

Abstract
Retroviral elements are found in abundance throughout the human genome but only rarely have alterations of endogenous genes by retroviral insertions been described. Herein we report that a human endogenous retrovirus (HERV) type C is inserted in the human growth factor gene pleiotrophin (PTN) between the 5′ untranslated and the coding region. This insert in the human genome expands the region relative to the murine gene. Studies with promoter-reporter constructs show that the HERV insert in the human PTN gene generates an additional promoter with trophoblast-specific activity. Due to this promoter function, fusion transcripts between HERV and the open reading frame of PTN (HERV-PTN) were detected in all normal human trophoblast cell cultures as early as 9 weeks after gestation (n = 7) and in all term placenta tissues (n = 5) but not in other normal adult tissues. Furthermore, only trophoblast-derived choriocarcinoma cell lines expressed HERV-PTN mRNA whereas tumor cell lines derived from the embryoblast (teratocarcinoma) or from other lineages failed to do so. We investigated the significance of HERV-PTN mRNA in a choriocarcinoma model by targeting this transcript with ribozymes and found that the depletion of HERV-PTN mRNA prevents human choriocarcinoma growth, invasion, and angiogenesis in mice. This suggests that the tissue-specific expression of PTN due to the HERV insertion in the human genome supports the highly aggressive growth of human choriocarcinoma and possibly of the human trophoblast.
Pleiotrophin (PTN) is a secreted heparin-binding polypeptide growth factor (1) with mitogenic (1–3) and transforming effects (3) on fibroblasts and growth factor activity on epithelial (2, 4, 5) and endothelial (2, 5, 6) cells. Furthermore, PTN induces the release of proteolytic enzymes from endothelial cells (7) and stimulates neurite outgrowth (8) and tube formation by endothelial cells in vitro (5) as well as angiogenesis in the rabbit corneal pocket assay (6). PTN gene expression is regulated in a time- and tissue-specific manner during rodent development and PTN mRNA is found at high levels in the central nervous system during the perinatal period, is down-regulated thereafter, and is present at low levels in a few adult tissues (1, 9–11). On the other hand, the PTN gene is up-regulated in several human tumor tissues and tumor cell lines (2) but little is known about the regulatory elements in this gene (12).
To understand the mechanisms that regulate expression of the human PTN gene, we studied 5′ ends of PTN mRNA isolated from various human tissues that express the PTN gene. To our surprise we found that all placenta samples, in contrast to brain, expressed PTN mRNA with 5′ exons that are homologous to a human endogenous retrovirus (HERV) and are spliced onto the intact open reading frame (ORF) of PTN. Upon analysis of human genomic DNA, we located the insertion of an HERV fragment into the intron region upstream of the ORF of the human PTN gene expanding this region relative to the ancestral PTN gene shared with other nonprimate species.
HERV-like particles were identified more than two decades ago in human oocytes, teratocarcinoma cells, and mammary carcinoma tissues, as well as in placenta, and retroviral transcripts were detected in a number of tissues and cell lines (13–17). These findings reflect the high number of HERV fragments found integrated throughout the human genome (several thousand copies). However, only one example of HERV germ-line insertion that induces changes of the expression pattern of a functional human gene product has been reported to date (18, 19): a C-type HERV was found integrated in reverse orientation into the 5′ flanking region of human amylase genes and was shown to function as an enhancer that confers additional salivary gland expression of amylase.
In the present paper, we demonstrate that germ-line insertion of an HERV fragment generates a phylogenetically new promoter within the human PTN gene. This insertion confers trophoblast-specific expression of functional PTN gene products. We evaluate the significance of this finding for the growth phenotype of human trophoblast-derived choriocarcinoma and discuss potential implications for the growth of the normal human trophoblast.
MATERIALS AND METHODS
Cell Lines and Growth Assays.
Human choriocarcinoma (JEG-3 and JAR), teratocarcinoma (PA-1), and adrenal carcinoma (SW-13) cells were from the American Type Culture Collection and were grown in Iscove’s modified medium (IMEM) with 10% fetal calf serum (FCS; Life Technologies, Gaithersburg, MD); human melanoma cells (1205LU; gift from M. Herlyn, Wistar Institute, Philadelphia) in KSFM/L15 medium mixed at a ratio of 3:1 (Life Technologies) and supplemented with 5% FCS. Primary cells grown from chorionic villus samples obtained for prenatal diagnostics were a gift of J. Simon (Georgetown University) and were kept in IMEM with 20% FCS. To determine the proliferation rates of differently modified JEG-3 cells, 2 × 104 cells were plated in triplicates into six-well plates and the number of cells was counted at different time intervals.
