

Abstract
The attenuated pseudorabies virus (PRV) strain Bartha contains several characterized mutations that affect its virulence and ability to spread through neural circuits. This strain contains a small genomic deletion that abrogates anterograde spread and is widely used as a retrograde-restricted neural circuit tracer. Previous studies showed that the retrograde-directed spread of PRV Bartha is slower than that of wild-type PRV. We used compartmented neuronal cultures to characterize the retrograde defect and identify the genetic basis of the phenotype. PRV Bartha is not impaired in retrograde axonal transport, but transneuronal spread among neurons is diminished. Repair of the UL21 locus with wild-type sequence restored efficient transneuronal spread both in vitro and in vivo. It is likely that mutations in the Bartha UL21 gene confer defects that affect infectious particle production, causing a delay in spread to presynaptic neurons and amplification of infection. These events manifest as slower kinetics of retrograde viral spread in a neural circuit.
Pseudorabies virus (PRV) is the causative agent of Aujeszky's disease in swine. It is a member of the Alphaherpesvirus subfamily of the Herpesviridae family, which includes the human pathogens herpes simplex virus and varicella-zoster virus. One facet of the alphaherpesvirus infectious cycle is conditional neuroinvasiveness. Upon initial inoculation of mucosal epithelium, infection spreads to the peripheral neurons innervating the mucosa via retrograde axonal transport. Here, viral latency is established that persists for the lifetime of the host (19). During occasional reactivation from latency, newly replicated viral particles are transported to the original site of infection via anterograde axonal transport, causing a recurring epithelial lesion. Transneuronal spread of infection from the peripheral nervous system to the central nervous system (CNS) is rare in the natural host. Remarkably, in susceptible nonnatural hosts, the spread of alphaherpesviruses almost invariably proceeds to the CNS, with lethal consequences (18, 31).
Because of their broad host range, self-amplifying character, and ability to spread directionally through synaptically connected circuits, alphaherpesviruses have been used successfully as neural circuit tracers (16). The most widely used tracing strains are PRV Bartha and various recombinants expressing reporter genes. The PRV Bartha strain was originally developed as a live vaccine against Aujeszky's disease in swine by serial passage in culture (1). PRV Bartha elicits protective immunity in swine without causing disease (25). Studies to understand the genetic basis of its attenuation and restricted pattern of nervous system infection have yielded considerable information regarding the mechanisms of alphaherpesvirus pathogenicity (20, 22, 27).
Several features make PRV Bartha particularly appealing for neural tracing studies. (i) It is attenuated, which allows the inoculated animals to live longer than those infected with a wild-type PRV strain; the extended survival time, in turn, enables extensive viral spread and labeling of the nervous system (2). (ii) Despite its attenuation, the virus replicates well in tissue culture cells (23). (iii) In vivo and in vitro studies have demonstrated that a small deletion in the unique short region of the genome, encompassing glycoprotein E (gE), gI, and US9 genes, renders the strain incapable of spread from an infected presynaptic cell to a postsynaptic cell (anterograde spread), with little effect on spread from a postsynaptic cell to a presynaptic cell (retrograde spread) (17, 30). Thus, infecting animals with PRV Bartha allows unambiguous interpretation of neural circuit architecture.
While studies performed with PRV Bartha have produced insight into the mechanisms of anterograde spread, no alphaherpesvirus mutants defective in retrograde transport and spread have been identified that do not affect virus replication. Previous in vivo studies have suggested that the kinetics of PRV Bartha retrograde spread through neural circuits are slower than those of the wild-type PRV Becker strain (8, 40). Therefore, we sought to characterize this defect and, by genomic repair, to improve the efficiency of retrograde- directed infection by PRV Bartha.
MATERIALS AND METHODS
Virus strains and cells.
PRV Becker is a laboratory wild-type strain; PRV GS443 encodes green fluorescent protein-tagged VP26 in the PRV Becker background (37). PRV Bartha is an attenuated vaccine strain (1); PRV 765 encodes red fluorescent protein-tagged VP26 in the PRV Bartha background (Ann Raldow, unpublished data). PRV 158 contains the unique long (UL) region of Bartha and the unique short (US) region of Becker (24). PRV BaBe is Becker containing the US deletion of Bartha (10). PRV 43/25 aB4 is Bartha with the wild-type PRV Kaplan sequence restoring BamHI fragment 4 and the US region (22). PRV 326 was constructed for this study and is PRV 43/25 aB4 with the US deletion of Bartha reintroduced. The strain was created by cotransfection of PRV 43/25 aB4 DNA and linearized pGS277, which contains a 9-kb PstI fragment from the Bartha US region, into PK15 cells. The black plaque assay was used to screen for and pick gE nonimmunoreactive plaques. Normal expression of genes upstream and downstream of the deletion was verified by Western blotting (not shown). All strains were propagated in PK15 (porcine kidney) cells, which were purchased from the American Type Culture Collection, and the virus titers were determined in PK15 cells. The cells were maintained in Dulbecco's modified Eagle medium supplemented with 10% fetal bovine serum and penicillin and streptomycin. Viral infections of PK15 cells were performed in Dulbecco's modified Eagle medium supplemented with 2% fetal bovine serum and penicillin and streptomycin.
Neuronal cultures.
Embryonic rat superior cervical ganglia (SCG) were isolated from Sprague-Dawley rats (Hilltop Labs, Inc., Scottdale, PA) on embryonic day 16 and cultured in compartmentalized cultures as described before (11, 12). The neuronal medium was changed every 3 days. All animal work pertaining to SCG dissection was done in accordance with the Institutional Animal Care and Use Committee of the Princeton University Research Board under protocol number 1691.
Antibodies and fluorescent dye.
The hybridoma producing the monoclonal antibody specific for VP5, the major capsid protein, was made by Alex Flood and the Princeton Molecular Biology Department monoclonal antibody facility. The lypophilic dye DiI was purchased from Molecular Probes and used to stain neurons. The dye was added to medium in the neurite compartment (N-compartment) following infections and incubated for 24 h. Alexa secondary fluorophore (Molecular Probes) was used at a dilution of 1:400.
Viral infection of compartmented neurons.
Infections of neurons were performed as described previously (11). To determine the efficiency of retrograde-directed infection of neurons, viral inoculum containing 105 PFU was added to the neurite compartment and adsorbed for 1 h in a humidified incubator at 37°C and 5% CO2. Inoculum was removed, and conditioned neuronal medium was returned to the neurite compartment following the adsorption period. At the appropriate time point, the contents of the soma compartment (S-compartment) were collected by scraping the surface of the dish with a gel-loading tip. To study the efficiency of neuron-to-cell spread of infection, PK15 cells were plated in the neurite compartment of 2-week-old compartmentalized neuronal cultures, and the medium of this compartment was supplemented with 1% fetal bovine serum. PK15 cells formed a confluent monolayer in the N-compartment 24 h after plating. At this point, viral inoculum was added to the soma compartment for 1 hour. Following adsorption, conditioned neuronal medium was returned to the S-compartment. The contents of the neurite and soma compartments were collected separately 24 h after infection.
Immunofluorescence.
