

Abstract
RNA silencing in plants is a natural defense system mechanism against invading nucleic acids such as viruses. Geminiviruses, a family of plant viruses characterized by a circular, single-stranded DNA genome, are thought to be both inducers and targets of RNA silencing. Some natural geminivirus-host interactions lead to symptom remission or host recovery, a process commonly associated with RNA silencing-mediated defense. Pepper golden mosaic virus (PepGMV)-infected pepper plants show a recovery phenotype, which has been associated with the presence of virus-derived small RNAs. The results presented here suggest that PepGMV is targeted by both posttranscriptional and transcriptional gene silencing mechanisms. Two types of virus-related small interfering RNAs (siRNAs) were detected: siRNAs of 21 to 22 nucleotides (nt) in size that are related to the coding regions (Rep, TrAP, REn, and movement protein genes) and a 24-nt population primarily associated to the intergenic regions. Methylation levels of the PepGMV A intergenic and coat protein (CP) coding region were measured by a bisulfite sequencing approach. An inverse correlation was observed between the methylation status of the intergenic region and the concentration of viral DNA and symptom severity. The intergenic region also showed a methylation profile conserved in all times analyzed. The CP region, on the other hand, did not show a defined profile, and its methylation density was significantly lower than the one found on the intergenic region. The participation of both PTGS and TGS mechanisms in host recovery is discussed.
Geminiviruses are small, single-stranded DNA viruses that cause economically important plant diseases worldwide. Recent reports have shown that geminivirus-infected plants can recover or show reduction of symptoms (1, 12, 45). Such processes have been correlated to RNA silencing mechanisms. RNA silencing is an ancient mechanism involved in different fundamental processes, such as gene regulation, de novo histone and DNA methylation, establishment of heterochromatin, defense against viruses, and control of transposon mobility (6, 13, 30, 53). Silencing pathways involve the cleavage of a double-stranded, or an imperfect stem-loop, RNA molecule into short 21 to 24 nucleotides (nt) RNAs by a Dicer enzyme. These RNAs, known as short interfering RNAs (siRNAs) and microRNAs, direct the silencing process in a sequence-specific manner (6). RNA silencing can occur at transcriptional (TGS, for transcriptional gene silencing) and posttranscriptional (PTGS) levels. Arabidopsis thaliana has evolved a diversity of RNA silencing pathways, small RNA classes, and Dicer-like (DCL) genes (32, 51). microRNA, trans-acting siRNA, and natural antisense transcript siRNA pathways (involving DCL1, DCL2, and DCL4 genes) are PTGS-related processes that play a crucial role in developmental gene regulation in plants (5, 52, 57). On the other hand, DCL3 produces 24-nt siRNAs, which operate at a nuclear level, guiding heterochromatin formation and transcriptional repression of transposon and DNA repeats in a TGS process (58). TGS in plants involves de novo, RNA-directed DNA methylation in all sequence contexts, not just in symmetrical CG dinucleotides (31, 35, 38). In addition to its endogenous functions, RNA silencing in plants provides an adaptive immune system for recognizing and inactivating viruses. The model for viral silencing is strongly supported by the fact that virus-related siRNAs accumulate in infected plant tissue (12, 14, 36, 49, 59). Furthermore, both RNA and DNA viruses encode distinct suppressors of RNA silencing that target different components of the system (53).
Small RNAs of 21, 22, and 24 nt have been reported in Nicotiana benthamiana and cassava plants infected with African cassava mosaic virus (ACMV) as well as in A. thaliana infected with Cabbage leaf curl virus (CaLCuV) (1, 50). Symptoms and viral RNA and DNA levels were not considerably affected in single, double, or triple DCL2, DCL3, and DCL4 mutant backgrounds in Cabbage leaf curl virus-infected Arabidopsis plants compared with wild-type infection (9). In addition, DNA methylation of ACMV and Tomato leaf curl virus genomes has been reported recently (7, 16). Altogether, these results suggest that both the PTGS and TGS pathways are involved in the defense against geminiviruses. The correlation of these pathways with biological processes related to the viral cycle has been reported very recently (14, 21, 43). Pepper golden mosaic virus (PepGMV) is a bipartite member of the genus Begomovirus and infects dicotyledonous crops such as pepper, tomato, tomatillo, and tobacco (34). PepGMV-infected pepper plants show a recovery phenotype accompanied by a decrease in viral DNA and RNA titers and the presence of virus-specific siRNAs (12); therefore, they represent an interesting system to study the silencing mechanisms acting on a plant-virus interaction.
MATERIALS AND METHODS
Plant inoculation and tissue harvest.
Pepper (Capsicum annuum L. var. Sonora Anaheim) seeds were germinated in a bioclimatic chamber at 26 to 28°C with a 16:8-h (light/dark) photoperiod. Plants were inoculated with PepGMV infectious dimeric clones (PepGMVAdimpBS and PepGMVBdimpBS) by biolistic bombardment with a low-pressure apparatus as described previously (12). We dissected the harvesting time in two stages: (i) symptomatic, in which two symptomatic leaves are present above the inoculated leaf at 10 days postinfection (dpi) (Fig. 1); and (ii) symptomatic recovered, in which six leaves (four symptomatic and two recovered) are present above the inoculated leaf at 15 dpi (Fig. 1).
FIG. 1.
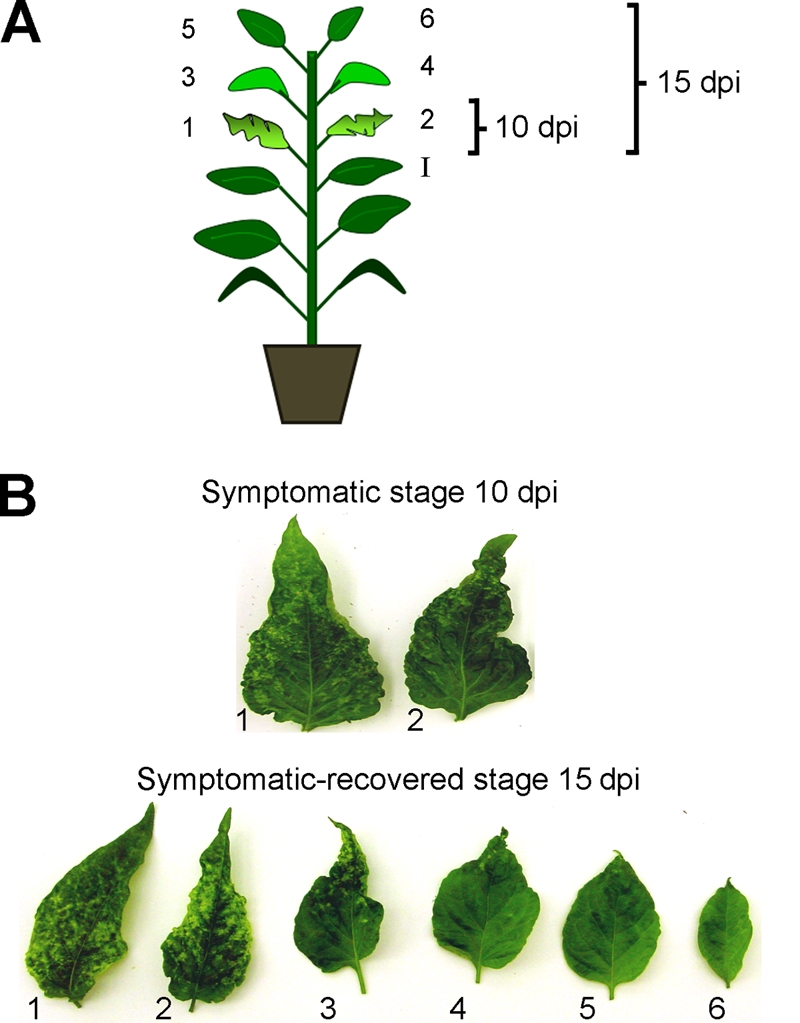
Stages of PepGMV infection used for characterization of the host recovery process. (A) Scheme of types of leaves and times of collection. I, inoculated leaf; 1 to 6, leaves that emerged after the inoculation and were collected for analysis. (B) Symptoms on the leaves collected from PepGMV-infected plants at 10 (symptomatic stage) and 15 (symptomatic/recovered stage) dpi.