Gene Structure Analysis.
Nonoverlapping phagemid P1 clones (Genome Systems, St. Louis) were used to complete the structural analysis of the 5′ untranslated region (5′-UTR) of the human PTN gene reported earlier (20, 21). P1 clones containing the upstream untranslated exon U1 (P203) or the first exon of the ORF, O1 (P2258), were obtained by PCR screening with specific primers. Long-range PCR (Expand long template PCR, Boehringer Mannheim) with P1 clone 2258 (containing O1) or with human genomic DNA and subcloning and sequencing of inserts was used to compile the structure of the human PTN gene. The nucleic acid sequence of the inserted promoter region and of HERV-derived exons UV3 and in part of UV2 were obtained from a genomic DNA fragment subcloned from a BamHI restriction library of P1 clone P2258 (containing O1), which was screened for positive clones with a UV3-specific probe. Furthermore, the 5′ rapid amplification of cDNA ends (RACE) PCR products of placenta cDNA were sequenced for comparison and to complement data from the genomic cloning.
5′-RACE PCR.
Two 5′-RACE cDNA libraries generated from human placenta and human brain mRNA (CLONTECH) served to generate PCR fragments of the 5′ ends of different PTN cDNAs. Nested primers derived from exon O1 were used as 3′ primers in the PCR, and an antisense oligonucleotide to the anchor sequence was used as a 5′ primer. PCR fragments were subcloned into TA-cloning vectors (Invitrogen) and inserts were sequenced.
Mapping of the Transcription Start Site by Primer Extension.
The avian myeloblastosis virus (AMV) reverse transcriptase primer extension system (Promega) was used with poly(A)+ RNA (9 μg) or total RNA (50 μg) as a template. RNA from JEG-3 and JAR choriocarcinoma cells (PTN-positive) or SW-13 cells (PTN-negative) was incubated with two UV3-specific nested primers designed to hybridize to the sequence stretch between positions −11154 and −11170 and positions −11168 and −11184, respectively (see Fig. 1B). After denaturation of the RNA for 30 min at 65°C, the primer hybridization reaction was incubated for 1 h at 52°C followed by a 1-h incubation at 42°C with AMV reverse transcriptase. The samples were then heated for 10 min at 90°C in formamide loading buffer and analyzed on a 6% sequencing gel. Sequencing reactions with each of the primers were used to read the position of the extended product.
Figure 1.
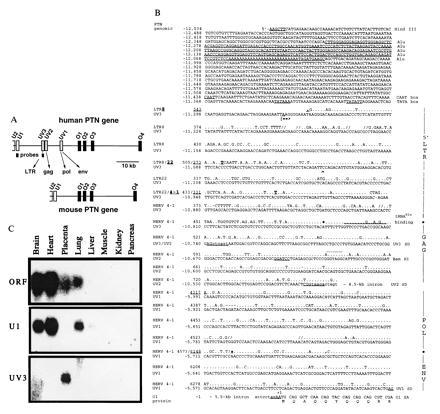
Insertion of HERV into the human PTN gene and tissue-specific distribution of HERV-PTN fusion transcripts. (A) Comparison of the genomic organization of the human and murine PTN genes. Homologous exons corresponding to the ORF (O1 to O4) or 5′-UTR (U2, U1) of the murine (24) and the human gene (20, 21), as well as the HERV-derived exons (UV3, UV2, and UV1) and the position of exon-specific probes, are shown. (B) Sequence comparison between the HERV insert in the human PTN gene and type C HERV fragments LTR8, LTR22, and HERV4-1 (22, 23) using the GCG program. Gaps (//) were introduced for optimal alignment. The PTN gene numbering system (GenBank accession nos. U71455U71455 and U71456U71456) on the left refers to the first nucleotide of the translation start codon as +1. The viral sequence positions were taken from the respective GenBank entries for LTR8, LTR22, and HERV4-1 (accession nos. M32219M32219, M32220M32220, and K02168K02168, respectively). A 280-nt Alu sequence in the promoter region and CAAT and TATA boxes are underlined and intronic sequence is denoted by lowercase type. The transcription start site (|==>) was mapped by primer extension. Similarity to retroviral elements is indicated on the right and the respective nucleotide positions separating the different elements are highlighted in the sequence. The tRNAGlu primer binding site, UV3 cDNA end (∧), restriction sites used for generation of the promoter-reporter constructs, and the N-terminal sequence of the PTN protein are shown. The splice donor (SD) sites of UV3, UV2, and UV1, as well as the splice acceptor (SA) in exon O1, are underlined and the retroviral SD of HERV 4–1 is indicated in addition (by dots above the sequence). (C) Northern blot analysis of mRNA from various human tissues using exon-specific probes.