Teflon trichambers (Tyler Research) were assembled on UV-sterilized Aclar (EM Sciences) strips; neurons were cultured and infected as described above. At 24 hours postinfection (hpi), all compartments were washed twice with phosphate-buffered saline (PBS) containing 3% bovine serum albumin (BSA) (PBS-BSA), chambers were gently lifted, and silicone grease was scraped off the Aclar strips. Samples were then fixed with 4% paraformaldehyde in PBS for 10 min. Fixative was washed away with three PBS-BSA rinses, after which the samples were permeabilized using 0.5% saponin and 3% BSA in PBS (PBS-BSA-SAP). Incubations with primary and secondary antibodies were performed for 1 hour in PBS-BSA-SAP. Following two rinses with PBS-BSA-SAP and one rinse with distilled water, the samples were mounted on glass slides using Aqua Poly/Mount (Polysciences). Images were taken on an inverted epifluorescence microscope (Nikon Eclipse TE300).
Live-cell imaging.
Dissociated SCG were cultured in glass-bottom dishes (MatTek Corporation, Ashland, MA) and infected with 105 PFU approximately 2 weeks postplating. At 16 hpi, the samples were placed in a humidified live-cell imaging chamber that provides 5% CO2 and constant temperature at 37°C (Live Cell Systems). Movies were captured on a Perkin-Elmer R30 spinning disk confocal microscope using ImageView software. Capsid movement was tracked and analyzed with ImageJ software (National Institutes of Health, Bethesda) using the MTrackJ plugin (created by E. Meijering).
In vivo experiments.
Fourteen adult male Sprague-Dawley rats (Harlan) weighing 300 to 350 g at the time of virus injection were used in the analysis. The experiments were performed in a laboratory dedicated to and approved for biosafety level 2+ experiments. Animals were moved to the facility 2 days prior to the virus injections and lived within the facility throughout the experiment. Photoperiod (12 h light; light on at 0700) and temperature (22 to 25°C) were standardized, and food and water were available ad libitum. The experiments conformed to the regulations mandated in the Guide for the Care and Use of Laboratory Animals (28a) and Biosafety in Microbiological and Biomedical Laboratories (37a). The experimental protocols were approved by the University of Pittsburgh IACUC, the Recombinant DNA Committee, and the Division of Environmental Health and Safety.
Each animal was anesthetized with isoflurane. The abdomen was shaved, and an incision was made through the skin and subjacent musculature. The stomach was gently removed from the peritoneal cavity, and the ventral wall was injected with PRV Bartha (1.7 × 109 PFU/ml) or PRV 326 (2 × 109 PFU/ml). A total of two microliters of virus was injected through three penetrations of the ventral wall of the stomach using a 10-μl Hamilton syringe with a beveled needle. At each of the three sites, the needle was inserted into the stomach wall at the greater curvature, and the tip of the needle was pushed to the hilus under visual guidance. Following injection of virus, the needle was left in place for a minimum of 2 min prior to removal to prevent reflux along the needle tract. We observed no reflux from any of the injection sites. The stomach was then returned to the peritoneal cavity, and the abdominal wall and skin were sutured using 4.0 silk suture. The animals recovered on a heating pad and were then returned to their home cage.
The experiments were terminated by transcardiac perfusion of anesthetized animals 48 (n = 7) or 72 (n = 7) hours following virus injection. Following deep sodium pentobarbital-induced anesthesia, the heart was exposed through a thoracotomy, a canula was inserted into the ascending aorta through the left ventricle, and the right atrium was slit. Approximately 100 ml of physiological saline was then infused under controlled pressure using a peristaltic pump to clear the vasculature of red blood cells. This was followed by infusion of approximately 400 ml of paraformaldehyde-lysine-periodate fixative (26). The brain and spinal cord of each animal were removed and postfixed in paraformaldehyde-lysine-periodate fixative for 2 or 3 days at 4°C, cryoprotected by immersion in 20% phosphate-buffered sucrose, and sectioned using a freezing microtome. The brain was sectioned serially in the coronal plane into six wells of cryoprotectant (39) at 35 μm/section, and the spinal cord was sectioned serially in the horizontal plane into four wells of cryoprotectant at 40 μm/section. Tissue was stored in cryoprotectant at −20°C until immunohistochemical processing to preserve antigenicity.
A minimum of one bin of brain and spinal cord tissue from each animal was processed for immunohistochemical localization of infected neurons using a rabbit polyclonal antiserum raised against acetone-inactivated PRV (Rb133). The specificity of this antiserum for localization of PRV-infected neurons in vivo was documented previously (6). A second bin of brain and spinal cord tissue from each animal was processed for immunocytochemical localization of immune cells of monocytic lineage using a mouse monoclonal antibody generated against the antigen ED1 (14). All antigens were localized using avidin-biotin immunoperoxidase procedures previously described (4). Essential reagents employed for these localizations included affinity-purified secondary antibodies (Jackson ImmunoResearch Laboratories) and the Vectastain Elite ABC kit (Vector Laboratories).
Quantification of viral spread in vivo.
The temporal kinetics of viral invasion of central autonomic circuitry were determined through a quantitative analysis of central cell groups previously shown to be synaptically linked to the parasympathetic (dorsal motor vagal nucleus [DMV]) and sympathetic (intermediolateral [IML] cell column) outflow to the stomach (6, 9, 33, 40). The organization of this circuitry is illustrated schematically (see Fig. 6). Cells were counted in sections through 25 cell groups that contribute to this circuit (see Table S1 in the supplemental material). The sections through each cell group selected for analysis were standardized to ensure comparable comparisons between animals. Cells within each cell group were counted using an image analysis system (StereoInvestigator version 7; Microbrightfield, Inc.) attached to a Nikon Optiphot 2 photomicroscope. The boundaries of each tissue section were traced, the entire section was systematically scanned using the 40× objective, and the position of each infected neuron within each section was recorded. Thus, both the distribution and number of neurons within each cell group were determined for each animal. For each brain region at each survival time (48 and 72 h), Student's t test was used to determine whether statistical differences in the number of infected neurons were produced by infection with PRV Bartha and PRV 326. Differences between groups were considered significant when the P value was ≤0.05. Representative examples of infected regions were photographed using an Olympus photomicroscope and assembled into figures using Adobe Illustrator and Photoshop software.
FIG. 6.