Total nucleic acids and low-molecular-weight (LMW) RNA extraction.
Total DNA or RNA from infected and mock-inoculated tissues was extracted as described previously (3) or with Trizol reagent (Life Technologies, Carlsbad, CA), respectively. For total DNA extraction, ∼200 mg of tissue was frozen in liquid nitrogen and ground using a mortar and pestle. Powdered tissue was mixed with 750 μl of 3NO buffer (7 M Urea, 0.35 M NaCl. 0.05 M Tris-HCl, 0.02 M EDTA, 1% sarcosyl) and incubated at 65°C for 10 min. Samples were centrifuged at a relative centrifugal force (RCF) of 16,000 for 2 min, and the supernatant was transferred to a new tube. Aqueous phase was extracted sequentially with 1 volume of phenol-chloroform-isoamylic alcohol (25:24:1, vol/vol/vol) and then with 1 volume of chloroform-isoamylic alcohol (24:1, vol/vol) followed by vortexing and centrifugation as described above. DNA was precipitated with 0.2 volumes of 10 M ammonium acetate and 1 volume of isopropanol. Samples were mixed and centrifuged for 20 min at 16,000 RCF. Tubes were decanted, and the pellet was washed with 500 μl of 70% ethanol and centrifuged for 5 min at 16,000 RCF. DNA pellets were air dried and redissolved in 50 μl of bidistilled water.
The LMW RNA fraction was prepared as described previously (35). Briefly, high-molecular-weight nucleic acids were precipitated from total RNA Trizol-extracted samples by adding polyethylene glycol (molecular weight, 8,000) and sodium chloride to final concentrations of 5% and 500 mM, respectively, followed by incubation on ice for 30 min. After the precipitate was removed by centrifugation at 9,000 rpm, the LMW nucleic acids remaining in the supernatant were extracted with 1 volume of phenol-chloroform-isoamylic alcohol (25:24:1, vol/vol/vol). Samples were vortexed and centrifuged at 9,000 rpm, and the aqueous phase was transferred to a new tube. This fraction was precipitated by adding 1/10 volume of 3 M sodium acetate, pH 5.0, and 3 volumes of absolute ethanol. After incubation for 2 h at −20°C, the precipitate was collected by centrifugation, washed with 70% ethanol, dried, and redissolved in bidistilled water. In all cases pools of four leaves for every condition were nitrogen frozen and ground to powder, and the resulting material was used for the respective nucleic acid extraction.
Preparation of PepGMV-specific PCR fragments.
Primers were designed to direct the amplification of 18 fragments (ca. 400 bp each) that spanned the entire PepGMV genome (Fig. 2 and Table 1). PCR products were loaded onto a 1.0% agarose gel, separated by electrophoresis, and transferred to Hybond N+ membrane (GE Healthcare, Little Chalfont, United Kingdom) by capillarity under alkaline conditions (0.4 N NaOH) and UV cross-linked.
FIG. 2.

Distribution of virus-derived siRNAs, isolated from symptomatic and recovered tissues, along the PepGMV genomic components. Eighteen PCR fragments (ca. 400 pb) covering the entire genome were immobilized in a membrane and hybridized with probes obtained by 5′ end-labeling of PepGMV-specific siRNAs isolated from tissues of symptomatic leaf 1-10 (S 1/10) and recovered leaf 6-15 (R 6/15) from two independent experiments. Hybridization signals were quantified with ImageJ software and graphed. The highest hybridization value in each set was scored as 100%. Asterisks indicate virus-derived genomic zones where major differences in siRNA concentrations between symptomatic and recovery stages were observed. Arrows represent the positions of the six viral genes (CP, Rep, TrAP, REn, NSP, and MP) on the genome; their correspondence with the PCR fragments is illustrated. Et-Br, ethidium bromide staining; A+B, hybridization using full-length viral components; mock, hybridization using small RNAs extracted from a mock-inoculated plant.
TABLE 1.
List of primers used to fragment PepGMV genomea
| PCR fragment | Primer name | Primer sequence (5′-3′) | Coordinates (nt) |
|---|---|---|---|
| A1 | A1F | CATTGTGGATATGTTAAGAAAATG | 2418-2441 |
| A1R | GAATTAAAGCGATAAATGCAGATG | 181-204 | |
| A1′ | A1′F | TGGTGTAGGACTCCAGCAGAGTC | 2494-2516 |
| A1′R | TAGGCCCACACCTTGGTCACCAAG | 145-168 | |
| A2 | A2F | GGACCACCAATTATTTGATTTTG | 79-101 |
| A2R | GTCACGTCAGAAATGCACATGAC | 457-479 | |
| A3 | A3F | CAGATGTGCCTAGASSATGTGAAG | 377-400 |
| A3R | CTAACCAATGCCTGCTCGTTGCTG | 768-791 | |
| A4 | A4F | CTGTGAAGAACGATTTGCGTGATC | 692-715 |
| A4R | CGTCATGTTGTATTTACATACG | 1071-1092 | |
| A5 | A5F | CATGATTCTCAAGTACATAATTTAC | 983-1007 |
| A5R | GACGACGCATAGACCTAAACTG | 1397-1418 | |
| A6 | A6F | GCCTGATGCACAGTGATGCTCTC | 1318-1340 |
| A6R | GTGGAGTATAACGTCATTGATG | 1705-1726 | |
| A7 | A7F | GTTGGACTGCCAGTCTTTTTG | 1631-1651 |
| A7R | CAAAGCTGGTGATCCGAAAACG | 2018-2039 | |
| A8 | A8F | GTTAAACGAGGATAATGGATAAGG | 1919-1942 |
| A8R | CCGATGGGTTCCCGCACTTGC | 2302-2322 | |
| A9 | A9F | CCCAGATTCACAATAATCACC | 2135-2155 |
| A9R | GGGACTCCAGGACTCCACAAC | 2535-2555 | |
| B1 | B1F | AAGGACAGGTGCCCTAATCG | 2557-2576 |
| B1R | CATGTATCAAAACTCAAGTCC | 339-359 | |
| B2 | B2F | GGTCCAGTAAAAACTCAAGTGTAG | 274-297 |
| B2R | GCTTAATATACGACCTGCTTC | 665-685 | |
| B3 | B3F | GCATACATGAAAACCAATATG | 563-583 |
| B3R | CATGACAGTGTCCTTCTCAAC | 949-969 | |
| B4 | B4F | GGTGTTGCATACATTTGATG | 828-847 |
| B4R | ATACATTTGTATCAGTCAATTAC | 1204-1226 | |
| B5 | B5F | CTAAAGCATCCACATTTGTATC | 1124-1145 |
| B5R | GTCAAGGGTTGGGCTTACAGGC | 1503-1524 | |
| B6 | B6F | CGAATCCCTTGAAGGACTACT | 1428-1448 |
| B6R | GGATAACGAGTCATTACAAGC | 1831-1851 | |
| B7 | B7F | GGTCCTTCAGTGAAAAGAATG | 1757-1777 |
| B7R | GCCGCACCTGTATATAAANC | 2154-2173 | |
| B8 | B8F | GAAAATATGCCGCAGCAGAAG | 2101-2121 |
| B8R | GAGTTGAGGGACTCCGCTGG | 2479-2498 | |
| B9 | B9F | GCAATGAGATGTCCTCTGAG | 2364-2383 |
| B9R | GATAAGGTTTAGCCAACGGTC | 151-171 | |
| U6 | GTCATCCTTGCGCAGGGGCCATGCTAAT |
Isolation, labeling of virus-derived small RNAs, and reverse Northern blot analysis.