Transcriptional Activity.
A 1.9-kb HindIII–BamHI (H, B) genomic fragment from P1 clone P2258 was used for these studies. This fragment starts upstream of the Alu region (position −12,534) and contains the TATA box and transcription start site of the HERV-PTN fusion transcripts and ends in exon UV2 (positions −10,640) (see Fig. 1). The fragment was cloned in both orientations into the pXP-1 promoterless luciferase reporter gene vector (25) and then used in transient transfection assays in different cell lines. For this, cells were plated overnight at 60–70% confluence in six-well plates and then transfected in Optimem (Life Technologies) with 1 μg of DNA per well using 7 μl of LipofectAmine (Life Technologies) for the JEG-3, JAR, and SW-13 cells and 2.5 μl of Transfectam (Pharmacia) for the 1205LU cells. After 5 h, transfection medium was replaced by fresh culture medium and the cells were incubated for another 24–36 h. Thereafter cells were harvested, washed, lysed in 0.25 M Tris·HCl (pH 7.8), and freeze–thawed three times, and 5–20 μl of the lysate was mixed with 350 μl of 0.1 M potassium phosphate/15 mM MgCl2/5 mM ATP at pH 7.8 and assayed for luciferase activity using 1 mM d-luciferin as a substrate. The promoter activity is shown as fold induction relative to the parent vector pXP-1. A cytomegalovirus-driven luciferase expression vector was used to control for transfection efficacy.
Depletion of PTN mRNA by Using Ribozyme Targeting.
The PTN-targeted ribozyme Rz261 (26) was expressed under the control of the tTA/heptameric operator binding site and a cytomegalovirus minimal promoter (27). For this purpose, the major portion of the luciferase gene and the simian virus 40 polyadenylylation site in the pUHC13-3 plasmid (27) were deleted by HindIII/HpaI digestion and replaced with the Rz261/bovine growth hormone polyadenylylation HindIII–PvuII fragment from the pRc/Rz261 expression vector (26). The remaining luciferase start codon was replaced by a SalI/ClaI/HindIII cassette to yield the construct pTET/Rz261. This ribozyme is designed to cleave PTN mRNA 3′ of nt 261 of the ORF (26). In JEG-3 cells, the ribozyme expression vector (pTET/Rz261, 0.5 μg), was cotransfected with the tTA expression vector [pUHG15-1 (27), 0.5 μg] and pRc/CMV (0.1 μg) to provide G418 resistance. After selection for stable integrants in the presence of G418 at 1 mg/ml, the cells were tested for PTN expression by Northern blot analysis.
Northern Blot Analysis.
Total RNA from cell lines or tissues was isolated with the RNA STAT-60 method (Tel-Test, Friendswood, TX), separated, and blotted as reported (2). In addition, a human multiple tissue Northern blot (CLONTECH) was used. PTN cDNA probes specific for the ORF (2) or 5′ untranslated exon U1 (287-nt fragment) or HERV-derived exon UV3 (257-nt fragment) were hybridized, washed, and autoradiographed for 48 h as described (2). After exposure, blots were stripped and reprobed. Glyceraldehyde-3-phosphate dehydrogenase (GAPDH) was used as a loading control.
RESULTS
Insertion of Retroviral Elements into the Human PTN Gene.
To elucidate the mechanisms that regulate expression of the human PTN gene, we examined the 5′ regions of mRNAs isolated from placenta and adult brain by 5′-RACE PCR. In particular, PTN expressed in placental tissues appeared of interest to us, since placenta is mostly derived from fetal tissues and a human cDNA clone was originally reported from the screening of a placental library (1). Furthermore, in situ hybridization (9) as well as Northern blot analysis (unpublished data) with rodent trophoblast tissues had failed to detect a signal for PTN in contrast to a strong signal in Northern blots with human placenta (see below). To our surprise, 10 of 11 5′-RACE PCR clones with mRNA from placenta contained novel 5′-UTRs that are distinct from the previously described 5′-UTR in human placental and brain cDNAs.