PRV 326 invades sympathetic and parasympathetic circuits at a faster rate than PRV Bartha does. The organization of polysynaptic circuits innervating the stomach is illustrated in the midsagittal schematic diagram of the rat brain. Virus injected into the ventral wall of the stomach invades the central nervous system through sympathetic and parasympathetic circuits. Virus injected into the stomach wall is retrogradely transported to the dorsal motor vagal complex (DVC) in the caudal brain stem through parasympathetic circuits. Infection of sympathetic neurons in the intermediolateral cell column (IML) of the thoracic spinal cord is temporally delayed compared to that resulting from invasion of parasympathetic circuits due to the need for first-order replication in sympathetic ganglia. First-order parasympathetic and sympathetic neurons are represented in black in the diagram. Neurons presynaptic to parasympathetic circuits are denoted in red, and those linked to the IML are represented in blue. Note that the paraventricular nucleus (PVN) of hypothalamus is synaptically linked to both. (A) The synaptic organization of circuits in the DVC is illustrated in coronal section. Neurons of the dorsal motor vagal nucleus (DMV) receive synaptic contact from neurons in the immediately adjacent nucleus of the solitary tract (nts) and the area postrema (AP). (B to E) The magnitude of retrograde transneuronal passage of PRV Bartha (B and D) and PRV 326 (C and E) through the DVC (B and C) and IML (D and E) 72 h following inoculation of the stomach is illustrated in the photomicrographs. PRV 326 exhibits more extensive retrograde transneuronal infection of the DVC than PRV Bartha does, and larger numbers of IML neurons are infected in the IML of PRV 326-infected animals. Abbreviations: A5, midbrain catecholamine cell group; BNST, bed nucleus of stria terminalis; CeA, central nucleus of the amygdala; IC, insular cortex; LHA, lateral hypothalamic area; PFC, prefrontal cortex; R, raphe; RVLM, rostroventrolateral medulla; VMM, ventromedial medulla.
RESULTS
PRV Bartha undergoes retrograde-directed axonal transport with wild-type PRV kinetics.
Our original hypothesis was that the observed delay in retrograde transneuronal spread of PRV Bartha resulted from inefficient axonal transport from neuronal termini toward the cell bodies. Possible defects leading to this phenotype might affect the processivity or rate of dynein-mediated retrograde transport of viral particles. Therefore, we used live-cell microscopy to characterize the movement of Bartha capsids in axons of dissociated embryonic rat SCG. We infected neurons with either PRV 765, which contains red fluorescent protein-tagged VP26 in the Bartha background, or PRV GS443, which contains green fluorescent protein-tagged VP26 in the PRV Becker background. We infected the cultures at a high multiplicity of infection (MOI) and imaged at 16 hpi (see the movies in the supplemental material). At this point, newly replicated and strongly fluorescing capsid puncta are easily detectable. Because the dissociated neurons establish synaptic connections in culture (32), retrograde-directed spread of infection can occur from an infected postsynaptic cell to a presynaptic cell; this feature enabled us to observe trafficking of capsids toward neuronal cell bodies.
We tracked retrograde movement of fluorescent puncta and measured the length of each run, which we define as a period of uninterrupted movement; in addition, we calculated the average velocity of the runs. Figure 1 shows the distribution and mean values of the measurements obtained. The average length of 124 runs by PRV 765 capsids was 5.11 μm, while the average length determined for 120 runs by PRV GS443 capsids was 4.47 μm; this difference was not significant by a two-sample T/P test (Student's t test, P = 0.475). The average velocity of PRV 765 capsids was 1.07 μm/s, which was comparable to the 1.09 μm/s measured for PRV GS443 capsids (P = 0.680). These data suggest that the retrograde intracellular trafficking of Bartha capsids occurs with wild-type PRV kinetics and cannot account for the observed delay in retrograde transneuronal spread.
FIG. 1.

Axonal retrograde transport kinetics. Dissociated SCG neurons were cultured on glass-bottom MatTek dishes for 2 weeks. Cultures were infected with PRV GS443 or PRV 765. The movies in the supplemental material were captured 16 h postinfection on a Perkin-Elmer R30 spinning disk confocal microscope. (A) Color-coded arrowheads track the movement of individual capsid puncta in 5-s intervals. (B) Distribution of capsid puncta run lengths is shown in box plots (n = 120). (C) Distribution of average run velocities of capsid puncta (n = 85). The open squares in box plots designate the minimum value, first quartile, median, and third quartile of the data set; filled squares are mean values.
In vitro time course of retrograde-directed neuronal infection.
We assessed whether the kinetic defect in neuronal infection by PRV Bartha can be recapitulated in vitro by performing infections of compartmented neuronal cultures. In this system, neuronal soma and axons are maintained in separate fluid environments. Therefore, inoculum can selectively be applied to axons, and the efficiency of retrograde-directed infection of cell bodies was ascertained by determining the titer of infectious virus in the soma (11). The sections of the trichamber are designated soma compartment (S-compartment, where the neurons are plated), methocel compartment (M-compartment, where viscous medium is placed), or neurite compartment (N-compartment, where axons emerge) (Fig. 2A).
FIG. 2.
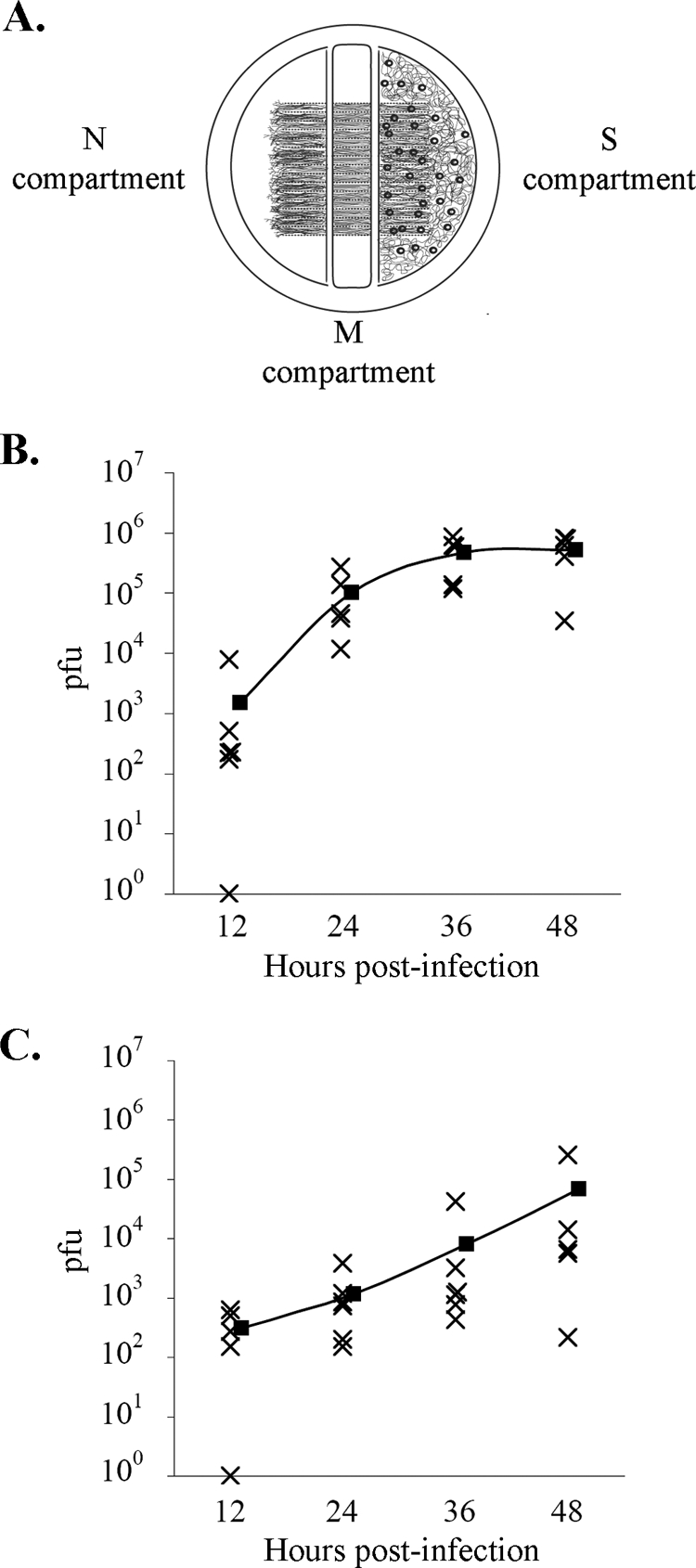
Retrograde infection time course in vitro. (A) Trichamber culture system for study of directional infection of neurons. Dissociated SCG neurons are plated in the S-compartment of the trichamber. Axonal growth is guided into the N-compartment by a series of grooves etched in the dish surface. Inoculum is applied to the N-compartment, and contents of the S-compartment are harvested, and the virus titers were determined. (B and C) Time course of retrograde neuronal infection by PRV Becker (B) or PRV Bartha (C). Filled squares indicate mean values at each time point.