Small-RNA extraction and labeling were performed as described previously (25). For small RNA isolation, ∼60 μg of a LMW RNA fraction derived from infected or mock-inoculated plants was denatured at 65°C for 10 min with 1 volume of loading dye (98% deionized formamide, 10 mM EDTA, pH 8.0, 1 mg/ml xylene cyanol, 1 mg/ml bromophenol blue) and separated on a 15% polyacrylamide-8 M urea gel in TBE buffer (89 mM Tris, 89 mM borate, 2 mM EDTA). Following electrophoresis, the gel was stained in a solution of 0.5 μg/ml ethidium bromide in TBE buffer. The small-RNA fraction (21 to 24 nt) was visualized under UV light using synthetic 21-, 24-, and 28-nt RNA oligonucleotides as size markers, and the relevant polyacrylamide gel piece was cut from the gel, crushed, and eluted in 500 μl of elution buffer (80% formamide, 40 mM PIPES [piperazine-N,N′-bis(2-ethanesulfonic acid)], pH 6.4, 1 mM EDTA, 400 mM NaCl) at 4°C overnight with gentle shaking. The eluted RNA was precipitated with 1 volume of isopropanol and 1 μl of glycogen (20 mg/ml) as a carrier. The pellet was dissolved in 400 μl of 20 mM Tris-HCl, pH 7.5, and 300 mM sodium acetate. Small RNA was reprecipitated by adding 1 ml of ethanol. The pellet was dried and dissolved in 40 μl of bidistilled water. The purified small RNAs (approximately 1 μg) were dephosphorylated and subsequently labeled in the presence of [γ-32P]ATP and RNase Out (Invitrogen) with 8 U of T4 polynucleotide kinase (Life Technologies). The labeled small RNAs were used for reverse Northern blotting as described previously (25). As a control for the assay (transfer and hybridization), the membranes were regenerated and hybridized against a 32P-labeled probe obtained from full-length clones of the PepGMV genome. The hybridization signals were scanned and quantified using a Storm phosphorimager and ImageJ free access software (http://rsb.info.nih.gov/ij/). The siRNA population does not present unspecific hybridization (data not shown). siRNA from PepGMV-infected plants does not hybridize with heterologous DNA (pBS plasmid or Pepper huasteco yellow vein virus [PHYVV] DNA), whereas siRNA from a noninoculated plant does not hybridize with the PepGMV genome.
Small-RNA Northern blot analysis.
Small-RNA Northern blot analysis was performed as described previously (1). The membranes containing LMW RNAs derived from infected pepper plants were probed with different PepGMV-derived genomic probes: A1′ (for the PepGMV DNA A intergenic region [IR]); A7 and A8 (Rep); A3 (coat protein [CP]); B1, B8, and B9 (IR of PepGMV DNA B); B3 and B4 (nuclear shuttle protein [NSP]); B6 (movement protein [MP]) and as loading control with a complementary U6 small nucleolar RNA oligonucleotide (Table 1). The viral genome probes were obtained by PCR and [α-32P]CTP label using a Redi Prime II kit (GE Healthcare). U6 RNA-complementary probe was also labeled in the presence of [γ-32P]ATP using T4 polynucleotide kinase (Life Technologies). Virus-derived RNA synthetic oligonucleotides of 21, 24, and 28 nt were used as molecular size markers. Hybridization was carried out overnight at 50°C as described previously (25). The hybridization signals were scanned and quantified as above.
Viral DNA quantification by real-time PCR.
The real-time quantitative PCR procedure and primers used for this assay were previously described (12).
Bisulfite treatment of DNA samples.
Bisulfite treatment was carried out following published protocols (19, 20). Briefly, DNA isolated from infected plants was digested with DraI endonuclease, followed by overnight treatment with proteinase K. DraI-proteinase K-treated DNA (0.5 to 1 μg) contained in 45 μl of water was denatured with 5.5 μl of 3 M NaOH at 37°C for 20 min. After a denaturalization step, samples were treated with 600 μl of freshly prepared sodium bisulfite solution (5.7 M sodium bisulfite, 10 mM hydroquinone), pH 5, and incubated for 4 to 16 h in darkness. After that, DNA was desalted with a Gene Clean Kit (QBioGene), and eluted with 50 μl of bidistilled water. DNA was desulfonated with 5.5 μl of 3 M NaOH at 37°C for 20 min, followed by precipitation, with 24 μl of 10 M ammonium acetate, 250 μl of ethanol, and 1.5 μl of glycogen (20 mg/ml) added as a carrier. The pellet was dissolved in 30 μl of 1 mM Tris-HCl, pH 8. In most instances, two or more independent, duplicate bisulfite experiments were performed.
MSPs.
Methylation-specific primers (MSPs) for PCR were designed following published parameters (29, 56). For methylated specific primers, it was assumed that the cytosines in symmetric sites (CpG and CpNpG) were methylated, whereas for the nonmethylated specific primers the assumption was that all cytosines were nonmethylated. Methylated bisulfite-treated DNA was amplified using the primers MF (5′-TTTTATAATATTATCGGATGGTCGC-3′; PepGMV coordinates, 2602 to 2613) and MR (5′-AAAACGATAAATACAAATACACGTA-3′; 175 to 199) under the following conditions: 30 cycles of 95°C for 30 s, 58°C for 30 s, and 72°C for 30 s, with an expected PCR product of 210 bp. Unmethylated bisulfite-treated DNA was amplified using the primers NMF (5′-TTTATAATATTATTGGATGGTTGTGA-3′; coordinates 2603 to 2615) and NMR (5′-AAAACAATAAATACAAATACACATA-3′; 175 to 199) under the following conditions: 30 cycles of 95°C for 30 s, 58°C for 30 s, and 72°C for 30 s, with an expected PCR product of 209 bp. One microliter of bisulfite-modified DNA was used for PCRs, and 10 μl of PCR product was analyzed on 2% agarose gels. To verify primer specificity and complete bisulfite-mediated conversion, methylated and nonmethylated PCR products were cloned in TOPO 2.1 vector (Life Technologies), and several clones were sequenced (3700 sequencer; Applied Biosystems).
Methylation density comparisons.
Bisulfite-modified DNA was used as a template for nonspecific or strand-specific (29, 56) PCRs. Nonspecific primers were designed with no cytosines in the 3′ ends; therefore, the primers will equally direct the amplification of both methylated and unmethylated viral DNA. The primers NSF (5′-CATTGTGGATATGTTAAGAAAT-3′; PepGMV coordinates 2418 to 2440) and NSR (5′-GAATTAAAGCGATAAATGCAGAT-3′; 182 to 204) were used under the following conditions: 30 cycles of 95°C for 30 s, 40°C for 30 s, and 72°C for 30 s. The expected PCR product size was 400 bp.
