

Abstract
Penetration of the endoplasmic reticulum (ER) membrane by polyomavirus (PyV) is a decisive step in virus entry. We showed previously that the ER-resident factor ERp29 induces the local unfolding of PyV to initiate the ER membrane penetration process. ERp29 contains an N-terminal thioredoxin domain (NTD) that mediates its dimerization and a novel C-terminal all-helical domain (CTD) whose function is unclear. The NTD-mediated dimerization of ERp29 is critical for its unfolding activity; whether the CTD plays any role in PyV unfolding is unknown. We now show that three hydrophobic residues within the last helix of the ERp29 CTD that were individually mutated to either lysine or alanine abolished ERp29's ability to stimulate PyV unfolding and infection. This effect was not due to global misfolding of the mutant proteins, as they dimerize and do not form aggregates or display increased protease sensitivity. Moreover, the mutant proteins stimulated secretion of the secretory protein thyroglobulin with an efficiency similar to that of wild-type ERp29. Using a cross-linking coimmunoprecipitation assay, we found that the physical interaction of the ERp29 CTD mutants with PyV is inefficient. Our data thus demonstrate that the ERp29 CTD plays a crucial role in PyV unfolding and infection, likely by serving as part of a substrate-binding domain.
Viruses co-opt host factors to enter cells and cause infection. The nature of the interactions between cellular factors and viral components during the infection process is often poorly characterized. The murine polyomavirus (MPyV), a nonenveloped virus, binds to the ganglioside receptor GD1a on the plasma membranes of host cells to initiate infection. It then traffics in a retrograde manner to reach the lumen of the endoplasmic reticulum (ER) (7, 20, 23). In the ER, PyV's major capsid protein, VP1, undergoes a conformational change in an unfolding reaction mediated by the ER-resident protein ERp29 (12), a putative escort chaperone structurally related to the protein disulfide isomerase (PDI) family (5, 13, 14). This conformational change generates a hydrophobic viral particle that enables it to bind, integrate, and perforate the ER membrane (12, 15), events postulated to initiate penetration of PyV across the ER membrane. Upon penetration of the ER membrane, PyV gains access to the nucleus, where transcription and replication of the viral genome ensue, leading to lytic infection and cell transformation. The mechanism by which a nonenveloped virus breaches a biological membrane, a decisive step in infection, remains enigmatic (24). Hence, in this study, we further characterized the nature of the ERp29-PyV interaction, an event that facilitates PyV penetration across the ER membrane.
ERp29 and its Drosophila melanogaster orthologue, Windbeutel (Wind), consist of an N-terminal thioredoxin-like domain (NTD) and a C-terminal domain (CTD) containing a novel, five-helix fold (9, 11). Whereas some PDI family members contain the redox-active CxxC motif in the thioredoxin domains, the ERp29 NTD lacks this motif and is redox inactive, retaining only the thioredoxin fold of multiple α-helices and β-sheets (5, 11, 13). The NTD mediates homodimerization of ERp29 (9, 11), which we previously found to be required to facilitate the unfolding of PyV and the secretion of the secretory protein thyroglobulin (Tg) (16). Dimerization of Wind is also mediated by its NTD and is similarly required for Wind's chaperone activity (1, 11, 19).
Although the function of the ERp29 CTD (which consists of helices α5 to α9) has not been established, Wind CTD has been demonstrated to play a critical role in Wind's chaperone activity, possibly serving as a substrate-binding site (1, 10). This finding raises the possibility that the ERp29 CTD may similarly play a crucial role during the PyV unfolding reaction. In the study described here, we assessed the role of the ERp29 CTD in PyV unfolding and infection. We found that hydrophobic residues in helix α9 of the ERp29 CTD mutated to lysine or alanine impaired ERp29's ability to promote virus unfolding and infection. This loss of activity cannot be attributed to defects in dimerization of the mutants or to global structural perturbations arising from the mutations. Instead, using a chemical cross-linking coimmunoprecipitation approach, we found that the helix α9 mutants of ERp29 form physical interactions with PyV that are weak compared to those of wild-type (WT) ERp29. Thus, the CTD of ERp29 likely provides a substrate-binding site for PyV that enables ERp29 to unfold the virus, which promotes viral penetration across the ER membrane, leading to infection.
MATERIALS AND METHODS
Reagents.
Crude MPyV, a polyclonal antibody against VP1, and NIH 3T3 cells were generously provided by T. Benjamin (Harvard Medical School, Boston, MA). Polyclonal anti-rat ERp29 antibody and pcDNA3.1(+)-rat ERp29 expression plasmid were provided by S. Mkrtchian. Polyclonal anti-Myc antibody and trypsin were purchased from Sigma-Aldrich (St. Louis, MO). Dulbecco's modified Eagle's medium (DMEM), penicillin-streptomycin, Opti-Mem, Lipofectamine 2000, and 0.05% trypsin-EDTA were purchased from Invitrogen (Carlsbad, CA). FetalClone III was purchased from HyClone (Logan, UT). Complete mini EDTA-free protease inhibitor cocktail tablets were purchased from Roche (Indianapolis, IN). The cross-linking reagent dithiobis[succinimidylpropionate] (DSP) was purchased from Pierce Biotechnology (Rockford, IL). Polyclonal antibodies against Tg were obtained from Dako (Denmark).
Mutagenesis.