We applied viral inoculum to the N-compartment and harvested the contents of the S-compartment at several time points. Twelve hours after the infection of axons, PRV Bartha titers in the soma compartment were comparable to the wild-type PRV Becker levels (Fig. 2B and C). A 2-log-unit difference in the mean titers of PRV Becker and Bartha, previously observed by Ch'ng and Enquist (11), was apparent at 24 hpi but decreased significantly by 48 hpi. These in vitro experiments recapitulate the kinetic delay of PRV Bartha infection in animal models. It is noteworthy that the range of titers of Bartha-infected samples increased over time, while titers of Becker samples remained closely clustered. This pattern indicates efficient primary infection with asynchrony in subsequent viral spread (secondary infection) among neurons in the S-compartment. Accordingly, we examined the efficiency of secondary transneuronal infection by PRV Bartha using immunofluorescence imaging.
PRV Bartha does not spread efficiently to second-order neurons in vitro.
We developed an immunofluorescence assay to test the hypothesis that PRV Bartha does not undergo efficient spread among S-compartment neurons. Not all neurons extend axons that reach the N-compartment; instead, axons often form connections with other neurons in the S-compartment. These cells cannot become directly infected by viral inoculum applied to the N-compartment but become infected only by secondary spread of infection from neurons that undergo primary infection (retrograde spread). We labeled the cells that extend axons across the full length of the trichamber by adding the lypophilic dye DiI to only the N-compartment. The dye diffuses laterally in the axonal membrane and reaches the cognate cell body, thereby labeling all neurons that can undergo primary infection. Approximately 15% of soma in the S-compartment of each sample exhibited DiI fluorescence (data not shown).
Twenty-four hours after infection of DiI-labeled axons, viral capsids were detected in the S-compartment via immunofluorescence. The number of cells exhibiting both DiI and capsid fluorescence was scored as cells that have undergone primary infection. Cells exhibiting capsid fluorescence only are a result of viral spread within the soma compartment and were scored as having undergone secondary infection. We counted all cells in randomly selected fields among three independent samples infected with either PRV Becker or PRV Bartha and calculated the percentage of cells with primary or secondary infection. PRV Bartha and PRV Becker infect equivalent numbers of cells via primary infection: of the 200 cells counted, 27% exhibited both DiI and capsid fluorescence in Bartha-infected samples, while 31% of cells were dually labeled in Becker-infected samples. However, of the 200 cells counted in Bartha-infected chambers, 16% exhibited capsid labeling and no DiI fluorescence, compared to 40% in Becker-infected samples (Fig. 3). These data indicate inefficient secondary infection by Bartha and support the hypothesis that the spread of infection in the soma compartment, but not the primary infection of neurons, is impaired in Bartha-infected neuronal cultures.
FIG. 3.

PRV Bartha does not spread efficiently to second-order neurons in vitro. DiI was added to the N-compartment immediately following infection with PRV Becker (A), PRV Bartha (B), or PRV 43/25 aB4 (C). Samples were processed for immunofluorescence against the major capsid protein VP5 at 24 hpi using a green secondary antibody. Arrows mark dually fluorescent cells. Arrowheads mark cells exhibiting VP5 fluorescence only. Bars = 40 μm. (D) The percentage of cells exhibiting both DiI and VP5 fluorescence is represented by filled bars; empty bars are cells exhibiting VP5 fluorescence only (n = 200). The chi-square test was used on raw data to determine significance. *, P < 0.05.
While PRV Bartha exhibits titers 100 times lower than PRV Becker upon retrograde infection, its efficiency of secondary spread is only 2 times lower than that of wild-type PRV (Fig. 2 and 3). This discrepancy is explained by the nature of our secondary spread assay, which measures the number of infected cells, and not infectious virions; immunofluorescence against the major capsid protein VP5 enables quantification of cells containing the viral antigen but does not distinguish between unincorporated VP5 protein, an empty capsid, or an infectious, fully assembled virion.
PRV Bartha repair strain PRV 43/25 aB4 is restored for retrograde spread.
To locate the mutation in the PRV Bartha genome that is responsible for the observed retrograde infection phenotype, we performed infections with several Bartha repair strains. PRV 158 contains the unique long region of Bartha and a repair of the Bartha US deletion with Becker sequence. Conversely, PRV BaBe contains the unique long region of Becker and harbors the same deletion in the US region that is found in Bartha. The mean titer of PRV 158 upon retrograde infection was equivalent to that of Bartha (4.77 × 104 PFU), while the mean titer of BaBe was equivalent to that of Becker (1.93 × 103 PFU). Therefore, the mutation responsible for the retrograde infection phenotype of PRV Bartha is located in its unique long region.
Several mutations in this region of the PRV Bartha genome have been characterized, namely, point mutations in glycoprotein C (36), glycoprotein M (15), Us3, and the intergenic UL20-UL21 region and three amino acid substitutions in the UL21 gene product (21). UL21 is a nonessential capsid-associated protein shown to play a role in genome processing and/or packaging (13). We hypothesized that the UL21 mutations found in Bartha may result in a delay of genome packaging, which may affect the rate of second-order spread of infection among neurons and amplification of infection. We therefore tested PRV 43/25 aB4, a strain derived from PRV Bartha via virulence rescue experiments (23). In this strain, the BamHI fragment 4 of the unique long region, encompassing UL21, has been repaired with wild-type PRV Kaplan sequence. Upon retrograde infection, the mean of PRV 43/25 aB4 titers was 5.60 × 104, equivalent to wild-type PRV levels. In addition, the efficiency of viral spread among neurons in the S-compartment, as assessed by immunofluorescence, improved significantly over that of PRV Bartha: of the 200 neurons counted, 32% had undergone primary infection, and 33% had undergone secondary infection (Fig. 3). PRV 43/25 aB4 also contains a repair of the Bartha US deletion; however, our data on retrograde infection with BaBe and individual analysis of gE, gI, and Us9 deletion mutants indicate that the gene products of the unique short region do not play a role in retrograde spread (Fig. 4) (11). Therefore, mutations in the UL21 locus are responsible for the reduced retrograde infection defect by Bartha.
FIG. 4.