Strand-specific primers direct the amplification of a 580-bp fragment corresponding to 337 bp of the PepGMV A IR, the first 183 bp of the CP coding region, and the first 60 bp of the Rep coding region. Primers were designed in regions without symmetric (CG and CNG) cytosines to amplify both methylated and nonmethylated molecules in the same PCR. For this assay the primers VCF (5′-TAAATATATTAAAAAAATATTTTTAC-3′; coordinates 2423 to 2448) and VCR (5′-TTTAGGTATATTTGGGTTTTTATA-3′; 367 to 390) were used under the following: 30 cycles of 95°C for 30 s, 50°C for 30 s, and 72°C for 45 s. The expected PCR product size was 580 bp. PCR products were cloned in TOPO 2.1 vector (Life Technologies), and 50 clones (for nonspecific PCR products) and 22 clones (for strand-specific PCR products) from each stage were sequenced (3700 Sequencer; Applied Biosystems).
RESULTS
PepGMV-derived siRNAs along the viral genome in symptomatic and recovered tissues.
The presence of siRNAs related to both components of PepGMV in symptomatic and recovery stages of infected pepper plants was previously reported (12). However, it was not clear if the siRNA populations in both stages were similar in terms of their origins (region of the viral genome) and relative concentrations. To answer these questions, we further dissected the viral infection by collecting independent leaf samples at two time points: (i) at 10 dpi when only two symptomatic leaves are present (10-dpi leaves 1 and 2, designated 1-10 and 2-10); and (ii) at 15 dpi when typically six new leaves, both symptomatic (1-15 and 2-15, corresponding to the symptomatic leaves already present at 10 dpi) and recovered (leaves 3-15, 4-15, 5-15, and 6-15, which show a gradient of symptoms typical of the recovery process, from mild symptoms to asymptomatic tissue) (Fig. 1) (see Materials and Methods). To determine which regions of the PepGMV genome were the sources of the siRNA populations, we designed a series of primers to direct the amplification of 18 PCR fragments (∼400 bp; partially overlapping) that covered the entire genome. The PCR fragments were immobilized on a nylon membrane and hybridized against gel-purified, 32P-labeled siRNA populations. In general, similar hybridization patterns were observed when siRNAs independently extracted from the first 7 leaves shown in Fig. 1 (from 1-10 to 5-15) were used as probes (data not shown). However, changes in the hybridization pattern were observed when siRNAs from a totally recovered leaf (6-15) were used. Figure 2 shows a comparison of the patterns obtained with the extreme samples: leaves 1-10 (severe symptoms) and 6-15 (symptomless). Quantification of hybridization signals showed that some regions corresponding to the Rep, TrAP, REn, and MP open reading frames (ORFs) and both noncoding IRs are preferential target zones (hot spots) for silencing mechanism(s) in symptomatic leaf 1-10. However, the siRNA population from leaf 6-15 showed a decrease (80 to 40%) in hybridization signals corresponding to both IRs (Fig. 2). These results suggested that IR-related transcripts might have different, independent regulation mechanisms.
Type and relative concentration of PepGMV-derived siRNAs.
The reverse-Northern strategy used above did not provide information about the siRNA population in terms of size and relative concentration. Therefore, we proceeded to analyze the virus-derived siRNAs present in the leaves shown in Fig. 1 using a Northern procedure and different virus fragments as probes. The results are shown in Fig. 3. When fragments corresponding to coding regions from both components (Rep, CP, NSP, and MP) were used as probes, the small RNAs detected were predominantly of 21 to 22 nt in size (PTGS hallmark) although a weak hybridization signal corresponding to 24-nt small RNAs (TGS hallmark) was also observed in some cases. However, when the probes corresponded to an IR, the opposite condition was observed: a strong hybridization signal was obtained for 24-nt small RNAs, whereas a weak signal corresponding to 21- to 22-nt siRNAs was observed specially with the IR DNA B probe. Although the siRNAs were detectable in all leaves analyzed (leaves 1-10 to 6-15), the concentrations varied, depending on virus origin and the leaf analyzed. siRNAs derived from both IRs and the NSP and MP genes were detected mostly in older symptomatic leaves, whereas younger leaves showed only a weak hybridization signal. For example, at 15 dpi, the levels of IR/NSP/MP-related siRNAs varied from the highest-detected concentration (arbitrarily set at 100%) in leaf 1-15 to around 20% for leaf 6-15. siRNAs corresponding to the CP and Rep genes presented a different pattern. Whereas the highest concentration (100%) was also detected in leaf 1-15, the lowest signal (40%) was detected in leaf 4-15. Leaves 5-15 and 6-15 showed intermediate concentrations of 40 to 70% for Rep and 40 to 60% for CP (Fig. 3). Virus replication has been associated to the apical tissue; therefore this increase in siRNA concentration corresponds to an increase in the transcription of viral genes in apical tissue, as previously reported (12).
FIG. 3.

Northern blot analysis of siRNAs from leaves collected as shown in Fig. 1. LMW RNA was fractionated by denaturing polyacrylamide gel electrophoresis and hybridized with several virus-derived DNA sequences (in parentheses, the corresponding PCR fragments used as probes, as shown in Fig. 2, are indicated) (see Materials and Methods). The membrane was also hybridized with a synthetic oligonucleotide complementary to small nucleolar RNA U6 as a loading control. U6-normalized, relative levels of viral siRNA were quantified, and values are shown for each region. RNA synthetic oligonucleotides of 21 and 24 nt were used as molecular weight markers. M, mock-inoculated plant; Et-Br, ethidium bromide staining of the polyacrylamide gel.
Symptoms and viral DNA levels are inversely correlated with viral DNA methylation.
The correlation between host recovery and a reduction in virus titers has been reported in several systems (12, 14, 15, 44). However, the mechanisms acting on this phenomenon are not clear. Since the presence of 24-nt siRNAs is considered a hallmark of a TGS mechanism, a possible correlation between viral DNA methylation and the recovery process was studied by analyzing both the concentration and methylation levels of viral DNA. We have previously shown that the relative concentration of viral DNA found in the symptomatic tissue was higher than that found in the leaves showing recovery (12). However, that study was carried out pooling several sets of leaves, unlike the leaf-by-leaf dissection described here. Therefore, we performed a viral DNA relative quantification in the eight leaf samples shown in Fig. 1 using a real-time PCR assay for both components. In general, the results confirmed previously reported data. The highest relative concentration of viral DNA was found in the oldest, highly symptomatic leaves (leaves 1-10 and 1-15; 100%), whereas the lowest values (20% and 15% for PepGMV DNAs A and B, respectively) were detected in leaf 6-15 (data not shown). Then, we proceeded to analyze the methylation status of PepGMV with a couple of strategies based on a bisulfite modification procedure, combined with PCR amplification and sequencing analysis (see Materials and Methods). For the first strategy, we designed two sets of MSPs. One set specifically recognized molecules derived from nonmethylated, modified DNA, whereas the second set recognized only methylated, unmodified DNA (Fig. 4A) (see Materials and Methods). Primer specificity and PCR efficiency were assessed by cloning and sequencing several independent clones from each PCR/primer pair assay. Sequence analysis showed that primers were indeed specific for either methylated or nonmethylated viral DNA. In addition, the analysis revealed, in the case of the methylated DNA, a great diversity of methylation patterns, demonstrating that the clones did not come from a single template. In the case of nonmethylated DNA, all clones obtained with the nonmethylation-specific primers showed a replacement of all cytosines (C residues) by thymines (T/U), demonstrating the efficiency of the bisulfite treatment. Total DNA from leaves 1-10 and 6-15 was then analyzed, and after quantification of the PCR products by densitometry, it was observed that in leaf 1-10 a higher proportion of the viral DNA was nonmethylated. Interestingly, in leaf 6-15, the nonmethylated/methylated ratio was inverted (Fig. 4B): a higher proportion of the viral DNA was methylated in recovered tissue. This result suggested an inverse correlation between viral DNA methylation and symptom severity/viral DNA concentration.