Mutagenesis of residues in the CTD of ERp29 was performed with the QuickChange II site-directed mutagenesis kit (Stratagene, La Jolla, CA). Expression plasmid pcDNA3.1(+)-ERp29 (cloning of the plasmid is described in reference 2) was used as a template, and polyacrylamide gel electrophoresis (PAGE)-purified mutagenic primers were ordered from Invitrogen (Carlsbad, CA). The various individual mutations in the ERp29 expression constructs were confirmed by DNA sequencing.
Cell culture and transfection.
NIH 3T3 cells were cultured at 37°C in DMEM supplemented with 10% FetalClone III and penicillin-streptomycin. To transiently transfect the WT and mutant ERp29 expression plasmids, complexes were prepared in Opti-Mem with Lipofectamine 2000. Prior to the addition of the transfection complexes, the cells were switched to DMEM plus 10% FetalClone III only. The transfection complexes were removed after 24 h, at which time the cells were returned to DMEM plus 10% FetalClone III and penicillin-streptomycin. At 48 h posttransfection, cells were harvested in 0.05% trypsin-EDTA for in vitro assays or were infected (see below).
Trypsin digestion assay.
Cell extracts were prepared by lysing cells transfected with WT or mutant ERp29 expression plasmids at 48 h posttransfection in a physiological buffer [150 mM potassium acetate (KOAc), 250 mM sucrose, 50 mM HEPES (pH 7.5), 2 mM Mg(OAc)2 with 1% Triton X-100]. Lysis was carried out for 30 min on ice, followed by centrifugation at 13,000 rpm to remove the cell debris. The resulting lysates were then analyzed for ERp29 expression by reducing sodium dodecyl sulfate (SDS)-PAGE followed by immunoblotting with a polyclonal antibody against ERp29. These lysates were used in the trypsin digestion assay as described previously (12).
Infection assay.
At 48 h posttransfection, one set of cells transfected with empty vector, the WT ERp29 expression plasmid, or the mutant ERp29 expression plasmid was harvested to analyze ERp29 expression by reducing SDS-PAGE followed by immunoblotting with anti-ERp29, while a duplicate set was washed with DMEM containing 10% FetalClone III only. Crude PyV (100 or 200 PFU/cell) was added to the medium and incubated for 2 h at 37°C, at which point the cells were changed to DMEM plus 10% FetalClone III and penicillin-streptomycin and allowed to incubate further at 37°C. The infected cells were plated at 24 h postinfection on glass coverslips at a density of 3 × 104 cells/well in a 6-well plate and allowed to incubate for 24 h longer. Cells were then fixed and stained with a rat monoclonal antibody against MPyV large T antigen and a rhodamine-conjugated donkey anti-rat immunoglobulin G (Jackson ImmunoResearch, West Grove, PA). Each condition depicted in Fig. 1B and C and Fig. 2B and D represents the average value of at least four independent experiments. In each experiment, 1,000 cells per condition were scored for the absence or presence of nuclear large T antigen expression, using standard fluorescence microscopy as described previously (23). An Eclipse TS100 epifluoresence microscope (Nikon, Melville, NY) equipped with a Texas Red emission filter and a 40× objective was used.
FIG. 1.

The L240K ERp29 mutant does not stimulate PyV unfolding and infection efficiently. (A) DTT and EGTA destabilize PyV. PyV was incubated with DTT and EGTA where indicated, followed by the addition of trypsin. The samples were subjected to reducing SDS-PAGE followed by immunoblotting with an antibody against VP1. (B) L240K ERp29 cannot unfold VP1. PyV was preincubated with extracts from mock-transfected NIH 3T3 cells or from cells overexpressing WT, L240K, K226E, L227S, E220Q, or R223A ERp29. Trypsin was then added to the reaction mixtures. The samples were analyzed by reducing SDS-PAGE and immunoblotting with antibodies against VP1 (top panel) or ERp29 (bottom panel). The asterisk (*) represents a partial degradation product of VP1a. (C) L240K ERp29 stimulates PyV infection inefficiently. Mock-transfected NIH 3T3 cells and cells overexpressing WT or L240K ERp29 were challenged with PyV (100 PFU/cell). Large-T-antigen expression was analyzed by standard fluorescence microscopy. (D) The experiment was the same as for panel C, except that cells were challenged with 200 PFU/cell. Data are means ± standard deviations.
FIG. 2.

Effects of the helix α9 ERp29 mutants on PyV unfolding and infection. (A) I246K and L247K ERp29 do not unfold VP1. PyV was preincubated with extracts from mock-transfected NIH 3T3 cells or from cells overexpressing WT, L244K, I246K, or L247K ERp29. Trypsin was then added to the reaction mixtures. The samples were analyzed by reducing SDS-PAGE and immunoblotting with antibodies against VP1 (top panel) or ERp29 (bottom panel). (B) I246K and L247K ERp29 stimulate PyV infection inefficiently. Mock-transfected NIH 3T3 cells and cells overexpressing WT, I246K, or L247K ERp29 were challenged with PyV (100 PFU/cell). Large-T-antigen expression was analyzed by standard fluorescence microscopy. (C) Alanine mutations at positions 240, 246, and 247 in helix α9 abrogate ERp29's unfolding activity. The experiment was the same as for panel A, except that extracts from cells overexpressing L240A, I246A, and L247A ERp29 were used. (D) Helix α9 alanine mutants of ERp29 do not stimulate PyV infection. The experiment was the same as for panel B, except that cells overexpressing L240A, I246A, and L247A ERp29 were used. Data are means ± standard deviations.