PRV 43/25 aB4 restores wild-type PRV retrograde titers. (A) Diagram of genomes used to map the PRV Bartha retrograde defect. (B) Viral inoculum was added to the N-compartment of neuronal cultures. The contents of the S-compartment were harvested, and virus titers were determined at 24 hpi. Retrograde titers achieved by each strain are shown by filled symbols (n = 4). Empty squares denote the mean values for each data set.
PRV 326 is a more efficient retrograde tracing strain in vitro and in vivo.
Our studies suggest that repair of the UL21 locus in PRV Bartha would result in a faster retrograde-restricted neural circuit tracer. We therefore constructed PRV 326 by reintroducing the US deletion of Bartha into the genome of PRV 43/25 aB4. In vitro analysis revealed that PRV 326 titers upon retrograde-directed infection of neurons exceed wild-type PRV levels, reaching 2.42 × 106 PFU (Fig. 5B). The strain's ability to undergo anterograde-directed, neuron-to-cell spread of infection was tested by infecting the neuronal soma and determining the titer of the contents of the neurite compartment, in which epithelial PK15 detector cells were plated and allowed to establish contact with the resident axons. PRV 326 was incapable of anterograde-directed, neuron-to-cell spread of infection (Fig. 5A), demonstrating its potential as a retrograde-restricted neural circuit tracer with rapid spread kinetics.
FIG. 5.

PRV 326 is defective for neuron-to-cell spread, but it undergoes efficient retrograde infection of neurons. (A) The epithelial PK15 detector cells were plated in the N-compartment of neuronal cultures prior to infection. Viral inoculum was applied to the S-compartment, the contents of the S- and N-compartments were harvested at 24 hpi (n = 7), and virus titers were determined. (B) Viral inoculum was adsorbed to axons in the N-compartment. The contents of the S-compartment were harvested at 24 hpi (n = 7), and virus titers were determined. Filled squares represent the mean value of each data set.
To characterize the temporal kinetics of PRV 326 spread through neural circuitry, we compared the invasiveness of PRV 326 and PRV Bartha after inoculation of the ventral wall of the stomach in rats. This model system has been used extensively in prior analyses of PRV neuroinvasiveness, including PRV Bartha (6, 7, 9, 33-35, 40). Each virus produced the pattern of transport predicted by prior investigations that have employed this experimental model to evaluate the invasiveness of PRV. However, the temporal kinetics of invasiveness differed substantially between strains, with PRV 326 invading the central autonomic circuits at a significantly faster rate than PRV Bartha. The data supporting these assertions are presented below.
Injection of PRV into the ventral wall of the stomach produces retrograde transneuronal infection of preautonomic circuits through the sympathetic and parasympathetic branches of the autonomic nervous system (Fig. 6). Infection of the brain through sympathetic pathways is delayed relative to parasympathetic pathways due to circuit architecture and the number of neurons that constitute the circuit. Virus is directly transported into the caudal brain stem through parasympathetic circuits and then passes transneuronally to infect other cell groups in the brain stem and forebrain. In contrast, brain stem and forebrain neurons antecedent to the sympathetic outflow become infected only after replication of virus in peripheral sympathetic ganglia and transneuronal infection of preganglionic neurons in the intermediolateral cell column of the thoracic and lumbar spinal cord. We designed our analysis to determine the progression of infection through both divisions of the autonomic nervous system and incorporated quantitative measures and statistical analysis. It is also important to note that circuits on the left side of the brain principally innervate parasympathetic outflow to the ventral surface of the stomach and that the inverse is true of polysynaptic circuits innervating the dorsal surface.
Analysis of the entire brain and spinal cord 48 hours following injection of PRV Bartha into the stomach revealed an infection of caudal brain stem parasympathetic neurons confined to the dorsal motor vagal nucleus and a total absence of infection of preganglionic sympathetic neurons in the spinal cord (see Table S1 in the supplemental material). In contrast, injection of the same volume and concentration of PRV 326 revealed a statistically significant increase in the number of DMV (parasympathetic) neurons and transneuronal infection of synaptically linked neurons in the immediately adjacent nucleus of the solitary tract (see Table S1 in the supplemental material). Similarly, the total absence of PRV Bartha infection in spinal cord contrasted with the presence of PRV 326-infected sympathetic preganglionic neurons in multiple IML segments of thoracic and lumbar segments of the cord.
The increased invasion of central circuits by PRV 326 compared to PRV Bartha observed 48 h postinoculation was even more apparent 72 h postinoculation (see Table S1 in the supplemental material). Statistically significant increases in the number of PRV 326-infected neurons were observed in the DMV (parasympathetic) and IML (sympathetic) cell groups, and there were also statistically significant increases in retrograde transneuronal infection of neurons synaptically linked to these cell groups. Quantitative comparisons of the numbers of infected neurons in the IML, DMV, and in selected synaptically linked populations of neurons are presented graphically in Fig. 7. Figure 8 illustrates maps of all these regions, and Fig. 9 shows photomicrographs of representative cases. The results were striking and unambiguous. For example, an average of 146 neurons were observed in multiple IML segments of the left thoracic and lumber spinal cord after infection with PRV Bartha compared to an average of 1,491 neurons in animals infected with PRV 326. Similarly, there was almost a twofold increase (122 versus 234) in the number of PRV 326-infected DMV neurons in animals compared to animals infected with PRV Bartha. The increased infection of the DMV and IML by PRV 326 was mirrored by statistically significant increases in the number of infected neurons in brain stem and forebrain cell groups known to project to one or both of these cell groups. For example, the paraventricular hypothalamic nucleus that projects to both the DMV and IML contained an average of 226 PRV 326-infected neurons compared to an average of only 6 neurons infected by PRV Bartha. Similarly, the central nucleus of the amygdala, which projects to the DMV but not to the IML contained an average of 64 neurons after PRV 326 inoculation compared to an average of 2 following equivalent inoculation with PRV Bartha. These increases and others documented in Table S1 in the supplemental material were consistent among animals and were statistically significant.
FIG. 7.
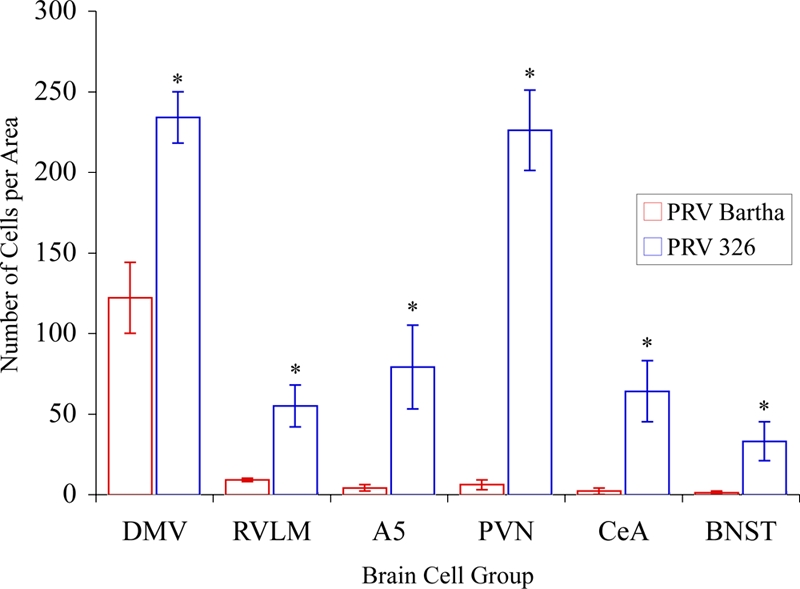
Quantitative analysis reveals a statistically significant increase of PRV 326 transport through preautonomic circuits compared to controls. The number of infected neurons observed in selected areas of the CNS 72 h following injection of PRV Bartha or PRV 326 into the stomach is illustrated. In each area, the number of PRV 326-infected neurons shows a statistically significant increase relative to PRV Bartha. The dramatic increase in the number of infected neurons in the PVN is likely related to the fact that this nucleus is synaptically linked to both the dorsal motor vagal complex (DVC) and IML, whereas the rostroventrolateral medulla (RVLM), midbrain catecholamine cell group (A5), central nucleus of the amygdala (CeA), and bed nucleus of stria terminalis (BNST) are selectively linked to the IML. Values that are statistically significantly different for PRV Bartha and PRV 326 are indicated with an asterisk (Student's t test, P ≤ 0.05).