FIG. 4.

Analysis of methylation in PepGMV DNA A noncoding (IR) and coding (CP) regions at symptomatic and recovered stages. (A) Primers used for methylation analysis and their expected PCR products. MF-MR, MSPs; NMF-NMR, nonmethylation-specific primers; VCF-VCR, virion complementary-strand-specific primers; NSF-NSR, nonspecific primers. (B) PCR products obtained using methylation-specific (M) and nonspecific (NM) primers from symptomatic (leaf 1-10) and recovered (leaf 6-15) tissues analyzed by electrophoresis in 2% agarose. L, 100-bp ladder. (C) Comparison of methylation levels of IR and CP sequences at symptomatic (10 dpi) and recovered (15 dpi) stages. DNA from three types of leaves was treated with bisulfite and amplified with the VCF-VCR primers. Twenty-two clones from each fragment were sequenced, and methylation levels were determined (methylated cytosines/total cytosines). (D) Distribution of methylation in symmetric (CG/CNG) and asymmetric (CHH) cytosines in the sequences analyzed in panel C; the number of cytosines in different sequence contexts for each segment is given.
Since this first strategy could present a possible bias because it relied on two sets of primers that might have different efficiencies in the PCR, we used a second approach with a different set of primers. The new primers (NSF and NSR) (Fig. 4A) were designed in a region with low cytosine content and with no cytosines in the last four to five bases of the 3′ end (see Materials and Methods). Therefore, these primers will equally direct the amplification (400 bp) of both modified (nonmethylated) and unmodified (methylated) viral DNA. The PCR assay was carried out using DNA extracts from leaves 1-10, 3-15, and 6-15. Fifty randomly selected clones from each case were sequenced and classified as methylated or nonmethylated. The results were similar to the first strategy: in symptomatic tissue (leaf 1-10) 40% of clones originated from methylated viral DNA, whereas in leaves showing mild symptoms (leaf 3-15) or no symptoms at all (leaf 6-15), the percentage of clones from methylated DNA increased to 58 and 65%, respectively (data not shown). Analysis of the sequences showed that cytosine methylation was found not only in the symmetric CG/CNG context but also in the asymmetric one (CHH).
Viral noncoding IR presents higher methylation levels than the CP coding region.
To verify if a coding region (CP) was similarly methylated as the IR, we designed an additional set of strand-specific primers to direct the amplification of a 580-bp fragment that includes the entire IR of PepGMV DNA A (337 nt) and 159 nt of the CP ORF (Fig. 4A) (see Materials and Methods). With this strategy, possible differences in methylation densities between two viral segments (IR and CP) could be evaluated in the same DNA molecule. Bisulfite-treated DNA from leaves 1-10, 3-15, and 6-15 was used as template for PCR, and 22 independent clones for each leaf extract were sequenced and analyzed. Differences in the methylation levels of IR and CP segments were evaluated by analyzing the density of methylated cytosines of all clones. The density was expressed as the percentage of the cytosines that remained unaltered after bisulfite treatment. Figure 4C shows that the percentage of cytosine methylation of the CP region was statistically lower (analysis of variance, P < 0.05) than the values found for the IR in all stages analyzed: 2.1 versus 9.1% (CP versus IR of DNA A) in leaf 1-10, 9.9 versus 24% in leaf 3-15, and 11.3 versus 21.2% in leaf 6-15. Furthermore, as mentioned before, there was no difference in the methylation efficiency of cytosines in a symmetric context (CG/CNG) versus an asymmetrical one (CHH) (Fig. 4D). The statistical analysis (analysis of variance, P < 0.05) also showed that the differences between the percentages observed for leaves 1-10 and 3-15 were significant. However, the differences between leaves 3-15 and 6-15 were not significant, suggesting a possible plateau in the methylation levels of virus in the recovered leaves (Fig. 4C). Finally, the comparison of the methylation patterns between the IR and CP segments showed that, in the first case, there is a pattern that is conserved in the three leaves analyzed: there are two areas with a higher percentage of methylation (“peaks”), resembling a bimodal distribution. The peaks are located on both sides of the conserved geminivirus stem-loop structure (Fig. 5). A sequence analysis of the high-methylation segments identified possible domains that might be important for the expression of the promoters found in the IR. On the other hand, the methylation pattern observed in the CP segment did not show conservation of methylated domains since the profile changed in the different leaves analyzed (Fig. 5). The different profiles observed in the two regions might suggest that the methylation in the IR has a biological significance, whereas the methylation of the CP region is due to a less precise, random mechanism or perhaps is a carryover from the methylation of the IR.
FIG. 5.

Distribution of methylation along the IR and CP segments. The frequency of methylation in each cytosine in both segments at all three stages analyzed (where, e.g., 1/10 is leaf 1-10) in the experiment shown in Fig. 4C was determined. Each bar represents the position of a cytosine in the IR/CP map (bottom), where important motifs are also identified. SL, stem-loop structure; TATA, TATA box for Rep and CP promoters; CLE, conserved late element in CP promoter. Twenty-two independent sequences were analyzed from each stage.
DISCUSSION
Virus-host interactions have been studied in several systems including Arabidopsis. However, processes such as host recovery have not been observed in geminivirus-infected Arabidopsis. Pepper is infected in nature by several well-characterized geminiviruses, and recovery has been reported even under field conditions. Here, we report a further characterization of the RNA silencing-associated processes acting in the host recovery process of the PepGMV-pepper interaction. The results of the leaf-by-leaf analysis confirmed a reduction of the viral DNA titers during the recovery process. Similar observations have been reported in other viral systems. However, unlike the reports with some cassava-infecting geminiviruses (14) but similar to the experiments with cucurbit-infecting viruses (21), the concentration of the siRNAs are lower in the recovered tissue than in the symptomatic one. It is possible that each virus-host combination might present its own characteristics. This idea is supported with recent results in our own lab with PHYVV and pepper plants. This combination also shows recovery; however, several differences in the siRNA populations and methylation levels have been observed (C. Hernández Zepeda and R. Rivera-Bustamante, unpublished data). We have also corroborated that the virus-associated siRNA populations are related to the entire genome although their concentrations are not homogeneous. Again, it seems that each virus or virus-host combination might present a particular pattern (14, 36, 45). For PepGMV, zones corresponding to transcripts of the Rep, TrAP, REn, and MP genes and both IRs seem to be preferential targets (hot spots) for a silencing mechanism in highly symptomatic tissue (Fig. 2). Rep, TrAP, REn, and MP are important for the virus cycle and are expressed early in the cycle (47). The fact that silencing mechanisms target these zones could allow the plant to establish a rapid and efficient defense response at an early stage. On the other hand, IRs contain cis regulatory elements (i.e., promoters and replication origin) (2, 17); therefore, the presence of IR-related siRNAs shows that the plant is also targeting regulatory elements.
