR4 agonist, monophosphoryl lipid A, attenuates the cytokine storm associated with respiratory syncytial virus vaccine-enhanced disease. Vaccine 245027-5035. [DOI] [PubMed] [Google Scholar]
- 8.Chang, S., A. Dolganiuc, and G. Szabo. 2007. Toll-like receptors 1 and 6 are involved in TLR2-mediated macrophage activation by hepatitis C virus core and NS3 proteins. J. Leukoc. Biol. 82479-487. [DOI] [PubMed] [Google Scholar]
- 9.Compton, T., E. A. Kurt-Jones, K. W. Boehme, J. Belko, E. Latz, D. T. Golenbock, and R. W. Finberg. 2003. Human cytomegalovirus activates inflammatory cytokine responses via CD14 and Toll-like receptor 2. J. Virol. 774588-4596. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Culley, F. J., A. M. Pennycook, J. S. Tregoning, T. Hussell, and P. J. Openshaw. 2006. Differential chemokine expression following respiratory virus infection reflects Th1- or Th2-biased immunopathology. J. Virol. 804521-4527. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Cyr, S. L., T. Jones, I. Stoica-Popescu, D. Burt, and B. J. Ward. 2007. C57Bl/6 mice are protected from respiratory syncytial virus (RSV) challenge and IL-5 associated pulmonary eosinophilic infiltrates following intranasal immunization with Protollin-eRSV vaccine. Vaccine 253228-3232. [DOI] [PubMed] [Google Scholar]
- 12.Demedts, I. K., K. R. Bracke, T. Maes, G. F. Joos, and G. G. Brusselle. 2006. Different roles for human lung dendritic cell subsets in pulmonary immune defense mechanisms. Am. J. Respir. Cell Mol. Biol. 35387-393. [DOI] [PubMed] [Google Scholar]
- 13.Ehl, S., R. Bischoff, T. Ostler, S. Vallbracht, J. Schulte-Monting, A. Poltorak, and M. Freudenberg. 2004. The role of Toll-like receptor 4 versus interleukin-12 in immunity to respiratory syncytial virus. Eur. J. Immunol. 341146-1153. [DOI] [PubMed] [Google Scholar]
- 14.Faisca, P., D. B. Tran Anh, A. Thomas, and D. Desmecht. 2006. Suppression of pattern-recognition receptor TLR4 sensing does not alter lung responses to pneumovirus infection. Microbes Infect. 8621-627. [DOI] [PubMed] [Google Scholar]
- 15.Fan, J., R. S. Frey, and A. B. Malik. 2003. TLR4 signaling induces TLR2 expression in endothelial cells via neutrophil NADPH oxidase. J. Clin. Investig. 1121234-1243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Fuentes, S., K. C. Tran, P. Luthra, M. N. Teng, and B. He. 2007. Function of the respiratory syncytial virus small hydrophobic protein. J. Virol. 818361-8366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Gias, E., S. U. Nielsen, L. A. Morgan, and G. L. Toms. 2008. Purification of human respiratory syncytial virus by ultracentrifugation in iodixanol density gradient. J. Virol. Methods 147328-332. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Gu, L., S. Tseng, R. M. Horner, C. Tam, M. Loda, and B. J. Rollins. 2000. Control of TH2 polarization by the chemokine monocyte chemoattractant protein-1. Nature 404407-411. [DOI] [PubMed] [Google Scholar]
- 19.Haeberle, H. A., R. Takizawa, A. Casola, A. R. Brasier, H. J. Dieterich, N. Van Rooijen, Z. Gatalica, and R. P. Garofalo. 2002. Respiratory syncytial virus-induced activation of nuclear factor-κB in the lung involves alveolar macrophages and toll-like receptor 4-dependent pathways. J. Infect. Dis. 1861199-1206. [DOI] [PubMed] [Google Scholar]