FIG. 8.

Distribution of infected neurons at three comparable levels of the neuraxis 72 h following injection of PRV Bartha or PRV 326 into the ventral wall of the stomach. The gray bars on the sagittal section at the top of the figure illustrate the positions of the coronal sections shown immediately below. The coronal sections sample the caudal brain stem at the level of the dorsal motor vagal complex (DMV) (nucleus of the solitary tract [nts] and area postrema [AP]), through the cardiovascular regulatory cell group linked to the sympathetic outflow (rostroventrolateral medulla [RVLM]), and through a level of the forebrain through the paraventricular nucleus (PVN) and central nucleus of the amygdala (CeA). Infected neurons were mapped by systematic examination of the section with a 40× objective. The position of each cell was recorded using an image analysis system. Each red dot represents an infected neuron. The maps demonstrate the dramatic increase in transport of PRV 326 relative to PRV Bartha in preautonomic circuits synaptically linked to the sympathetic and parasympathetic outflow.
FIG. 9.

The increase in the magnitude of infection of preautonomic circuits resulting from infection with PRV 326 compared to PRV Bartha is striking and unambiguous. The extent of infection of four cell groups following identical inoculation of PRV Bartha (A, C, E, and G) or PRV 326 (B, D, F, and H) is illustrated. Comparisons of infection in the rostral portion of the nucleus of the solitary tract (A and B), rostroventrolateral medulla (C and D), midbrain catecholamine cell group (E and F), and paraventricular nucleus (G and H) are illustrated. Each comparison reveals larger numbers of infected neurons, a finding that was confirmed by statistical analysis. The yellow areas in the schematic insets illustrate the locations of the areas that were photographed. The third ventricle of the hypothalamus (3V) is indicated. The magnification of all images in this figure is the same. Bar = 100 μm.
We have previously demonstrated that immune cells of monocytic lineage (ED1+) invade areas of viral replication at advanced stages of infection (33). That analysis demonstrated that ED1+ cells invade the DMV approximately 70 h after injection of PRV Bartha into the ventral stomach wall and an increase in number with advancing survival. Staining of tissue for the ED1 antigen in the present study confirmed this finding for PRV Bartha-infected animals and revealed more extensive extravasation of ED1+ cells from the vasculature at this survival interval (72 h) in PRV 326-infected animals (Fig. 10). The extent of invasion of ED1+ cells in the PRV 326-infected animals was more consistent with that observed 90 to 96 h after injection of PRV Bartha (33). This finding is consistent with the increased invasiveness of PRV 326 relative to PRV Bartha.
FIG. 10.

Virus-induced recruitment of immune cells into the brain is increased in PRV 326-infected animals. The extent of ED1+ immune cell recruitment into the dorsal motor vagal complex 72 h after infection with PRV Bartha (A) or PRV 326 (B) is illustrated. Note the larger number of immunopositive cells within the DMV and immediately adjacent nucleus of the solitary tract (nts) of PRV 326-infected animals. The area postrema (AP) is indicated. The yellow area in the schematic inset illustrates the location of the area illustrated in the photomicrographs. The magnification of both images is the same. Bar = 100 μm.
DISCUSSION
In vivo retrograde infection by PRV Bartha of neural circuitry innervating the eye and stomach is slow compared to infection by wild-type PRV Becker (6, 40). We employed live-cell imaging and single particle tracking techniques to characterize this phenotype in vitro. Our analysis revealed that the average intra-axonal retrograde-directed run length and velocity of fluorescently tagged Bartha capsids were equivalent to those of fluorescent Becker capsids. Using the modified Campenot neuronal culture system, we recapitulated in vitro the strain's kinetic delay observed in vivo. Furthermore, by fluorescently labeling all neurons capable of undergoing primary infection in compartmented neuronal cultures, we measured the efficiency of viral spread from an infected presynaptic neuron to an uninfected postsynaptic neuron.
We mapped the retrograde defect of PRV Bartha to the UL21 locus, which contains seven point mutations (21). Michael and colleagues showed that these mutations result in inefficient tegument assembly in PRV Bartha (28). Such a defect may diminish the infectivity of transmitted virions. In addition, the product of UL21 has been implicated in the production of encapsidated infectious particles. Mutagenesis of the gene in PRV leads to impaired cleavage of the concatemeric viral genome into single-unit lengths (13). Furthermore, the complete absence of UL21 protein in PRV results in an increase in the number of empty capsids (38). As genome cleavage and encapsidation are linked processes in alphaherpesviruses, inefficient DNA processing may lead to a delay in nucleocapsid assembly, which in turn may lead to delayed transneuronal transmission of infectious particles. Our preliminary data indeed reveal an abundance of empty capsids in the nuclei of PRV Bartha-infected neurons (not shown). However, a detailed ultrastructural study is needed to determine the significance of these observations.
PRV Bartha is known to replicate well in most nonneuronal cell lines. The single step growth kinetics of the virus are similar to those of wild-type PRV Becker, except that the final titers achieved by PRV Bartha are typically 1 log unit higher than the wild-type PRV levels. One explanation for these observations is that simultaneous infection of all cells may mask the defect in nucleocapsid assembly because viral transmission from an infected cell to an uninfected cell is not required for amplification of infection. We attempted to detect any delays in replication by performing low-MOI infections of epithelial PK15 cells. Under these conditions, the efficiency of viral spread influences the rate of viral amplification. However, we detected no difference between PRV Bartha and PRV Becker, except that PRV Bartha reached titers 1 log unit higher than those of the wild-type virus (data not shown).
An alternate explanation is that PRV UL21 is not required for efficient infectious particle assembly in epithelial cells, and its function is cell type specific. This hypothesis has been suggested previously (23, 38). Ch'ng and Enquist have previously reported that PRV Bartha achieves wild-type PRV levels upon high-MOI infection of dissociated S-compartment neurons at 24 h postinfection (11). However, simultaneous infection of all neurons precludes detection of spread delays, and input inoculum applied to the cell bodies obscures subsequent measurements even after citrate inactivation, as few de novo infectious particles are produced per neuron. In our retrograde infection assay, input inoculum is confined to the N-compartment and therefore does not affect the quantification of infectious units in the S-compartment. Additionally, only 15% of the plated neurons extend axons that reach the N-compartment and undergo primary infection (unpublished observations), which effectively establishes a low MOI in the S-compartment. These conditions enabled us to detect the PRV Bartha replication defect in neurons.