The involvement of two types of siRNAs and the respective Dicer enzymes (e.g., DCL2, DCL3, and DCL4) has been suggested in some geminivirus-plant systems (1, 9, 50). However, a correlation between the siRNA types and a specific genome region or biological process, such as recovery, has not been reported. The siRNA population derived from the IR showed some differences with the population derived from coding sequences. First, the IR- and ORF-related small RNAs seem to have different kinetics. Second, IR-related siRNAs were primarily 24 nt in size, whereas the population originating from the ORF segments was mainly of 21 to 22 nt. These results suggest an independent biogenesis and/or regulation for both types of siRNAs (IR versus ORF). Transcripts through the IR, in both polarities, have been detected in PepGMV-infected plants and other systems (22; also H. Shimada-Beltrán and R. Rivera-Bustamante, unpublished data). Therefore, it is possible that these IR transcripts with a different, independent regulation are involved in the biogenesis for the IR siRNAs. It is possible that this biogenesis pathway could be similar to the one described in A. thaliana, where it has been shown that RNA-dependent RNA polymerase 2 (RDR2), DICER-LIKE 3 (DCL3), and RNA polymerase IVa (Pol IVa) proteins are all required for generating siRNAs from endogenous loci that direct RNA-directed DNA methylation (23, 27, 37, 41, 58). On the other hand, AGO4, domains-rearranged methyltransferase 2 (DRM2), the SWI/SNF-like chromatin remodeling complex defective in RNA-directed DNA methylation (DRD1), and RNA Pol IVb are required for the actual process of RNA-directed DNA methylation (11, 27, 60). In this system, it has been suggested that methylated regions of genomic DNA provide the template, either directly or indirectly, for Pol IVa and that the resultant Pol IVa transcripts are copied by RDR2 to generate double-stranded RNA (dsRNA). Subsequently, DCL3 processes the dsRNA into 24-nt siRNAs that are incorporated into an AGO4 effector complex that directs the de novo DNA methylation of homologous loci in association with Pol IVb, DRM2, and DRD1 (28, 40).
Taking all of these observations into consideration, it seems that viral coding regions could be targeted by a PTGS system, leading to virus-derived transcript degradation, whereas noncoding IRs could be transcriptionally downregulated by a TGS system through methylation modifications.
Geminivirus genomes replicate in infected plant cells through double-stranded DNA intermediates that are assembled into nucleosomes (24, 39, 48). These replicating intermediaries represent excellent targets for a TGS pathway. Several studies support a possible role of methylation as an important plant defense strategy against geminivirus. For example, in vitro methylation of geminiviral DNA drastically reduced its infectivity (10, 18). Transgenic reporter genes driven by geminiviral promoters can be transcriptionally silenced (due to hypermethylation of the inserted viral sequences) after infection of the transgenic plants with the homologous virus (7, 46). TGS signals generated by a dsRNA construct can repress the homologous promoter of an episomal, replicating viral genome (42). In addition, transgenic plants developed to express a TGS signal (siRNAs related to the IR of ACMV) showed, after inoculation, an accelerated recovery that correlated with the presence of both types of viral siRNAs, i.e., those 21 and 24 nt in length. This suggested that although the plants were developed for TGS signals, the inoculated plant is still able to respond with both defense mechanisms, TGS and PTGS (50), which is to the results being reported here.
Methylation-deficient mutant Arabidopsis plants are hypersusceptible to geminivirus infection, and hypersusceptibility is correlated with reduced levels of viral promoter cytosine methylation (43). These results suggest that geminivirus promoters (as transgenes or within the viral genome) are not inherently different from plant promoters and can, during infection, be negatively regulated through a TGS mechanism (50).
The analysis of the methylation results can be summarized as follows. First, the levels of methylation of viral DNA increase as the recovery process progress (Fig. 4 and 5). Second, whereas the methylation of the CP 5′ segment seemed to be random, the IR methylation showed a pattern conserved in the three stages analyzed (Fig. 5). Some of the preferentially methylated elements included the conserved late element and the iterons (Rep binding sites) (2). Third, DNA used for bisulfite sequencing was extracted using a procedure that enriches double-stranded DNA (3). Two strategies were used to eliminate the possibility of artifacts due to primer design. The first analysis used specific primers for either methylated or nonmethylated viral DNA (Fig. 4). The second assay used nonspecific primers (to direct the amplification of both methylated and nonmethylated viral DNA) (data not shown). No obvious differences were detected in either analysis in terms of the patterns and levels of methylation. In addition, no differences were observed in the methylation rate of symmetric and asymmetric cytosines. In plants, RNA-directed methylation occurs in both symmetric and asymmetric cytosine contexts. Symmetric methylation (CG/CNG) can be perpetuated by DNA methyltransferases that recognize hemimethylated DNA after replication. Asymmetric methylation (CHH) is considered a measure of de novo methylation because, to be maintained, it requires the continuous presence of the RNA that triggered the initial response (4, 26, 27). In the PepGMV DNA A IR, 77% (61 out of 79) of the cytosines are asymmetric. This supports the idea that the virus is transcriptionally repressed preferentially through a de novo methylation process of the IRs. A recent report with Tomato leaf curl virus found no differences in methylation between a coding region and the IR; unfortunately, the classes of siRNA were not analyzed, and the number of clones sequenced was rather low (7). Nevertheless, as mentioned before, each virus/system might present a particular response. In both cases, however, it is clear that a subpopulation of the virus remains unmethylated due to a still unknown process. This allows the virus to remain infectious and, in the case of recovery, induce again a symptomatic stage under certain conditions or treatments (e.g., trimming).
In our system, the recovery phenotype is associated with a reduction of viral DNA titers, a reduction of viral RNA levels (12), and an increase in the methylation levels of the viral IR. In this matter, it has been shown that TrAP possesses a silencing suppressor activity (8, 54), and this activity has been correlated with repression of the host protein adenosine kinase (ADK) (55). ADK is a kinase involved in the regulation of the endogenous methylation cycle and perhaps in the defense against DNA virus. Recently, experiments with a Beet curly top virus mutant in L2, the TrAP-equivalent protein, showed that viral DNA isolated from tissue of an infected Arabidopsis plant that showed recovery presented hypermethylation in viral promoter regions (43). It is possible that in our PepGMV-pepper recovery system, the plant is able to counteract the TrAP-mediated inhibition of ADK to reduce the interference with the methylation process, thereby resulting in a high percentage of methylation of the viral genome in the recovered tissue. Interestingly, plants infected with PHYVV, another begomovirus, also show recovery under our conditions. However, plants doubly infected with PepGMV and PHYVV, a mixture commonly found in nature (33, 34), do not present recovery (12). It is possible that in the presence of these two TrAPs the plant is unable to counteract their action. This hypothesis and the relationship of ADK with both recovery and mixed infections are currently under study.
Acknowledgments
E.A.R.-N. acknowledges fellowship support from Conacyt-Mexico. This research was supported by a Conacyt grant (49784-Z/2005) to R.F.R.-B.
We thank Marco Garcia-Neria for his help with statistical analysis.
Footnotes
Published ahead of print on 19 November 2008.