Chemical cross-linking to determine the oligomerization state of ERp29 mutants.
Cell extracts from cells transfected with WT ERp29 or mutant ERp29 expression plasmids were prepared as in the trypsin digestion assay (see above), except that the lysis conditions also included a protease inhibitor cocktail. The extracts were incubated with the cross-linking reagent DSP for 30 min at room temperature, after which 50 mM Tris (pH 7.5) was added for 10 min on ice to quench the reaction. Samples were analyzed by nonreducing SDS-PAGE followed by immunoblotting with an antibody against ERp29.
Proteolytic analysis to detect gross misfolding of ERp29 mutants.
Cell extracts from cells transfected with WT ERp29 or mutant ERp29 expression plasmids were prepared as in the trypsin digestion assay (see above). The cell extracts were incubated with trypsin for 30 min on ice, and then the reactions were quenched by the addition of TLCK (Nα-p-tosyl-l-lysine chloromethyl ketone) for 10 min on ice. Samples were analyzed by reducing SDS-PAGE followed by immunoblotting with an antibody against ERp29.
Size exclusion chromatography to detect aggregation of ERp29 mutants.
Cell extracts from cells transfected with WT ERp29 or mutant ERp29 expression plasmids were prepared as in the trypsin digestion assay (see above), except that the lysis conditions also included a protease inhibitor cocktail. The ERp29 species in the resulting extracts were analyzed individually, using the Bio-Rad BioLogic Duo-Flow fast-performance liquid chromatography system equipped with a Bio-Sil SEC 250 gel filtration column (Bio-Rad, Hercules, CA). Following sample injection, the column was washed isotonically with buffer containing 150 mM KOAc, 50 mM HEPES [pH 7.5], and 2 mM Mg(OAc)2, and then 0.5-ml fractions were collected. For each even-numbered fraction, 50 μl was analyzed by reducing SDS-PAGE followed by immunoblotting with ERp29 antibody. The column was calibrated using the Bio-Rad gel filtration standard.
Tg secretion assay.
According to a protocol previously described (2), FRTL-5 rat thyroid epithelial cells were transiently transfected with an empty vector, a WT ERp29 expression plasmid, or mutant ERp29 expression plasmids and were metabolically labeled, and the secreted Tg was immunoprecipitated from the culture medium. The immune complexes were resolved by SDS-PAGE and analyzed by autoradiography to detect Tg, while cell lysates were analyzed for ERp29 expression by SDS-PAGE followed by immunoblotting with an ERp29 antibody. Densitometry was used to quantify Tg from the autoradiographs, and the relative amounts of secreted Tg were normalized to that of ERp29 expression for each condition. A two-tailed t test was used.
Cross-linking/coimmunoprecipitation assay.
Cell extracts were prepared by lysing cells transfected with the WT or mutant ERp29 expression plasmids at 48 h posttransfection in a buffer containing 150 mM KOAc, 50 mM HEPES (pH 7.5), 4 mM MgCl2, 1% Big Chap deoxy (EMD Biosciences, La Jolla, CA), and a protease inhibitor cocktail. Lysis was carried out for 30 min on ice, followed by centrifugation at 13,000 rpm to remove the cell debris. The resulting extracts were then incubated with crude PyV for 30 min at 37°C. The reaction mixtures were next incubated with the cross-linking reagent DSP for 30 min at room temperature and then quenched with 100 mM Tris (pH 7.5) on ice for 10 min. After the quenched reaction mixtures were diluted into an immunoprecipitation buffer (150 mM KOAc, 30 mM Tris [pH 7.5], 4 mM MgCl2, 0.08% sodium deoxycholate, and protease inhibitor cocktail), a control polyclonal Myc antibody or a polyclonal ERp29 antibody was incubated with the reaction mixtures at 4°C overnight. Protein A-agarose beads (Invitrogen) were then added to the samples and rotated at 4°C for 15 min. The beads were washed three times in a wash buffer (150 mM KOAc, 30 mM Tris [pH 7.5], 4 mM MgCl2) and then resuspended and boiled in a sample buffer containing 2% SDS and β-mercaptoethanol to cleave any cross-linked species. The samples were analyzed by reducing SDS-PAGE followed by immunoblotting with a VP1 antibody or an ERp29 antibody. A horseradish peroxidase-conjugated secondary antibody, Clean-Blot immunoprecipitation detection reagent (Pierce), was used to eliminate detection of the heavy and light chains from the antibody used for the immunoprecipitation.
RESULTS
The L240K mutant of ERp29 is defective in PyV unfolding and infection.