Sequence comparisons revealed that the novel 5′ exons contained in the PTN mRNA from placenta are highly homologous to different regions of HERV type C (22, 23, 28). Analysis of human genomic DNA revealed that the HERV fragment is inserted in sense orientation into the intron region immediately upstream of the ORF of the human PTN gene expanding this region relative to the murine gene (Fig. 1A). Low-stringency Southern blot analysis confirmed insertion of HERV also in the rhesus monkey genome and showed the lack thereof in murine genomic DNA (unpublished data). The most 5′ HERV-derived PTN exon (UV3) is homologous to the viral 5′ long terminal repeat region and the downstream UV2 and UV1 exons are homologous to regions of the HERV gag, pol, and env pseudogenes (70, 85, and 80% identity, respectively; Fig. 1B) (22, 23). Unlike infectious C-type viruses encountered today, all of which contain a tRNAPro primer binding site in their DNA (22), this prehistoric virus contains a tRNAGlu primer binding site as its signature (Fig. 1B).
Expression of HERV-PTN Fusion Transcripts and of PTN Protein.
The three HERV-derived exons are directly spliced to the first exon in the ORF of the PTN gene (O1) using splice donor sites distinct from the site reported in the retrovirus (Fig. 1B) (22). The HERV-PTN fusion transcripts were present in placental cDNA at a ratio of 8:1:1 for UV3, UV2, and UV1, respectively, based on the number of clones obtained from the 5′-RACE PCR analysis. Exon U1 spliced to O1 was found in only 1 of 11 placental 5′-RACE PCR clones. This predominant expression of the HERV-derived exons in placenta contrasted with the lack of expression of these exons in brain; all seven 5′-RACE PCR clones obtained from brain contained only exon U1 directly spliced to O1. Northern blot analysis with exon-specific probes confirmed this striking difference between placenta and brain (Fig. 1C). Other retroviral transcripts reported from placental tissues (17) did not show cross-hybridization signals.
We detected HERV-PTN fusion transcripts not only in human term placenta tissue (n = 5) but also in the trophoblast of a fetus stillborn after 15 weeks of gestation, in primary cultures of cells grown from trophoblast biopsies obtained for prenatal diagnostics (9–12 weeks of gestation; n = 7), and in trophoblast-derived human choriocarcinoma cell lines (Fig. 2A). In addition to Northern blot analysis, RNase protection studies and reverse transcription-coupled PCR confirmed the presence of HERV-PTN fusion transcripts and the lack of U1 exon usage in JEG-3 and JAR choriocarcinoma cells (data not shown). In contrast to the use of HERV-derived 5′ exons in the trophoblast and choriocarcinoma, PTN mRNA from other embryonic or adult tissues and tumor cells contained U1 as their first 5′-UTR exon (Figs. 1C and 2A), implying that these transcripts originate from the promoter upstream of U1 (12).
Figure 2.
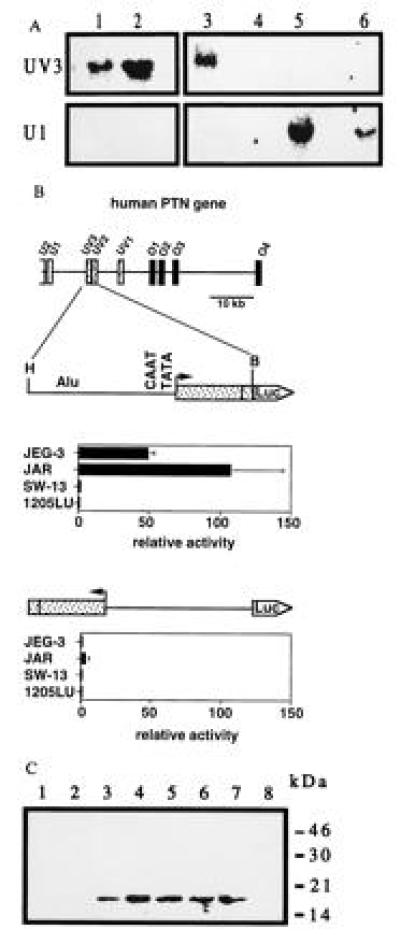
Expression of the PTN gene in various human cell lines and promoter activity of the HERV insert. (A) Northern blot analysis of total RNA from choriocarcinoma cells JEG-3 and JAR (lanes 1 and 2), a chorion biopsy from a stillborn fetus (lane 3), adrenal carcinoma SW-13 (lane 4), melanoma 1205LU (lane 5), and teratocarcinoma PA-1 cells (lane 6) using exon-specific probes. (B) Transcriptional activity of a HindIII–BamHI (H, B) genomic fragment inserted into the promoterless pXP-1 reporter vector (25) in sense and antisense orientation. Data are the mean ± SD of triplicate determinations and are representative of at least two transient transfection experiments for each cell line. Luciferase activity, normalized to the protein content, is expressed relative to that obtained with the pXP-1 vector. Western blot for PTN present in the culture medium of JEG-3 cells. Proteins present in medium conditioned by JEG-3 cells were concentrated and partially purified by heparin-affinity chromatography using a NaCl step gradient of 0.9 to 1.5 M (lanes 1–8) and analyzed as described (4).