- 20.Hancock, G. E., K. M. Heers, K. S. Pryharski, J. D. Smith, and L. Tiberio. 2003. Adjuvants recognized by toll-like receptors inhibit the induction of polarized type 2 T cell responses by natural attachment (G) protein of respiratory syncytial virus. Vaccine 214348-4358. [DOI] [PubMed] [Google Scholar]
- 21.Haynes, L. M., D. D. Moore, E. A. Kurt-Jones, R. W. Finberg, L. J. Anderson, and R. A. Tripp. 2001. Involvement of toll-like receptor 4 in innate immunity to respiratory syncytial virus. J. Virol. 7510730-10737. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Iwasaki, A., and R. Medzhitov. 2004. Toll-like receptor control of the adaptive immune responses. Nat. Immunol. 5987-995. [DOI] [PubMed] [Google Scholar]
- 23.Janssen, R., J. Pennings, H. Hodemaekers, A. Buisman, M. van Oosten, L. de Rond, K. Ozturk, J. Dormans, T. Kimman, and B. Hoebee. 2007. Host transcription profiles upon primary respiratory syncytial virus infection. J. Virol. 815958-5967. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Jones, A., J. M. Qui, E. Bataki, H. Elphick, S. Ritson, G. S. Evans, and M. L. Everard. 2002. Neutrophil survival is prolonged in the airways of healthy infants and infants with RSV bronchiolitis. Eur. Respir. J. 20651-657. [DOI] [PubMed] [Google Scholar]
- 25.Kawai, T., and S. Akira. 2007. TLR signaling. Semin. Immunol. 1924-32. [DOI] [PubMed] [Google Scholar]
- 26.Kong, X., H. San Juan, M. Kumar, A. K. Behera, A. Mohapatra, G. R. Hellermann, S. Mane, R. F. Lockey, and S. S. Mohapatra. 2003. Respiratory syncytial virus infection activates STAT signaling in human epithelial cells. Biochem. Biophys. Res. Commun. 306616-622. [DOI] [PubMed] [Google Scholar]
- 27.Kurt-Jones, E. A., M. Chan, S. Zhou, J. Wang, G. Reed, R. Bronson, M. M. Arnold, D. M. Knipe, and R. W. Finberg. 2004. Herpes simplex virus 1 interaction with Toll-like receptor 2 contributes to lethal encephalitis. Proc. Natl. Acad. Sci. USA 1011315-1320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Kurt-Jones, E. A., L. Popova, L. Kwinn, L. M. Haynes, L. P. Jones, R. A. Tripp, E. E. Walsh, M. W. Freeman, D. T. Golenbock, L. J. Anderson, and R. W. Finberg. 2000. Pattern recognition receptors TLR4 and CD14 mediate response to respiratory syncytial virus. Nat. Immunol. 1398-401. [DOI] [PubMed] [Google Scholar]
- 29.Lee, A. H., J. H. Hong, and Y. S. Seo. 2000. Tumour necrosis factor-alpha and interferon-gamma synergistically activate the RANTES promoter through nuclear factor κB and interferon regulatory factor 1 (IRF-1) transcription factors. Biochem. J. 350131-138. [PMC free article] [PubMed] [Google Scholar]
- 30.Liu, P., M. Jamaluddin, K. Li, R. P. Garofalo, A. Casola, and A. R. Brasier. 2007. Retinoic acid-inducible gene I mediates early antiviral response and Toll-like receptor 3 expression in respiratory syncytial virus-infected airway epithelial cells. J. Virol. 811401-1411. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Matsuda, K., H. Tsutsumi, S. Sone, Y. Yoto, K. Oya, Y. Okamoto, P. L. Ogra, and S. Chiba. 1996. Characteristics of IL-6 and TNF-alpha production by respiratory syncytial virus-infected macrophages in the neonate. J. Med. Virol. 48199-203. [DOI] [PubMed] [Google Scholar]