Upon reaching the ER lumen, the PDI-like ER chaperone ERp29 induces a conformational change to VP1, generating a hydrophobic viral particle that binds and perforates the ER membrane (12, 15). These events are thought to initiate penetration of PyV across the ER membrane into the cytosol, a step required for virus infection. The unfolding of VP1 by ERp29 was shown, using a limited-proteolysis approach (12). In this assay, the reducing agent dithiothreitol (DTT) and the calcium chelator EGTA were incubated with PyV to cause partial destabilization of the VP1 coat, which is stabilized by intermolecular disulfide bonds and internally bound calcium ions (3, 21). DTT and EGTA likely mimic the activities of ER reductases (e.g., PDI) (8) and calcium-binding proteins (e.g., calnexin) that would normally act on the virus. Thus, when PyV was incubated with DTT and EGTA, followed by the addition of trypsin, a VP1-derived product called VP1a was generated (Fig. 1A, compare lane 4 to lane 2), consistent with previous observations (12, 15). PyV incubated with DTT, EGTA, and an extract from mock-transfected NIH 3T3 cells, followed by the addition of trypsin, also produced VP1a (Fig. 1B, top panel, lane 1) (12, 16). Partial degradation of VP1a was also observed (Fig. 1B, top panel), likely due to nonspecific activities in the extract. Importantly, treatment of PyV with an extract from cells overexpressing ERp29 (Fig. 1B, bottom panel, lane 2) results in the efficient unfolding of VP1 such that previously hidden tryptic cleavage sites are exposed and cleaved to produce a defined smaller fragment, VP1b (Fig. 1B, top panel, lane 2) (12, 16). Formation of the VP1b fragment was shown previously to result from the exposure of the VP1 C-terminal arm (12). As structural studies have shown that the VP1 C-terminal arms form interactions with neighboring VP1 pentamers to stabilize the virus structure (21), externalization of the C-terminal arm by ERp29 likely contributes to the disassembly of the virus.
The ERp29 monomer comprises a single NTD and a CTD with a novel all-helical fold (9, 14). We showed previously that NTD-mediated ERp29 dimerization is required for PyV unfolding and infection (16). The role of the ERp29 CTD in PyV unfolding and infection, however, remains unclear. Studies of the ERp29 D. melanogaster orthologue, Wind, have shown that five residues in the CTD of Wind (E212, R215, R218, L219, and L232) contribute to its ability to chaperone the secretory substrate, Pipe, from the ER to the Golgi compartment (1). Furthermore, when fused to the NTD of Wind, the CTD of mouse ERp29 can functionally replace the corresponding Wind domain, and mutations corresponding to the ERp29 CTD residues (E220Q, R223A, K226E, L227S, and L240K) disrupt the chaperone activity of the Wind-ERp29 hybrid (10, 11). We therefore asked whether these residues in ERp29 CTD might play a role in mediating PyV unfolding. When PyV was incubated with extracts from cells expressing the L240K mutant (Fig. 1B, bottom panel, lane 3), a very low level of VP1b was found (Fig. 1B, top panel, lane 3). By contrast, extracts containing either the K226E or L227S mutant (Fig. 1B, bottom panel, lanes 5 and 6) potently induced VP1b formation (Fig. 1B, top panel, compare lanes 5 and 6 to lane 4). Extracts containing the E220Q or R223A mutant (Fig. 1B, bottom panel, lanes 8 and 9) also induced VP1b formation, albeit with less efficiency than WT ERp29 (Fig. 1B, top panel, compare lanes 8 and 9 to lane 7). We concluded that L240 of the ERp29 CTD is required for PyV unfolding. Because the L240K ERp29 exhibited the most severe defect in the virus-unfolding reaction, we focused our studies on this mutant.
As overexpression of WT ERp29 in cells stimulates PyV infection (16), we asked whether L240 of the ERp29 CTD facilitates this process. In cells incubated with PyV (100 PFU/cell), overexpression of WT ERp29, but not the L240K ERp29 mutant, stimulated PyV infection, which was measured as a function of large-T-antigen expression normalized to that for mock-transfected cells (Fig. 1C). Similarly, in cells incubated with a higher level of virus (200 PFU/cell), overexpression of WT ERp29 promoted infection, while overexpression of the L240K mutant did so only partially (Fig. 1D). We concluded that residue L240 of ERp29 plays an important role in stimulating PyV infection, consistent with its role in mediating virus unfolding.
Residues in helix α9 of ERp29 are required for VP1 unfolding and PyV infection.
The L240 residue lies in the ninth and final α-helix of ERp29, whereas residues 220, 223, 226, and 227 lie in the eighth α-helix (9, 11). We therefore asked if ERp29's unfolding activity requires other residues in helix α9 and designed additional mutations in helix α9 to test this hypothesis. To this end, the remaining three hydrophobic residues in helix α9 were mutated to a lysine residue (i.e., L244K, I246K, and L247K), and the resulting mutants were expressed in NIH 3T3 cells. Extracts prepared from those cells (Fig. 2A, bottom panel) were incubated with PyV. When exposed to trypsin, PyV treated with an extract from cells expressing L244K, but not I246K and L247K ERp29, was efficiently unfolded, as assessed by the appearance of VP1b (Fig. 2A, top panel, compare lane 3 to lanes 4 and 5). We concluded that in addition to L240, I246 and L247 are required for PyV unfolding. Furthermore, overexpression of WT ERp29, but not of I246K or L247K ERp29, stimulated virus infection (Fig. 2B), consistent with the unfolding defects of these mutants (Fig. 2A). Our results thus demonstrated that L240, I246, and L247 in helix α9 of ERp29 CTD are critical for chaperone activity.
As the lysine substitutions may disrupt the global folding of the mutant proteins, we introduced a more conservative mutation at these sites by generating the L240A, I246A, and L247A ERp29 mutants and analyzed their abilities to promote virus unfolding and infection. When incubated with PyV, extracts from cells expressing WT ERp29, but not L240A, I246A, and L247A ERp29, stimulated VP1b formation (Fig. 2C, compare lane 2 to lanes 3, 4, and 5). Moreover, overexpression of WT ERp29, but not L240A, I246A, and L247A ERp29, stimulated infection (Fig. 2D). As mutations of L240, I246, and L247 to alanine are less likely than lysine to cause significant structural perturbations in the CTD, these findings support the conclusion that helix α9 is required for ERp29 to unfold PyV and stimulate infection.