Western blot analysis showed that the PTN protein is produced and released into the culture medium by choriocarcinoma cells (Fig. 2C). This protein was mitogenically active and stimulated colony formation of an indicator cell line, SW-13 cells (2, 4) (data not shown).
Promoter Function Due to the HERV Insertion.
Primer extension with primers targeted within the most 5′ UV3 exon of the HERV insert mapped the start site of the HERV-PTN transcripts to an adenosine residue 39 nt downstream of putative CAAT and TATA boxes (see Fig. 1B). Presence of these elements at the transcription start site suggested that the HERV insertion might have generated an additional promoter in the intron immediately upstream of the coding region of the human PTN gene. To investigate whether this putative promoter was responsible for the trophoblast-specific expression of HERV-PTN, we performed transient transfection assays with promoter-reporter constructs. Upstream of a luciferase reporter gene, we inserted a genomic fragment that starts 1.5 kb upstream of the transcription start site, contains the CAAT and TATA boxes, and extends downstream of the start site into exon UV2 (Figs. 1B and 2B). Transcriptional activity of the resulting construct was observed exclusively in human choriocarcinoma cells (JEG-3 and JAR cells) and only when the HERV insert was oriented as in vivo. Luciferase activity in JEG-3 and JAR cells transfected with the HERV promoter-reporter construct in sense orientation was 50- to 100-fold that of cells transfected with the promoterless vector alone. Deletion of the Alu element did not affect this transcriptional activity (data not shown). Only background activity was detected in PTN-positive human melanoma (1205LU) or in PTN-negative adrenal carcinoma (SW-13) cells (Fig. 2B). We conclude from these data that the HERV insertion in the human PTN gene generates a functional promoter that confers high tissue-specific expression.
Depletion of HERV-PTN with Ribozymes to Evaluate its Biological Role.
We investigated the biological significance of the expression of HERV-PTN in trophoblast-derived tissues using JEG-3 choriocarcinoma cells as a model system. Tumor growth of these cells in experimental animals mimics the highly invasive and angiogenic growth phenotype of the normal human trophoblast (29) and clinical choriocarcinoma, and we hypothesized that one of the contributing factors to this phenotype could be the expression of HERV-PTN. To address this hypothesis, we examined the effects of reducing the abundance of HERV-PTN transcripts in JEG-3 cells by stable expression of a PTN-targeted ribozyme (26). A vector (pTET/Rz261) with high transcriptional activity in these cells (unpublished data) was used to express the ribozyme.
Northern blot analysis revealed that ribozyme expression reduced the amount of HERV-PTN mRNA in JEG-3 cells to background levels (Fig. 3A). No difference in the proliferation rate of PTN-depleted versus control cells was apparent in vitro (Fig. 3B), suggesting that the cells do not require PTN as an autocrine growth factor even though they secrete the protein in a biologically active form (see above). However, a marked difference in the growth phenotype of PTN-depleted versus control cells was observed after xenografting tumor cells into athymic nude mice. In an initial study, we implanted the tumor cells into their “natural” intraabdominal environment to observe their orthotopic growth behavior. The control cells formed large tumor masses that invaded the abdominal organs within 2–3 weeks, whereas only a few small seedings of PTN-depleted tumor cells were detected in the abdomen at the end of the study (n = 5 and n = 4 animals, respectively). Parallel results were obtained after subcutaneous injection of tumor cells. In contrast to control cells, which grew rapidly into highly angiogenic tumors (n = 7; Figs. 3C and 4 A and B), no tumor growth was observed with PTN-depleted cells (n = 9; Figs. 3C and 4 C and D). These observations indicate that PTN is an essential and rate-limiting factor for choriocarcinoma growth, invasion, and angiogenesis in vivo.