REFERENCES
- 1.Akbergenov, R., A. Si-Ammour, T. Blevins, I. Amin, C. Kutter, H. Vanderschuren, P. Zhang, W. Gruissem, F. Meins, T. Hohn, and M. M. Pooggin. 2006. Molecular characterization of geminivirus-derived small RNAs in different plant species. Nucleic Acids Res. 34462-471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Argüello-Astorga, G. R., R. G. Guevara-González, L. R. Herrera-Estrella, and R. F. Rivera-Bustamante. 1994. Geminivirus replication origins have a group-specific organization of iterative elements: a model for replication. Virology 20390-100. [DOI] [PubMed] [Google Scholar]
- 3.Ascencio-Ibañez, J. T., and S. B. Settlage. 2007. DNA abrasion onto plants is an effective method for geminivirus infection and virus-induced gene silencing. J. Virol. Methods 142198-203. [DOI] [PubMed] [Google Scholar]
- 4.Aufsatz, W., M. F. Mette, J. van der Winden, A. J. M. Matzke, and M. Matzke. 2002. RNA-directed DNA methylation in Arabidopsis. Proc. Natl. Acad. Sci. USA 9916499-16506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Bartel, D. P. 2004. MicroRNAs: Genomics, biogenesis, mechanism, and function. Cell 116281-297. [DOI] [PubMed] [Google Scholar]
- 6.Baulcombe, D. 2004. RNA silencing in plants. Nature 431356-363. [DOI] [PubMed] [Google Scholar]
- 7.Bian, X. Y., M. S. Rasheed, M. J. Seemanpillai, and M. A. Rezaian. 2006. Analysis of silencing escape of Tomato leaf curl virus: an evaluation of the role of DNA methylation. Mol. Plant-Microbe Interact. 19614-624. [DOI] [PubMed] [Google Scholar]
- 8.Bisaro, D. M. 2006. Silencing suppression by geminivirus proteins. Virology 344158-168. [DOI] [PubMed] [Google Scholar]
- 9.Blevins, T., R. Rajeswaran, P. V. Shivaprasad, D. Beknazariants, A. Si-Ammour, H. S. Park, F. Vazquez, D. Robertson, F. Meins, T. Hohn, and M. M. Pooggin. 2006. Four plant Dicers mediate viral small RNA biogenesis and DNA virus induced silencing. Nucleic Acids Res. 346233-6246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Brough, C. L., W. E. Gardiner, N. M. Inamdar, X. Y. Zhang, M. Ehrlich, and D. M. Bisaro. 1992. DNA methylation inhibits propagation of tomato golden mosaic virus DNA in transfected protoplasts. Plant Mol. Biol. 18703-712. [DOI] [PubMed] [Google Scholar]
- 11.Cao, X., and S. Jacobsen. 2002. Role of the Arabidopsis DRM methyltransferases in de novo DNA methylation and gene silencing. Curr. Biol. 121138-1144. [DOI] [PubMed] [Google Scholar]
- 12.Carrillo-Tripp, J., E. Lozoya-Gloria, and R. F. Rivera-Bustamante. 2007. Symptom remission and specific resistance of pepper plants after infection by Pepper golden mosaic virus. Phytopathology 9751-57. [DOI] [PubMed] [Google Scholar]
- 13.Carrington, J. C., and V. Ambros. 2003. Role of microRNAs in plant and animal development. Science 301336-338. [DOI] [PubMed] [Google Scholar]
- 14.Chellappan, P., R. Vanitharani, and C. M. Fauquet. 2004. Short interfering RNA accumulation correlates with host recovery in DNA virus-infected hosts, and gene silencing targets specific viral sequences. J. Virol. 787465-7477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Covey, S. N., N. S. Al-Kaff, A. Langara, and D. S. Turner. 1997. Plants combat infection by gene silencing. Nature 385781-782. [Google Scholar]
- 16.Dogar, A. M. 2006. RNAi dependent epigenetic marks on a geminivirus promoter. Virol. J. 35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Eagle, P. A., B. M. Orozco, and L. Hanley-Bowdoin. 1994. A DNA sequence required for geminivirus replication also mediates transcriptional regulation. Plant Cell 61157-1170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Ermak, G., U. Paszkowski, M. Wohlmuth, O. M. Scheid, and J. Paszkowski. 1993. Cytosine methylation inhibits replication of African cassava mosaic virus by two distinct mechanisms. Nucleic Acids Res. 213445-3450. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Frommer, M., L. E. McDonald, D. S. Millar, C. M. Collis, F. Watt, G. W. Grigg, P. L. Molloy, and C. L. Paul. 1992. A genomic sequencing protocol that yields a positive display of 5-methylcytosine residues in individual DNA strands. Proc. Natl. Acad. Sci. USA 891827-1831. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Grunau, C., S. J. Clark, and A. Rosenthal. 2001. Bisulfite genomic sequencing: systematic investigation of critical experimental parameters. Nucleic Acids Res. 29:e65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Hagen, C., M. R. Rojas, T. Kon, and R. L. Gilbertson. 2008. Recovery from Cucurbit leaf crumple virus (family Geminiviridae, genus Begomovirus) infection is an adaptive antiviral response associated with changes in viral small RNAs. Phytopathology 981029-1037. [DOI] [PubMed] [Google Scholar]
- 22.Hanley-Bowdoin, L., J. S. Elmer, and S. G. Rogers. 1989. Functional expression of the leftward open reading frames of the A component of tomato golden mosaic virus in transgenic tobacco plants. Plant Cell 11057-1067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Herr, A. J., M. B. Jensen, T. Dalmay, and D. C. Baulcombe. 2005. RNA polymerase IV directs silencing of endogenous DNA. Science 308118-120. [DOI] [PubMed] [Google Scholar]
- 24.Heyraud, F., V. Matzeit, M. Kammann, S. Schaefer, J. Schell, and B. Gronenborn. 1993. Identification of the initiation sequence for viral-strand DNA synthesis of wheat dwarf virus. EMBO J. 124445-4452. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Hutvágner, G., L. Mlynárová, and J. P. Nap. 2000. Detailed characterization of the posttranscriptional gene-silencing-related small RNA in a GUS gene-silenced tobacco. RNA 61445-1454. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Jones, L., F. Ratcliff, and D. F. Baulcombe. 2001. RNA-directed transcriptional gene silencing in plants can be inherited independently of the RNA trigger and requires Met1 for maintenance. Curr. Biol. 11747-757. [DOI] [PubMed] [Google Scholar]
- 27.Kanno, T., W. Aufsatz, E. Jaligot, M. F. Mette, M. Matzke, and A. J. M. Matzke. 2005. A SNF2-like protein facilitates dynamic control of DNA methylation. EMBO Rep. 6649-654. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Li, C. F., O. Pontes, M. El-Shami, I. R. Henderson, Y. V. Bernatavichute, S. W. L. Chan, T. Lagrange, C. S. Pikaard, and S. E. Jacobsen. 2006. An ARGONAUTE4-containing nuclear processing center colocalized with Cajal bodies in Arabidopsis thaliana. Cell 12693-106. [DOI] [PubMed] [Google Scholar]
- 29.Li, L. C., and R. Dahiya. 2002. MethPrimer: designing primers for methylation PCRs. Bioinformatics 181427-1431. [DOI] [PubMed] [Google Scholar]
- 30.Lippman, Z., and R. Martienssen. 2004. The role of RNA interference in heterochromatic silencing. Nature 431364-370. [DOI] [PubMed] [Google Scholar]
- 31.Matzke, M. A., and J. A. Birchler. 2005. RNAi-mediated pathways in the nucleus. Nat. Rev. Genet. 624-35. [DOI] [PubMed] [Google Scholar]