- 32.Matsukawa, A., C. M. Hogaboam, N. W. Lukacs, P. M. Lincoln, R. M. Strieter, and S. L. Kunkel. 1999. Endogenous monocyte chemoattractant protein-1 (MCP-1) protects mice in a model of acute septic peritonitis: cross-talk between MCP-1 and leukotriene B4. J. Immunol. 1636148-6154. [PubMed] [Google Scholar]
- 33.Mayer, A. K., M. Muehmer, J. Mages, K. Gueinzius, C. Hess, K. Heeg, R. Bals, R. Lang, and A. H. Dalpke. 2007. Differential recognition of TLR-dependent microbial ligands in human bronchial epithelial cells. J. Immunol. 1783134-3142. [DOI] [PubMed] [Google Scholar]
- 34.Megiovanni, A. M., F. Sanchez, M. Robledo-Sarmiento, C. Morel, J. C. Gluckman, and S. Boudaly. 2006. Polymorphonuclear neutrophils deliver activation signals and antigenic molecules to dendritic cells: a new link between leukocytes upstream of T lymphocytes. J. Leukoc. Biol. 79977-988. [DOI] [PubMed] [Google Scholar]
- 35.Ogra, P. L. 2004. Respiratory syncytial virus: the virus, the disease and the immune response. Paediatr Respir. Rev. 5(Suppl. A)S119-S126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Openshaw, P. J., F. J. Culley, and W. Olszewska. 2001. Immunopathogenesis of vaccine-enhanced RSV disease. Vaccine 20(Suppl. 1)S27-S31. [DOI] [PubMed] [Google Scholar]
- 37.Openshaw, P. J., G. S. Dean, and F. J. Culley. 2003. Links between respiratory syncytial virus bronchiolitis and childhood asthma: clinical and research approaches. Pediatr. Infect. Dis. J. 22S58-S64. [DOI] [PubMed] [Google Scholar]
- 38.Rassa, J. C., J. L. Meyers, Y. Zhang, R. Kudaravalli, and S. R. Ross. 2002. Murine retroviruses activate B cells via interaction with toll-like receptor 4. Proc. Natl. Acad. Sci. USA 992281-2286. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Rudd, B. D., M. A. Schaller, J. J. Smit, S. L. Kunkel, R. Neupane, L. Kelley, A. A. Berlin, and N. W. Lukacs. 2007. MyD88-mediated instructive signals in dendritic cells regulate pulmonary immune responses during respiratory virus infection. J. Immunol. 1785820-5827. [DOI] [PubMed] [Google Scholar]
- 40.Rudd, B. D., J. J. Smit, R. A. Flavell, L. Alexopoulou, M. A. Schaller, A. Gruber, A. A. Berlin, and N. W. Lukacs. 2006. Deletion of TLR3 alters the pulmonary immune environment and mucus production during respiratory syncytial virus infection. J. Immunol. 1761937-1942. [DOI] [PubMed] [Google Scholar]
- 41.Schlender, J., V. Hornung, S. Finke, M. Gunthner-Biller, S. Marozin, K. Brzozka, S. Moghim, S. Endres, G. Hartmann, and K. K. Conzelmann. 2005. Inhibition of toll-like receptor 7- and 9-mediated α/β interferon production in human plasmacytoid dendritic cells by respiratory syncytial virus and measles virus. J. Virol. 795507-5515. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Stark, J. M., V. Godding, J. B. Sedgwick, and W. W. Busse. 1996. Respiratory syncytial virus infection enhances neutrophil and eosinophil adhesion to cultured respiratory epithelial cells. Roles of CD18 and intercellular adhesion molecule-1. J. Immunol. 1564774-4782. [PubMed] [Google Scholar]
- 43.Stark, J. M., S. A. McDowell, V. Koenigsknecht, D. R. Prows, J. E. Leikauf, A. M. Le Vine, and G. D. Leikauf. 2002. Genetic susceptibility to respiratory syncytial virus infection in inbred mice. J. Med. Virol. 6792-100. [DOI] [PubMed] [Google Scholar]