Figure 3.
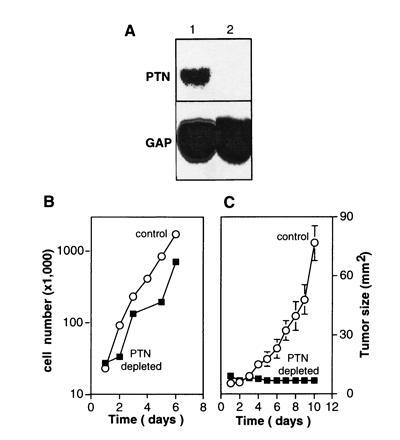
Effect of the depletion of HERV-PTN mRNA from JEG-3 choriocarcinoma cells. (A) Northern blot analysis of total RNA from control JEG-3 cells (lane 1) and JEG-3 cells expressing a ribozyme targeted to PTN mRNA (lane 2) is shown. (B) Proliferation in vitro of control and PTN mRNA-depleted JEG-3 cells. (C) Growth curves of subcutaneous tumors in athymic nude mice. Two million control or PTN mRNA-depleted JEG-3 cells were inoculated. The data represent tumor sizes (mean ± SD).
Figure 4.
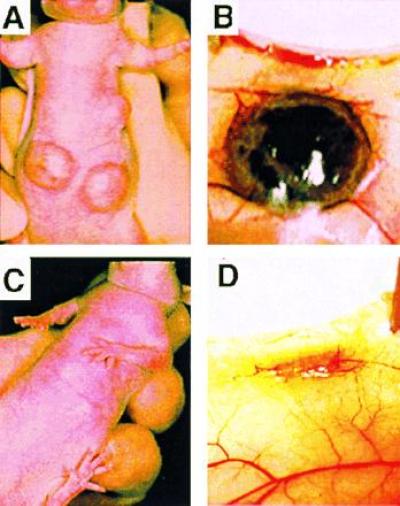
Representative subcutaneous tumors grown from two million control (A and B) or PTN mRNA-depleted (C and D) JEG-3 cells 2 weeks after injection of cells into athymic nude mice.
DISCUSSION
We report herein that integration of an HERV element into the human PTN gene generates a novel tissue-specific promoter not present in the common ancestral gene from which the primate and murine PTN genes descended. Transcriptional activity of this promoter results in the expression of HERV-PTN fusion transcripts specifically in human trophoblast-derived normal and tumor cells. The integration site of the HERV fragment shows several features indicative of inserted retroelements, i.e., location at the 5′ region of a gene (30), presence of an Alu sequence of 280 nt and of a stretch of 26 adenosine residues at its 5′ end (16) (Fig. 1B).
The HERV found in the human PTN gene represents a noninfective replication-defective prototype of a retrovirus (22) that integrated into ancestral DNA before the divergence of apes (including human predecessors) and Old World monkeys more than 25 million years ago (31). It is quite surprising that altered expression of human genes due to the insertion of retroviral elements appears to be an extremely rare event (see Introduction), bearing in mind the high number of HERV fragments found integrated throughout the human genome (several thousand copies) (16, 18, 19). On the other hand, an alteration of a gene expression pattern will only penetrate during phylogenesis, if a better survival chance is associated. Our functional studies show that ribozyme-mediated depletion of HERV-PTN mRNA in human choriocarcinoma cells reverses their highly aggressive growth phenotype in an in vivo model. From this it is tempting to speculate that the expression of HERV-PTN could be one of the factors that enhances the invasive growth phenotype also of the normal human trophoblast and that this was advantageous during phylogenesis. In line with this speculation, the superficial less-invasive implantation of the murine trophoblast (29) coincides with the lack of trophoblast-specific PTN gene expression.