The success of neural tracing studies is dependent on replication and transneuronal passage of virus through the nervous system. Our findings clearly demonstrate that repair of the mutations present in the UL21 locus of PRV Bartha increases the temporal kinetics of viral transport through neural circuits. These data have important implications for analysis of complex neural systems. Here, efficient transport of virus is integral to the ability to define all components of circuits that may extend throughout the full extent of the brain and spinal cord and that may differ in the number of synaptic contacts between neurons. The latter feature of neural circuitry is particularly important for avoiding false-negative results (e.g., not infecting neurons that are involved in the circuit). Strong evidence supports the conclusion that the progression of infection through a circuit depends on both the infectious dose and the number of synaptic connections between neurons (3, 5). To illustrate the latter point, it is useful to consider the findings of O'Donnell and colleagues (29) who used PRV to define the organization of parallel circuits between the basal forebrain and thalamus. The authors noted that a small subset of neurons known to be involved in this circuitry were not infected. Because the neurons were shown to be permissive to infection by PRV in other studies, the authors hypothesized that the observed lack of infection was due to the established sparse projections of their axons in this circuitry. It will be important to determine whether the improved transport kinetics of PRV 326 through neural circuits can resolve issues such as that noted in the O'Donnell study.
The potential influence of repairing the mutations of the UL21 gene upon the virulence and cytotoxicity of PRV 326 also merits attention. Several studies suggest that UL21 contributes to the virulence of wild-type virus and that the mutations in this gene in the PRV Bartha genome contribute to its attenuated phenotype (13, 21). We did not observe any increased cytopathogenicity compared to PRV Bartha following infection of central autonomic circuits with PRV 326. The pattern of transport of this virus recapitulated that observed with PRV Bartha and other deletion mutants that are transported only retrogradely through this circuitry. However, we did note that animals infected with PRV 326 exhibited more pronounced symptoms of infection (e.g., oronasal secretions) indicative of stress than PRV Bartha-infected animals at the same time postinoculation did. In addition, PRV 326-infected animals lost more weight (an average of 46 g versus 10 g) during the last day of the experiment than their PRV Bartha-infected counterparts did. It will be important to examine longer survival times and the transport of virus through different circuits (e.g., following intracerebral injection) to determine the full impact of these observations on the utility of PRV 326 as a neural tracer. Nevertheless, our data provide further insight into the function of the UL21 locus in viral invasiveness and confirm the findings of Klupp and colleagues regarding its role in virulence (21).
Supplementary Material
Acknowledgments
We acknowledge support from the National Institutes of Health (grant R01 33506 [to L.W.E.] and grant NCRR P40 RR0118604 [to J.P.C., P. L. Strick, and L.W.E.]). L.W.E. acknowledges support from the Center for Behavioral Neuroscience Viral Tract Tracing Core at Georgia State University through the STC Program of the National Science Foundation under agreement IBN-9876754 to L.W.E. and Tim Bartness. M.G.L. was supported by The American Cancer Society Eastern Division-Mercer Board Postdoctoral Fellowship (PF-08-264-01-MBC).
We acknowledge Peggy Bisher for help with electron microscopy.
Footnotes
Published ahead of print on 19 November 2008.
Supplemental material for this article may be found at http://jvi.asm.org/.
REFERENCES
- 1.Bartha, A. 1961. Experimental reduction of virulence of Aujeszky's disease virus. Magy. Allatorv. Lapja 1642-45. [Google Scholar]
- 2.Brittle, E. E., A. E. Reynolds, and L. W. Enquist. 2004. Two modes of pseudorabies virus neuroinvasion and lethality in mice. J. Virol. 7812951-12963. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Card, J. P., J. R. Dubin, M. E. Whealy, and L. W. Enquist. 1995. Influence of infectious dose upon productive replication and transynaptic passage of pseudorabies virus in rat central nervous system. J. Neurovirol. 1349-358. [DOI] [PubMed] [Google Scholar]
- 4.Card, J. P., and L. W. Enquist. 1999. Transneuronal circuit analysis with pseudorabies viruses, vol. 1. John Wiley & Sons, San Diego, CA.
- 5.Card, J. P., L. W. Enquist, and R. Y. Moore. 1999. Neuroinvasiveness of pseudorabies virus injected intracerebrally is dependent on viral concentration and terminal field density. J. Comp. Neurol. 407438-452. [DOI] [PubMed] [Google Scholar]
- 6.Card, J. P., L. Rinaman, J. S. Schwaber, R. R. Miselis, M. E. Whealy, A. K. Robbins, and L. W. Enquist. 1990. Neurotropic properties of pseudorabies virus: uptake and transneuronal passage in the rat central nervous system. J. Neurosci. 101974-1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Card, J. P., P. Levitt, M. Y. Gluhovsky, and L. Rinaman. 2005. Early experience modifies the postnatal assembly of autonomic emotional motor circuits in rats. J. Neurosci. 259102-9111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Card, J. P., M. E. Whealy, A. K. Robbins, R. Y. Moore, and L. W. Enquist. 1991. Two alphaherpesvirus strains are transported differentially in the rodent visual system. Neuron 6957-969. [DOI] [PubMed] [Google Scholar]
- 9.Card, J. P., L. Rinaman, R. B. Lynn, B.-H. Lee, R. P. Meade, R. R. Miselis, and L. W. Enquist. 1993. Pseudorabies virus infection of the rat central nervous system: ultrastructural characterization of viral replication, transport, and pathogenesis. J. Neurosci. 132515-2539. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Card, J. P., M. E. Whealy, A. K. Robbins, and L. W. Enquist. 1992. Pseudorabies virus envelope glycoprotein gI influences both neurotropism and virulence during infection of the rat visual system. J. Virol. 663032-3041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Ch'ng, T. H., and L. W. Enquist. 2005. Neuron-to-cell spread of pseudorabies virus in a compartmented neuronal culture system. J. Virol. 7910875-10889. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Ch'ng, T. H., E. A. Flood, and L. W. Enquist. 2005. Culturing primary and transformed neuronal cells for studying pseudorabies virus infection. Methods Mol. Biol. 292299-316. [DOI] [PubMed] [Google Scholar]
- 13.de Wind, N., F. Wagenaar, J. Pol, T. Kimman, and A. Berns. 1992. The pseudorabies virus homology of the herpes simplex virus UL21 gene product is a capsid protein which is involved in capsid maturation. J. Virol. 667096-7103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Dijkstra, C. D., E. A. Dopp, P. Joling, and G. Kraal. 1985. The heterogeneity of mononuclear phagocytes in lymphoid organs: distinct macrophage subpopulations in the rat recognized by monoclonal antibodies ED1, ED2 and ED3. Immunology 54589-599. [PMC free article] [PubMed] [Google Scholar]