The helix α9 ERp29 mutants dimerize efficiently and are not globally misfolded.
We next assessed whether the alanine substitution mutants display global structural perturbations that may disrupt their activities. Size exclusion column chromatography was used to test for aggregation of the ERp29 mutants. Aggregated proteins typically migrate through a size exclusion column faster than the corresponding WT proteins, due to their larger radii. We compared the migration of WT ERp29 and the helix α9 alanine mutants over a size exclusion column and found the elution profiles of the L240A, I246A, and L247A mutants to be similar to that of WT ERp29 (Fig. 3A). Importantly, no WT ERp29 or helix α9 alanine mutant proteins were detected in the early fractions, where large protein complexes formed by aggregation are expected to elute. Although the resolution of this column does not allow the oligomerization state of the WT and mutant ERp29 species to be determined, we can minimally conclude that the introduction of mutations into ERp29 helix α9 does not lead to aggregation of the protein.
FIG. 3.

The helix α9 ERp29 mutants are not globally misfolded. (A) The helix α9 ERp29 alanine mutants migrate similarly to WT ERp29 by size exclusion chromatography. Extracts from cells overexpressing WT, L240A, I246A, or L247A ERp29 were individually subjected to size exclusion column chromatography. The column was washed isotonically with a physiological buffer containing no sucrose. Fractions were continuously collected from the column, and the even-numbered fractions were analyzed by reducing SDS-PAGE followed by immunoblotting with an antibody against ERp29. The column was calibrated using a gel filtration standard. (B) The helix α9 ERp29 alanine mutants dimerize efficiently. Extracts from NIH 3T3 cells overexpressing WT, L240A, I246A, or L247A ERp29 were incubated with various concentrations of the cross-linking reagent DSP. The cross-linking reaction was quenched, and the samples were analyzed by nonreducing SDS-PAGE followed by immunoblotting with an antibody against ERp29. (C) The helix α9 ERp29 alanine mutants have protease sensitivity similar to that of WT ERp29. Extracts from cells overexpressing WT, L240A, I246A, or L247A ERp29 were incubated with various concentrations of trypsin. The digestion reactions were quenched, and the samples were analyzed by reducing SDS-PAGE followed by immunoblotting with an antibody against ERp29.
As reported previously, dimerization of ERp29 (via its NTD) is required to trigger PyV unfolding and infection (16). Thus, any potential block in dimerization of the L240, I246, and L247 mutants could explain their impaired activities. As the size exclusion column used to detect the formation of ERp29 aggregates cannot accurately resolve ERp29 monomers and dimers, we employed a previously established chemical cross-linking assay (16) to analyze the oligomerization state of the helix α9 mutants. Extracts from cells expressing WT, L240A, I246A, or L247A ERp29 were treated with the cross-linking reagent DSP, which has been shown to stabilize the ∼60-kDa dimer of WT ERp29 (9, 16). Using increasing concentrations of the cross-linker, we found that the L240A, I246A, and L247A mutants each formed the dimerized species efficiently and similarly to WT ERp29 (Fig. 3B, compare lanes 5 to 8, 9 to 12, and 13 to 16 to lanes 1 to 4). The L240K, I246K, and L247K mutants also dimerized as efficiently as WT ERp29 (data not shown). These results demonstrate that the defective activity of the helix α9 ERp29 mutants is not due to an inability to dimerize. The finding that mutations in the ERp29 CTD have no effect on ERp29 dimerization is consistent with previous structural analyses of ERp29 and Wind dimers that indicated that the two CTDs in an ERp29 dimer are separated and oriented in opposite directions (9, 11).
To further establish that the helix α9 ERp29 mutants are not misfolded globally, we asked whether these mutant proteins are more sensitive to protease digestion. Extracts from cells over-expressing WT, L240A, I246A, or L247A ERp29 were incubated with increasing concentrations of the general protease trypsin, and the samples were analyzed by SDS-PAGE followed by immunoblotting with an ERp29 antibody. Similar to WT ERp29, the alanine mutants remained resistant to trypsin digestion at lower trypsin concentrations (Fig. 3C, compare lanes 1 to 3 to lanes 5 to 7, 9 to 11, and 13 to 15); at a higher trypsin concentration, both WT ERp29 and the alanine mutants are sensitive to digestion (Fig. 3C, compare lanes 4, 8, 12, and 16). These findings indicate that the mutants are not misfolded significantly. Collectively, our analyses of the helix α9 ERp29 mutants by size exclusion chromatography, chemical cross-linking, and proteolysis indicated that these mutants are not grossly misfolded. Thus, the inability of these mutants to stimulate unfolding of VP1 or PyV infection likely reflects a requirement for ERp29 helix α9 in these processes and not general structural defects.
Tg secretion is not affected significantly by mutations in helix α9 of ERp29.