Formation of the trophoblast is one of the early differentiation events occurring in the developing embryo after it organizes into an inner and outer cell mass; the trophoblast evolves during the first weeks from the outer cell mass. Interestingly, teratocarcinoma (PA-1) cells that stem from the undifferentiated embryoblast (32), as well as adult tissues and tumor cells derived from different germ layers, utilize the PTN promoter that is located in a region in common with the murine gene (i.e., upstream of U1; Figs. 1 A and C and 2A; ref. 12). In contrast, the phylogenetically novel HERV-PTN transcription unit is active in human trophoblast-derived normal and tumor cells and tissues, and our data suggest that activation of this promoter occurs early during the formation of the trophoblast.
In conclusion, a distinct role of retroviral integration into somatic cells for the development of solid tumors or leukemia was demonstrated in rodents (mouse mammary tumor virus and Moloney murine leukemia virus), birds (avian leukosis virus), and cats (feline leukemia virus) (33). We show herein that trophoblast-specific expression of the human PTN gene is due to the germ-line insertion of a retroviral fragment that generates a phylogenetically novel promoter in the human gene. The resulting gene products appear to be responsible for the aggressive and invasive growth phenotype of human choriocarcinoma and possibly also of the human trophoblast.
Acknowledgments
We thank K. Alrustamani, A. Jaetsch, and V. Harris for help with the experiments; M. Hughes (National Institutes of Health Genome Center), K. Smith (National Cancer Institute), M. Avigan, H. J. List, and R. Schlegel (Georgetown University) for comments and suggestions; K. W. Brocklehurst (Washington, DC) for editorial assistance; J. Simon and his colleagues (Georgetown University) for providing tissue samples and cultured cells; H. Bujard (Heidelberg, Germany) for the plasmids pUHC13-3 and pUHG15-1. The studies were supported in part by Special Program of Research Excellence Grants CA58185 from the National Cancer Institute and DAMD 17–94-J-4445 from the U.S. Army Medical Research Material Command Breast Cancer Program to A.W.
Footnotes
References
- 1.Li Y S, Milner P G, Chauhan A K, Watson M A, Hoffman R M, Kodner C M, Milbrandt J, Deuel T F. Science. 1990;250:1690–1694. doi: 10.1126/science.2270483. [DOI] [PubMed] [Google Scholar]
- 2.Fang W J, Hartmann N, Chow D, Riegel A T, Wellstein A. J Biol Chem. 1992;267:25889–25897. [PubMed] [Google Scholar]
- 3.Chauhan A K, Li Y S, Deuel T F. Proc Natl Acad Sci USA. 1993;90:679–682. doi: 10.1073/pnas.90.2.679. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Wellstein A, Fang W J, Khatri A, Lu Y, Swain S S, Dickson R B, Sasse J, Riegel A T, Lippman M E. J Biol Chem. 1992;267:2582–2587. [PubMed] [Google Scholar]
- 5.Laaroubi K, Delbe J, Vacherot F, Desgranges P, Tardieu M, Jaye M, Barritault D, Courty J. Growth Factors. 1994;10:89–98. doi: 10.3109/08977199409010982. [DOI] [PubMed] [Google Scholar]
- 6.Courty J, Dauchel M C, Caruelle D, Perderiset M, Barritault D. Biochem Biophys Res Commun. 1991;180:145–151. doi: 10.1016/s0006-291x(05)81267-7. [DOI] [PubMed] [Google Scholar]
- 7.Kojima S, Inui T, Muramatsu H, Kimuara H, Sakakibara S, Muramatsu T. Biochem Biophys Res Commun. 1995;216:574–581. doi: 10.1006/bbrc.1995.2661. [DOI] [PubMed] [Google Scholar]
- 8.Rauvala H. EMBO J. 1989;8:2933–2941. doi: 10.1002/j.1460-2075.1989.tb08443.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Vanderwinden J M, Mailleux P, Schiffmann S N, Vanderhaeghen J J. Anat Embryol. 1992;186:387–406. doi: 10.1007/BF00185989. [DOI] [PubMed] [Google Scholar]