- 32.Meins, F., A. Si-Ammour, and T. Blevins. 2005. RNA silencing systems and their relevance to plant development. Ann. Rev. Cell Dev. Biol. 21297-318. [DOI] [PubMed] [Google Scholar]
- 33.Méndez-Lozano, J., R. F. Rivera-Bustamante, C. M. Fauquet, and R. De la Torre-Almaraz. 2001. Pepper huasteco virus and Pepper golden mosaic virus are geminiviruses affecting tomatillo (Physalis ixocarpa) crops in Mexico. Plant Dis. 851291. [DOI] [PubMed] [Google Scholar]
- 34.Méndez-Lozano, J., I. Torres-Pacheco, C. M. Fauquet, and R. F. Rivera-Bustamante. 2003. Interactions between geminiviruses in a naturally occurring mixture: Pepper huasteco virus and Pepper golden mosaic virus. Phytopathology 93270-277. [DOI] [PubMed] [Google Scholar]
- 35.Mette, M. F., W. Aufsatz, J. van der Winden, M. A. Matzke, and A. J. M. Matzke. 2000. Transcriptional silencing and promoter methylation triggered by double-stranded RNA. EMBO J. 195194-5201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Molnar, A., T. Csorba, L. Lakatos, E. Varallyay, C. Lacomme, and J. Burgyan. 2005. Plant virus-derived small interfering RNAs originate predominantly from highly structured single-stranded viral RNAs. J. Virol. 797812-7818. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Onodera, Y., J. R. Haag, T. Ream, P. C. Nunes, O. Pontes, and C. S. Pikaard. 2005. Plant nuclear RNA polymerase IV mediates siRNA and DNA methylation-dependent heterochromatin formation. Cell 120613-622. [DOI] [PubMed] [Google Scholar]
- 38.Pelissier, T., S. Thalmeir, D. Kempe, H. L. Sanger, and M. Wassenegger. 1999. Heavy de novo methylation at symmetrical and non-symmetrical sites is a hallmark of RNA-directed DNA methylation. Nucleic Acids Res. 271625-1634. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Pilartz, M., and H. Jeske. 1992. Abutilon mosaic geminivirus double-stranded DNA is packed into minichromosomes. Virology 189800-802. [DOI] [PubMed] [Google Scholar]
- 40.Pontes, O., C. F. Li, P. C. Nunes, J. Haag, T. Ream, A. Vitins, S. E. Jacobsen, and C. S. Pikaard. 2006. The Arabidopsis chromatin-modifying nuclear siRNA pathway involves a nucleolar RNA processing center. Cell 12679-92. [DOI] [PubMed] [Google Scholar]
- 41.Pontier, D., G. Yahubyan, D. Vega, A. Bulski, J. Saez-Vasquez, M. A. Hakimi, S. Lerbs-Mache, V. Colot, and T. Lagrange. 2005. Reinforcement of silencing at transposons and highly repeated sequences requires the concerted action of two distinct RNA polymerases IV in Arabidopsis. Genes Dev. 192030-2040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Pooggin, M., and T. Hohn. 2003. RNAi targeting of DNA virus in plants. Nat.Biotechnol. 21131-132. [DOI] [PubMed] [Google Scholar]
- 43.Raja, P., B. C. Sanville, R. C. Buchmann, and D. M. Bisaro. 2008. Viral genome methylation as an epigenetic defense against geminiviruses. J. Virol. 828997-9007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Ratcliff, F., B. D. Harrison, and D. C. Baulcombe. 1997. A similarity between viral defense and gene silencing in plants. Science 2761558-1560. [DOI] [PubMed] [Google Scholar]
- 45.Ribeiro, S. G., H. Lohuis, R. Goldbach, and M. Prins. 2007. Tomato chlorotic mottle virus is a target of RNA silencing but the presence of specific short interfering RNAs does not guarantee resistance in transgenic plants. J. Virol. 811563-1573. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Seemanpillai, M., I. Dry, J. Randles, and A. Rezaian. 2003. Transcriptional silencing of geminiviral promoter-driven transgenes following homologous virus infection. Mol. Plant-Microbe Interact. 16429-438. [DOI] [PubMed] [Google Scholar]
- 47.Shimada-Beltrán, H., and R. F. Rivera-Bustamante. 2007. Early and late gene expression in pepper huasteco yellow vein virus. J. Gen. Virol. 883145-3153. [DOI] [PubMed] [Google Scholar]
- 48.Stenger, D. C., G. N. Revington, M. C. Stevenson, and D. M. Bisaro. 1991. Replicational release of geminivirus genomes from tandemly repeated copies: evidence for rolling-circle replication of a plant viral DNA. Proc. Nat. Acad. Sci. USA 888029-8033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Szittya, G., A. Molnar, D. Silhavy, C. Hornyik, and J. Burgyan. 2002. Short defective interfering RNAs of tombusviruses are not targeted but trigger post-transcriptional gene silencing against their helper virus. Plant Cell 14359-372. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Vanderschuren, H., R. Akbergenov, M. M. Pooggin, T. Hohn, W. Gruissem, and P. Zhang. 2007. Transgenic cassava resistance to African cassava mosaic virus is enhanced by viral DNA-A bidirectional promoter-derived siRNAs. Plant Mol. Biol. 64549-557. [DOI] [PubMed] [Google Scholar]
- 51.Vaucheret, H. 2006. Post-transcriptional small RNA pathways in plants: mechanisms and regulations. Genes Dev. 20759-771. [DOI] [PubMed] [Google Scholar]
- 52.Vazquez, F., H. Vaucheret, R. Rajagopalan, C. Lepers, V. Gasciolli, A. C. Mallory, J. L. Hilbert, D. P. Bartel, and P. Crete. 2004. Endogenous trans-acting siRNAs regulate the accumulation of Arabidopsis mRNAs. Mol. Cell 1669-79. [DOI] [PubMed] [Google Scholar]
- 53.Voinnet, O. 2005. Induction and suppression of RNA silencing: Insights from viral infections. Nat. Rev. Genet. 6206-220. [DOI] [PubMed] [Google Scholar]
- 54.Voinnet, O., Y. M. Pinto, and D. C. Baulcombe. 1999. Suppression of gene silencing: A general strategy used by diverse DNA and RNA viruses of plants. Proc. Natl. Acad. Sci. USA 9614147-14152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Wang, H., K. J. Buckley, X. J. Yang, R. C. Buchmann, and D. M. Bisaro. 2005. Adenosine kinase inhibition and suppression of RNA silencing by geminivirus AL2 and L2 proteins. J. Virol. 797410-7418. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Warnecke, P. M., C. Stirzaker, J. Song, C. Grunau, J. R. Melki, and S. J. Clark. 2002. Identification and resolution of artifacts in bisulfite sequencing. Methods 27101-107. [DOI] [PubMed] [Google Scholar]
- 57.Xie, Z. X., E. Allen, A. Wilken, and J. C. Carrington. 2005. DICER-LIKE 4 functions in trans-acting small interfering RNA biogenesis and vegetative phase change in Arabidopsis thaliana. Proc. Natl. Acad. Sci. USA 10212984-12989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Xie, Z. X., L. K. Johansen, A. M. Gustafson, K. D. Kasschau, A. D. Lellis, D. Zilberman, S. E. Jacobsen, and J. C. Carrington. 2004. Genetic and functional diversification of small RNA pathways in plants. PLoS Biol. 2642-652. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Zhang, P., H. Vanderschuren, J. Futterer, and W. Gruissem. 2005. Resistance to cassava mosaic disease in transgenic cassava expressing antisense RNAs targeting virus replication genes. Plant Biotechnol. J. 3385-397. [DOI] [PubMed] [Google Scholar]
- 60.Zilberman, D., X. F. Cao, L. K. Johansen, Z. X. Xie, J. C. Carrington, and S. E. Jacobsen. 2004. Role of Arabidopsis ARGONAUTE4 in RNA-directed DNA methylation triggered by inverted repeats. Curr. Biol. 141214-1220. [DOI] [PubMed] [Google Scholar]