In addition to its role in PyV infection, ERp29 promotes the efficient secretion of Tg from cells by escorting Tg through the secretory pathway and out of the cell (2, 17). As this escort activity also requires ERp29 dimerization (16), we asked whether the helix α9 lysine mutants might be defective in Tg secretion as well. To address this question, WT ERp29 and the L240K, I246K, and L247K ERp29 mutants were transfected into FRTL-5 rat thyrocytes. At 48 h posttransfection, cells were metabolically labeled with [35S]methionine-[35S]cysteine for 1 h, followed by replacement of the medium with normal medium. After 6 additional hours (to enable Tg secretion), the medium was collected. Total secreted Tg was isolated from the medium by immunoprecipitation, using a Tg antibody; the sample was subjected to autoradiography; and the intensity of the band corresponding to Tg was quantified for each sample. Interestingly, we found that overexpression of each of the L240K, I246K, and L247K ERp29 mutants stimulated Tg secretion with only a small reduction in efficiency compared to that stimulated by the overexpression of WT ERp29 (Fig. 4). That there is no significant difference in the escort activities of the helix α9 mutants compared to that of WT ERp29 supports our claim that the mutants are not grossly misfolded. Instead, helix α9 appears to be uniquely required for the PyV-related chaperone activity of ERp29 and not for its escort activity on Tg.
FIG. 4.
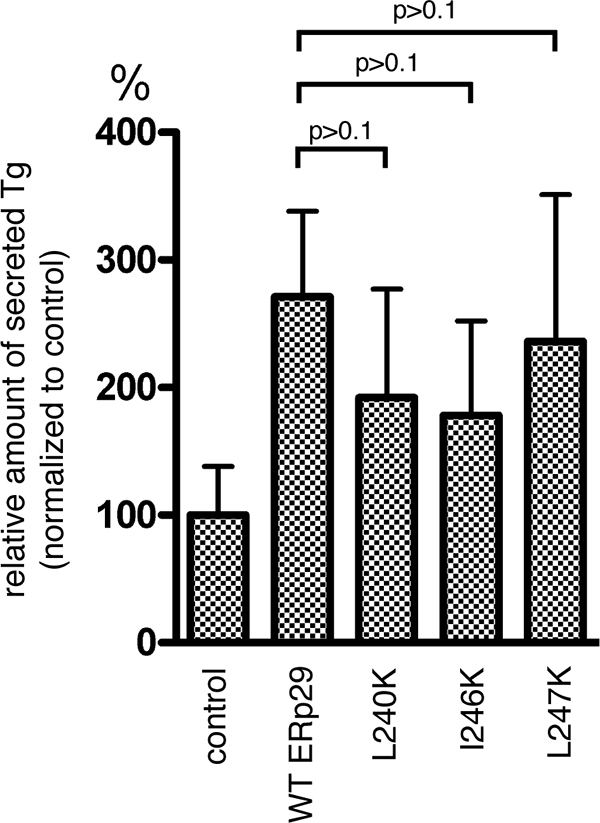
The helix α9 ERp29 mutants do not significantly affect Tg secretion. Mock-transfected FRTL-5 cells or cells overexpressing WT, L240K, I246K, or L247K ERp29 were labeled metabolically. Secreted Tg was immunoprecipitated from the culture media, and the immune complexes were analyzed by SDS-PAGE followed by autoradiography to detect Tg and by immunoblotting with an ERp29 antibody to detect ERp29 (data not shown). The amount of secreted Tg was quantified and normalized to that of ERp29 expression. Data are means ± standard errors.
The ERp29 helix α9 mutants bind less efficiently to PyV than WT ERp29.
The helix α9 ERp29 mutants failed to promote unfolding of the PyV VP1 or to stimulate infection. As these mutants dimerize and do not appear to have any gross folding defects, one possible explanation for their reduced activities is their inability to bind to the substrate. We therefore designed an assay to establish an ERp29-PyV interaction in order to assess the impact of mutating helix α9 of the ERp29 CTD on this interaction. As the physical interaction between ERp29 and PyV is expected to be transient, the cross-linking reagent DSP was added to a reaction mixture in which PyV was preincubated with extracts from cells overexpressing WT, L240A, I246A, or L247A ERp29. The different ERp29 species were subsequently immunoprecipitated, the immune complexes were reduced to cleave any cross-linked species, and the samples were analyzed for the presence of VP1 by immunoblotting. We found that when the sample was incubated with an ERp29 antibody, but not with the control Myc antibody, a significant level of PyV coprecipitated with WT ERp29 (Fig. 5, compare lane 7 to lane 6). This finding establishes for the first time a physical interaction between WT ERp29 and PyV. Importantly, significantly less PyV coimmunoprecipitated with the helix α9 mutants, the L240A, I246A, and L247A mutants, than with WT ERp29 (Fig. 5, top panel, compare lanes 8 to 10 to lane 7), suggesting that the hydrophobic residues of ERp29 helix α9 contribute to the physical interaction of ERp29 with PyV. This finding provides a mechanistic explanation for the inability of the helix α9 mutants to stimulate VP1 unfolding and PyV infection. Thus, the ERp29 CTD plays a crucial role in binding to PyV, facilitating the viral unfolding event that promotes penetration of PyV across the ER membrane.
FIG. 5.

The helix α9 ERp29 mutants bind less efficiently than WT ERp29 to PyV. PyV was incubated with extracts from cells expressing WT, L240A, I246A, or L247A ERp29. The cross-linking reagent DSP was added to the reaction mixtures. After the reaction was quenched, the WT and mutant ERp29 proteins were immunoprecipitated with an ERp29 antibody or a control Myc antibody, and the immune complexes were analyzed by reducing SDS-PAGE followed by immunoblotting with an antibody against ERp29 or against VP1 to detect PyV. The asterisk (*) represents an unidentified protein that is precipitated by the Myc antibody. IP, immunoprecipitation.