- 10.Böhlen P, Kovesdi I. Prog Growth Factor Res. 1991;3:143–157. doi: 10.1016/s0955-2235(05)80005-5. [DOI] [PubMed] [Google Scholar]
- 11.Kurtz A, Schulte A M, Wellstein A. Crit Rev Oncog. 1995;6:151–177. [PubMed] [Google Scholar]
- 12.Li Y S, Hoffman R M, Le Beau M M, Espinosa R, Jenkins N A, Gilbert D J, Copeland N G, Deuel T F. J Biol Chem. 1992;267:26011–26016. [PubMed] [Google Scholar]
- 13.Kalter S S, Helmke R J, Heberling R L, Panigel M, Fowler A K, Strickland J E, Hellman A. J Natl Cancer Inst. 1973;50:1081–1084. doi: 10.1093/jnci/50.4.1081. [DOI] [PubMed] [Google Scholar]
- 14.Kalter S S, Helmke R J, Panigel M, Heberling R L, Felsburg P J, Axelrod L R. Science. 1973;179:1332–1333. doi: 10.1126/science.179.4080.1332. [DOI] [PubMed] [Google Scholar]
- 15.Kalter S S, Heberling R L, Smith G C, Helmke R J. J Natl Cancer Inst. 1975;55:735–736. doi: 10.1093/jnci/55.3.735. [DOI] [PubMed] [Google Scholar]
- 16.Wilkinson D A, Mager D L, Leong J A C. In: The Retroviridiae. Levy J A, editor. New York: Plenum; 1994. pp. 465–535. [Google Scholar]
- 17.Loewer R, Loewer J, Kurth R. Proc Natl Acad Sci USA. 1996;93:5177–5184. doi: 10.1073/pnas.93.11.5177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Samuelson L C, Wiebauer K, Snow C M, Meisler M H. Mol Cell Biol. 1990;10:2513–2520. doi: 10.1128/mcb.10.6.2513. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Ting C N, Rosenberg M P, Snow C M, Samuelson L C, Meisler M H. Genes Dev. 1992;6:1457–1465. doi: 10.1101/gad.6.8.1457. [DOI] [PubMed] [Google Scholar]
- 20.Lai S, Schulte A M, Wellstein A, Riegel A T. Gene. 1995;153:301–302. doi: 10.1016/0378-1119(94)00674-h. [DOI] [PubMed] [Google Scholar]
- 21.Lai S, Czubayko F, Riegel A T, Wellstein A. Biochem Biophys Res Commun. 1992;187:1113–1122. doi: 10.1016/0006-291x(92)91312-e. [DOI] [PubMed] [Google Scholar]
- 22.Repaske R, Steele P E, O’Neill R R, Rabson A B, Martin M A. J Virol. 1985;54:764–772. doi: 10.1128/jvi.54.3.764-772.1985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Tomita N, Horii A, Doi S, Yikouchi H, Ogawa M, Mori T, Matsubara K. Biochem Biophys Res Commun. 1990;166:1–10. doi: 10.1016/0006-291x(90)91904-7. [DOI] [PubMed] [Google Scholar]
- 24.Katoh K, Takeshita S, Sato M, Ito T, Amann E. DNA Cell Biol. 1992;11:735–743. doi: 10.1089/dna.1992.11.735. [DOI] [PubMed] [Google Scholar]
- 25.Nordeen S K. BioTechniques. 1988;6:454–457. [PubMed] [Google Scholar]
- 26.Czubayko F, Riegel A T, Wellstein A. J Biol Chem. 1994;269:21358–21363. [PubMed] [Google Scholar]
- 27.Gossen M, Bujard H. Proc Natl Acad Sci USA. 1992;89:5547–5551. doi: 10.1073/pnas.89.12.5547. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Steele P E, Rabson A B, Bryan T, Martin M A. Science. 1984;225:943–947. doi: 10.1126/science.6089336. [DOI] [PubMed] [Google Scholar]
- 29.Cross J C, Werb Z, Fisher S J. Science. 1994;266:1508–1518. doi: 10.1126/science.7985020. [DOI] [PubMed] [Google Scholar]
- 30.Rohdewohld H, Weiher H, Reik W, Jaenisch R, Breindl M. J Virol. 1987;61:336–343. doi: 10.1128/jvi.61.2.336-343.1987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Shih A, Coutavas E E, Rush M G. Virology. 1991;182:495–502. doi: 10.1016/0042-6822(91)90590-8. [DOI] [PubMed] [Google Scholar]
- 32.Stewart T A, Mintz B. Proc Natl Acad Sci USA. 1981;78:6314–6318. doi: 10.1073/pnas.78.10.6314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Fan H. In: The Retroviridiae. Levy J A, editor. New York: Plenum; 1994. pp. 313–353. [Google Scholar]