- 44.Sukkar, M. B., S. Xie, N. M. Khorasani, O. M. Kon, R. Stanbridge, R. Issa, and K. F. Chung. 2006. Toll-like receptor 2, 3, and 4 expression and function in human airway smooth muscle. J. Allergy Clin. Immunol. 118641-648. [DOI] [PubMed] [Google Scholar]
- 45.Suzuki, K., T. Suda, T. Naito, K. Ide, K. Chida, and H. Nakamura. 2005. Impaired toll-like receptor 9 expression in alveolar macrophages with no sensitivity to CpG DNA. Am. J. Respir. Crit. Care Med. 171707-713. [DOI] [PubMed] [Google Scholar]
- 46.Syed, M. M., N. K. Phulwani, and T. Kielian. 2007. Tumor necrosis factor-alpha (TNF-alpha) regulates Toll-like receptor 2 (TLR2) expression in microglia. J. Neurochem. 1031461-1471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Taylor, G., E. J. Stott, M. Hughes, and A. P. Collins. 1984. Respiratory syncytial virus infection in mice. Infect. Immun. 43649-655. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Tekkanat, K. K., H. Maassab, A. Miller, A. A. Berlin, S. L. Kunkel, and N. W. Lukacs. 2002. RANTES (CCL5) production during primary respiratory syncytial virus infection exacerbates airway disease. Eur. J. Immunol. 323276-3284. [DOI] [PubMed] [Google Scholar]
- 49.Tripp, R. A., C. Oshansky, and R. Alvarez. 2005. Cytokines and respiratory syncytial virus infection. Proc. Am. Thorac. Soc. 2147-149. [DOI] [PubMed] [Google Scholar]
- 50.Tulic, M. K., R. J. Hurrelbrink, C. M. Prele, I. A. Laing, J. W. Upham, P. Le Souef, P. D. Sly, and P. G. Holt. 2007. TLR4 polymorphisms mediate impaired responses to respiratory syncytial virus and lipopolysaccharide. J. Immunol. 179132-140. [DOI] [PubMed] [Google Scholar]
- 51.van der Sluijs, K. F., L. van Elden, M. Nijhuis, R. Schuurman, S. Florquin, H. M. Jansen, R. Lutter, and T. van der Poll. 2003. Toll-like receptor 4 is not involved in host defense against respiratory tract infection with Sendai virus. Immunol. Lett. 89201-206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Wang, S. Z., and K. D. Forsyth. 2000. The interaction of neutrophils with respiratory epithelial cells in viral infection. Respirology 51-10. [DOI] [PubMed] [Google Scholar]
- 53.Wang, S. Z., H. Xu, A. Wraith, J. J. Bowden, J. H. Alpers, and K. D. Forsyth. 1998. Neutrophils induce damage to respiratory epithelial cells infected with respiratory syncytial virus. Eur. Respir. J. 12612-618. [DOI] [PubMed] [Google Scholar]
- 54.Wu, Q., R. J. Martin, S. Lafasto, B. J. Efaw, J. G. Rino, R. J. Harbeck, and H. W. Chu. 2008. Toll-like receptor 2 down-regulation in mouse allergic lungs decreases mycoplasma clearance. Am. J. Respir. Crit. Care Med. 177720-729 [DOI] [PubMed] [Google Scholar]
- 55.Yamamoto, M., S. Sato, K. Mori, K. Hoshino, O. Takeuchi, K. Takeda, and S. Akira. 2002. Cutting edge: a novel Toll/IL-1 receptor domain-containing adapter that preferentially activates the IFN-beta promoter in the Toll-like receptor signaling. J. Immunol. 1696668-6672. [DOI] [PubMed] [Google Scholar]
- 56.Zhou, S., E. A. Kurt-Jones, L. Mandell, A. Cerny, M. Chan, D. T. Golenbock, and R. W. Finberg. 2005. MyD88 is critical for the development of innate and adaptive immunity during acute lymphocytic choriomeningitis virus infection. Eur. J. Immunol. 35822-830. [DOI] [PubMed] [Google Scholar]