DISCUSSION
Viruses promote their own survival and proliferation in the host cell by hijacking cellular machineries that normally serve physiological functions. For example, during entry, many nonenveloped viruses co-opt the properties or activities of various host receptors, proteases, or chaperones to facilitate viral penetration across a host limiting membrane and gain access to the cytosol (24). Defining the mechanisms by which viruses divert normal physiological processes to achieve pathological ends is fundamental to understanding the nature of the host-pathogen interaction. Here, we characterized a host chaperone, ERp29, which is co-opted by the MPyV to facilitate its penetration across the ER membrane. The results of this study demonstrate that the hydrophobic residues in the last α-helix of ERp29 CTD (i.e., L240, I246, and L247 in helix α9) are critical to ERp29's pathogen-related activities, likely due to a role for the CTD in substrate binding.
Although differences in the structure of the rat ERp29 CTD as determined by nuclear magnetic resonance (9) and the crystal structures of Wind (11, 19) and human ERp29 (submitted recently to the Protein Data Bank [accession number 2qc7]) make the specific relative orientation of helices α8 and α9 uncertain, all of these structures at least indicate that ERp29 L240 (Wind L232) is surface exposed, suggesting that it is available to interact with a substrate or potential cofactors. ERp29 I246 and L247 (Wind I238 and L239, respectively) are likely buried within the helix bundle; their mutations may affect the positioning of L240 or the local integrity of helix α9 and thereby indirectly affect the interaction of ERp29 with PyV or a cofactor. Intriguingly, ERp29 residue L244 (Wind L236), which is not required for PyV unfolding, is exposed on the opposite surface of the CTD from L240, suggesting that PyV binds exclusively to one side of the ERp29 CTD. As we demonstrated previously that the C-terminal arms of the VP1 pentamers are exposed in an ERp29-dependent manner (12), we suspect that helix α9 of the ERp29 CTD likely makes contact with this region of the viral capsid. Of note, ERp29 helix α9 does not appear to mediate the binding and secretion of Tg, indicating that the mechanisms by which ERp29 engages its physiological and foreign substrates are distinct.
It remains unclear whether one CTD of the ERp29 dimer binds to PyV or whether the two CTDs of dimeric ERp29 simultaneously act on the virus. Additionally, whether ERp29 interacts with PyV using only its CTD or in cooperation with the NTD is a key question. Interestingly, Wind has been shown to bind to its substrate Pipe via its NTD and helix α8/α9 of the CTD (1, 19). Thus, while a major function of the ERp29 NTD is to mediate dimerization of ERp29, which we showed previously is required for ERp29 activity (16), this domain may play an additional role in binding to PyV. Future studies will further clarify the substrate-binding roles of the ERp29 NTD and CTD.
ERp29 is one of several proteins in the PDI family of ER chaperones that are manipulated by viruses and other pathogens. For example, in addition to ERp29 being required by PyV, downregulation of canonical PDI inhibits PyV infection (8). Downregulation of PDI and the PDI-like protein ERp72 affects the infection of cells by the related PyV simian virus 40 (SV40) in opposing manners, presumably through mechanisms related to their protein-folding and isomerase activities (18). Furthermore, the PDI-like protein ERp57 promotes the disulfide bond rearrangements of the SV40 capsid in the ER, an event that likely initiates SV40 uncoating (18). Bacterial toxins, such as cholera toxin, also hijack PDI-related proteins during their intracellular trafficking (6, 22). Why PDI-like proteins are uniquely targeted by pathogens and pathogenic factors is not known. These ER chaperones display a myriad of cellular activities. They function normally to ensure the proper folding of newly synthesized proteins by catalyzing disulfide bond formation and isomerization reactions. Furthermore, the PDI-like proteins participate in the ER quality control process, in which they act to refold misfolded substrates or to unfold them in preparation for removal from the ER (4). Hence, the ability of PDI-like proteins to perform a variety of functions coupled with the diversity of substrates on which they act likely contribute to the tendency of PDI-like proteins to be co-opted by viruses and toxins during entry.
Acknowledgments
B.T. is a Burroughs Wellcome Fund Investigator in Pathogenesis of Infectious Disease and is supported by the NIH (RO1-AI064296).
Footnotes
Published ahead of print on 19 November 2008.