- 15.Dijkstra, J. M., T. C. Mettenleiter, and B. G. Klupp. 1997. Intracellular processing of pseudorabies virus glycoprotein M (gM): gM of strain Bartha lacks N-glycosylation. Virology 237113-122. [DOI] [PubMed] [Google Scholar]
- 16.Ekstrand, M. I., L. W. Enquist, and L. E. Pomeranz. 2008. The alpha-herpesviruses: molecular pathfinders in nervous system circuits. Trends Mol. Med. 14134-140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Enquist, L. W. 2002. Exploiting circuit-specific spread of pseudorabies virus in the central nervous system: insights to pathogenesis and circuit tracers. J. Infect. Dis. 186(Suppl. 2)S209-S214. [DOI] [PubMed] [Google Scholar]
- 18.Goodpasture, E. W., and O. Teague. 1923. Transmission of the virus of herpes fibrils along nerves in experimentally infected rabbits. J. Med. Res. 44139-184. [PMC free article] [PubMed] [Google Scholar]
- 19.Hill, T. J., and H. J. Field. 1973. The interaction of herpes simplex virus with cultures of peripheral nervous tissue: an electron microscopic study. J. Gen. Virol. 21123-133. [DOI] [PubMed] [Google Scholar]
- 20.Klupp, B. G., H. Kern, and T. C. Mettenleiter. 1992. The virulence-determining genomic BamHI fragment 4 of pseudorabies virus contains genes corresponding to the UL15 (partial), UL18, UL19, UL20, and UL21 genes of herpes simplex virus and a putative origin of replication. Virology 191900-908. [DOI] [PubMed] [Google Scholar]
- 21.Klupp, B. G., B. Lomniczi, N. Visser, W. Fuchs, and T. C. Mettenleiter. 1995. Mutations affecting the UL21 gene contribute to avirulence of pseudorabies virus vaccine strain Bartha. Virology 212466-473. [DOI] [PubMed] [Google Scholar]
- 22.Lomniczi, B., S. Watanabe, T. Ben-Porat, and A. S. Kaplan. 1984. Genetic basis of the neurovirulence of pseudorabies virus. J. Virol. 52198-205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Lomniczi, B., S. Watanabe, T. Ben-Porat, and A. S. Kaplan. 1987. Genome location and identification of functions defective in the Bartha vaccine strain of pseudorabies virus. J. Virol. 61796-801. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Lyman, M. G., G. L. Demmin, and B. W. Banfield. 2003. The attenuated pseudorabies virus strain Bartha fails to package the tegument proteins Us3 and VP22. J. Virol. 771403-1414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.McFerran, J. B., and C. Dow. 1975. Studies on immunisation of pigs with the Bartha strain of Aujeszky's disease virus. Res. Vet. Sci. 1917-22. [PubMed] [Google Scholar]
- 26.McLean, I. W., and P. K. Nakane. 1974. Periodate-lysine-paraformaldehyde fixative. A new fixative for immunoelectron microscopy. J. Histochem. Cytochem. 221077-1083. [DOI] [PubMed] [Google Scholar]
- 27.Mettenleiter, T. C., L. Zsak, A. S. Kaplan, T. Ben-Porat, and B. Lomniczi. 1987. Role of a structural glycoprotein of pseudorabies in virus virulence. J. Virol. 614030-4032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Michael, K., B. G. Klupp, A. Karger, and T. C. Mettenleiter. 2007. Efficient incorporation of tegument proteins pUL46, pUL49, and pUS3 into pseudorabies virus particles depends on the presence of pUL21. J. Virol. 811048-1051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28a.National Resource Council. 1996. Guide for the care and use of laboratory animals. National Academy Press, Washington, DC.
- 29.O'Donnell, P., A. Lavin, L. W. Enquist, A. A. Grace, and J. P. Card. 1997. Interconnected parallel circuits between rat nucleus accumbens and thalamus revealed by retrograde transynaptic transport of pseudorabies virus. J. Neurosci. 172143-2167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Pickard, G. E., C. A. Smeraski, C. C. Tomlinson, B. W. Banfield, J. Kaufman, C. L. Wilcox, L. W. Enquist, and P. J. Sollars. 2002. Intravitreal injection of the attenuated pseudorabies virus PRV Bartha results in infection of the hamster suprachiasmatic nucleus only by retrograde transsynaptic transport via autonomic circuits. J. Neurosci. 222701-2710. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Pomeranz, L. E., A. E. Reynolds, and C. J. Hengartner. 2005. Molecular biology of pseudorabies virus: impact on neurovirology and veterinary medicine. Microbiol. Mol. Biol. Rev. 69462-500. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Potter, D. D., S. C. Landis, S. G. Matsumoto, and E. J. Furshpan. 1986. Synaptic functions in rat sympathetic neurons in microcultures. II. Adrenergic/cholinergic dual status and plasticity. J. Neurosci. 61080-1098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Rinaman, L., J. P. Card, and L. W. Enquist. 1993. Spatiotemporal responses of astrocytes, ramified microglia, and brain macrophages to central neuronal infection with pseudorabies virus. J. Neurosci. 13685-702. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Rinaman, L., P. Levitt, and J. P. Card. 2000. Progressive postnatal assembly of limbic-autonomic circuits revealed by central transneuronal transport of pseudorabies virus. J. Neurosci. 202731-2741. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Rinaman, L., M. R. Roesch, and J. P. Card. 1999. Retrograde transynaptic pseudorabies virus infection of central autonomic circuits in neonatal rats. Dev. Brain Res. 114207-216. [DOI] [PubMed] [Google Scholar]
- 36.Robbins, A. K., J. P. Ryan, M. E. Whealy, and L. W. Enquist. 1989. The gene encoding the gIII envelope protein of pseudorabies virus vaccine strain Bartha contains a mutation affecting protein localization. J. Virol. 63250-258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Smith, G. A., S. P. Gross, and L. W. Enquist. 2001. Herpesviruses use bidirectional fast-axonal transport to spread in sensory neurons. Proc. Natl. Acad. Sci. USA 133466-3470. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37a.U.S. Department of Health and Human Services. 1999. Biosafety in microbiological and biomedical laboratories. U.S. Department of Health and Human Services publication no. (CDC) 88-8395. U.S. Department of Health and Human Services, Washington, DC.
- 38.Wagenaar, F., J. M. Pol, N. de Wind, and T. G. Kimman. 2001. Deletion of the UL21 gene in pseudorabies virus results in the formation of DNA-deprived capsids: an electron microscopy study. Vet. Res. 3247-54. [DOI] [PubMed] [Google Scholar]
- 39.Watson, R. E., S. T. Wiegand, R. W. Clough, and G. E. Hoffman. 1986. Use of cryoprotectant to maintain long-term peptide immunoreactivity and tissue morphology. Peptides 7155-159. [DOI] [PubMed] [Google Scholar]
- 40.Yang, M., J. P. Card, R. S. Tirabassi, R. R. Miselis, and L. W. Enquist. 1999. Retrograde, transneuronal spread of pseudorabies virus in defined neuronal circuitry of the rat brain is facilitated by gE mutations that reduce virulence. J. Virol. 734350-4359. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.