REFERENCES
- 1.Barnewitz, K., C. Guo, M. Sevvana, Q. Ma, G. M. Sheldrick, H. D. Söling, and D. M. Ferrari. 2004. Mapping of a substrate binding site in the protein disulfide isomerase-related chaperone Wind based on protein function and crystal structure. J. Biol. Chem. 27939829-39837. [DOI] [PubMed] [Google Scholar]
- 2.Baryshev, M., E. Sargsyan, and S. Mkrtchian. 2006. ERp29 is an essential endoplasmic reticulum factor regulating secretion of thyroglobulin. Biochem. Biophys. Res. Commun. 340617-624. [DOI] [PubMed] [Google Scholar]
- 3.Brady, J. N., V. D. Winston, and R. A. Consigli. 1978. Characterization of a DNA-protein complex and capsomere subunits derived from polyoma virus by treatment with ethyleneglycol-bis-N,N′-tetraacetic acid and dithiothreitol. J. Virol. 27193-204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Ellgaard, L., and A. Helenius. 2003. Quality control in the endoplasmic reticulum. Nat. Rev. Mol. Cell Biol. 4181-191. [DOI] [PubMed] [Google Scholar]
- 5.Ferrari, D. M., P. Nguyen Van, H. D. Kratzin, and H. D. Söling. 1998. ERp28, a human endoplasmic-reticulum-lumenal protein, is a member of the protein disulfide isomerase family but lacks a CXXC thioredoxin-box motif. Eur. J. Biochem. 255570-579. [DOI] [PubMed] [Google Scholar]
- 6.Forster, M. L., K. Sivick, Y. N. Park, P. Arvan, W. I. Lencer, and B. Tsai. 2006. Protein disulfide isomerase-like proteins play opposing roles during retrotranslocation. J. Cell Biol. 173853-859. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Gilbert, J., and T. Benjamin. 2004. Uptake pathway of polyomavirus via ganglioside GD1a. J. Virol. 7812259-12267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Gilbert, J. M., W. Ou, J. Silver, and T. Benjamin. 2006. Downregulation of protein disulfide isomerase inhibits infection by the mouse polyomavirus. J. Virol. 8010868-10870. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Liepinsh, E., M. Baryshev, A. Sharipo, M. Ingelman-Sundberg, G. Otting, and S. Mkrtchian. 2001. Thioredoxin fold as homodimerization module in the putative chaperone ERp29: NMR structures of the domains and experimental model of the 51 kDa dimer. Structure 9457-471. [DOI] [PubMed] [Google Scholar]
- 10.Lippert, U., D. Diao, N. N. Barak, and D. M. Ferrari. 2007. Conserved structural and functional properties of D-domain containing redox-active and -inactive protein disulfide isomerase-related protein chaperones. J. Biol. Chem. 28211213-11220. [DOI] [PubMed] [Google Scholar]
- 11.Ma, Q., C. Guo, K. Barnewitz, G. M. Sheldrick, H. D. Söling, I. Usón, and D. M. Ferrari. 2003. Crystal structure and functional analysis of Drosophila Wind, a protein-disulfide isomerase-related protein. J. Biol. Chem. 27844600-44607. [DOI] [PubMed] [Google Scholar]
- 12.Magnuson, B., E. K. Rainey, T. Benjamin, M. Baryshev, S. Mkrtchian, and B. Tsai. 2005. ERp29 triggers a conformational change in polyomavirus to stimulate membrane binding. Mol. Cell 28289-300. [DOI] [PubMed] [Google Scholar]
- 13.Mkrtchian, S., M. Baryshev, O. Matvijenko, A. Sharipo, T. Sandalova, G. Schneider, and M. Ingelman-Sundberg. 1998. Oligomerization properties of ERp29, an endoplasmic reticulum stress protein. FEBS Lett. 431322-326. [DOI] [PubMed] [Google Scholar]
- 14.Mkrtchian, S., C. Fang, U. Hellman, and M. Ingelman-Sundberg. 1998. A stress-inducible rat liver endoplasmic reticulum protein, ERp29. Eur. J. Biochem. 251304-313. [DOI] [PubMed] [Google Scholar]
- 15.Rainey-Barger, E. K., B. Magnuson, and B. Tsai. 2007. A chaperone-activated nonenveloped virus perforates the physiologically relevant endoplasmic reticulum membrane. J. Virol. 8112996-13004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Rainey-Barger, E. K., S. Mkrtchian, and B. Tsai. 2007. Dimerization of ERp29, a PDI-like protein, is essential for its diverse functions. Mol. Biol. Cell 181253-1260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Sargsyan, E., M. Baryshev, L. Szekely, A. Sharipo, and S. Mkrtchian. 2002. Identification of ERp29, an endoplasmic reticulum lumenal protein, as a new member of the thyroglobulin folding complex. J. Biol. Chem. 27717009-17015. [DOI] [PubMed] [Google Scholar]
- 18.Schelhaas, M., J. Malmström, L. Pelkmans, J. Haugstetter, L. Ellgaard, K. Grünewald, and A. Helenius. 2007. Simian virus 40 depends on ER protein folding and quality control factors for entry into host cells. Cell 131516-529. [DOI] [PubMed] [Google Scholar]
- 19.Sevvana, M., M. Biadene, Q. Ma, C. Guo, H. Söling, G. M. Sheldrick, and D. M. Ferrari. 2006. Structural elucidation of the PDI-related chaperone Wind with the help of mutants. Acta Cryst. D 62589-594. [DOI] [PubMed] [Google Scholar]
- 20.Smith, A. E., H. Lilie, and A. Helenius. 2003. Ganglioside-dependent cell attachment and endocytosis of murine polyomavirus-like particles. FEBS Lett. 555199-203. [DOI] [PubMed] [Google Scholar]
- 21.Stehle, T., Y. Yan, T. L. Benjamin, and S. C. Harrison. 1994. Structure of murine polyomavirus complexed with an oligosaccharide receptor fragment. Nature 369160-163. [DOI] [PubMed] [Google Scholar]
- 22.Tsai, B., C. Rodighiero, W. I. Lencer, and T. A. Rapoport. 2001. Protein disulfide isomerase acts as a redox-dependent chaperone to unfold cholera toxin. Cell 104937-948. [DOI] [PubMed] [Google Scholar]
- 23.Tsai, B., J. M. Gilbert, T. Stehle, W. Lencer, T. L. Benjamin, and T. A. Rapoport. 2003. Gangliosides are receptors for murine polyoma virus and SV40. EMBO J. 224346-4355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Tsai, B. 2007. Penetration of nonenveloped viruses into the cytoplasm. Annu. Rev. Cell Dev. Biol. 2323-43. [DOI] [PubMed] [Google Scholar]