

Abstract
Murine gammaherpesvirus 68 (MHV68) establishes a lifelong infection in mice and is used as a model pathogen to study the role of viral and host factors in chronic infection. The maintenance of chronic MHV68 infection, at least in some latency reservoirs, appears to be dependent on the capacity of the virus to reactivate from latency in vivo. However, the signals that lead to MHV68 reactivation in vivo are not well characterized. Toll-like receptors (TLRs), by recognizing the specific patterns of microbial components, play an essential role in the activation of innate immunity. In the present study, we investigated the capacity of TLR ligands to induce MHV68 reactivation, both in vitro and in vivo. The stimulation of latently infected B cell lines with ligands for TLRs 3, 4, 5, and 9 enhanced MHV68 reactivation; the ex vivo stimulation of latently infected primary splenocytes, recovered from infected mice, with poly(I:C), lipopolysaccharide, flagellin, or CpG DNA led to early B-cell activation, B-cell proliferation, and a significant increase in the frequency of latently infected cells reactivating the virus. In vivo TLR stimulation also induced B-cell activation and MHV68 reactivation, resulting in heightened levels of virus replication in the lungs which correlated with an increase in MHV68-specific CD8+ T-cell responses. Importantly, TLR stimulation also led to an increase in MHV68 latency, as evidenced by an increase in viral genome-positive cells 2 weeks post-in vivo stimulation by specific TLR ligands. Thus, these data demonstrate that TLR stimulation can drive MHV68 reactivation from latency and suggests that periodic pathogen exposure may contribute to the homeostatic maintenance of chronic gammaherpesvirus infection through stimulating virus reactivation and reseeding latency reservoirs.
Gammaherpesviruses are characterized by their capacity to establish lifelong latent infection within host lymphocytes. Virus reactivation is thought to be necessary for transmission of the virus to new hosts and may also be required to maintain reservoirs of latently infected cells in the chronically infected host (8, 19, 30, 52, 53). The switch between latency and the lytic cycle for the human gammaherpesviruses Epstein-Barr virus (EBV) and Kaposi's sarcoma-associated herpesvirus (KSHV) has been extensively characterized in established latently infected cell lines in vitro (18, 45, 61). The initiation of the EBV lytic cycle can be stimulated by several different reagents, including anti-immunoglobulin (anti-Ig), calcium ionophore, sodium butyrate, and tetradecanoyl phorbol acetate (TPA) (45). KSHV reactivation can also be induced by stimulation with phorbol esters and sodium butyrate (9, 37).
Toll-like receptors (TLRs) are important pattern recognition receptors in innate immunity. Following TLR engagement by ligands of microbial origin, dendritic cells undergo maturation, which in turn activates the adaptive immune response. Pathogen-associated molecular patterns (PAMPs) recognized by TLRs can come from bacteria, fungi, protozoans, insects, or viruses. TLR1/2 and -2/6 recognize bacterial components (e.g., lipoproteins) and components from yeast (e.g., zymosan) (40, 48-51), TLR4 recognizes lipopolysaccharides (LPS) (44), and TLR5 recognizes flagellin (20). TLRs that sense viral PAMPs include TLR3 recognition of double-stranded RNA (1), TLR7 and TLR8 (14, 21, 22, 26, 34) recognition of single-stranded RNA (ssRNA), and TLR9 sensing of unmethylated CpG DNA (12, 23, 28, 33).
The engagement of TLR ligands, as well as heterologous viral infections, can trigger the reactivation of latent infections. Signaling through TLR2, -4, or -9 enhances viral replication from human immunodeficiency virus type 1 (HIV-1) latently infected mast cells (46). LPS can also induce KSHV reactivation from BCBL-1 cells (38) and the reactivation of latent murine cytomegalovirus (CMV) (13). HIV-1 infection of the primary effusion lymphoma cell lines BC-3 and BCBL-1 triggers KSHV reactivation (36, 57). CMV superinfection of cell lines latently infected with EBV (BJAB-B1 and P3HR-1) induces the reactivation of EBV (3).
Although EBV and KSHV reactivation has been extensively studied in tissue culture, little is known regarding the triggers of gammaherpesvirus reactivation in vivo. Murine gammaherpesvirus 68 (MHV68) is closely related to EBV and KSHV. The infection of mice with MHV68 provides a tractable small-animal model in which to study the pathogenesis of gammaherpesviruses in vivo. The ectopic expression of the MHV68 orf50 gene product RTA (the lytic transactivator) in an MHV68 latently infected lymphoma B-cell line, S11, can drive the expression of both early and late viral genes and the production of lytic virus (55, 63). We have previously demonstrated that, like EBV and KSHV, TPA and anti-Ig can stimulate MHV68 reactivation from MHV68 latently infected B-cell lines. In addition, we have shown that the ex vivo stimulation of latently infected splenocytes with anti-Ig/anti-CD40, as well as LPS stimulation, enhances MHV68 reactivation (39).
In the present study, we investigated the role of TLR stimulation in inducing MHV68 reactivation. We demonstrate that the stimulation of MHV68 latently infected B-cell lines with ligands for TLRs 3, 4, 5, and 9 increases the output viral titer. The ex vivo stimulation of primary splenocytes latently infected with MHV68 with these same TLR ligands led to B-cell activation and proliferation and an increase in the frequency of cells reactivating the virus. Lastly, the administration of LPS or CpG DNA in vivo at 42 days postinfection (dpi) triggered an increase in MHV68 reactivation, early B-cell activation, and CD8+ T-cell responses. MHV68 reactivation in the spleen was accompanied by an increase in virus replication in the lungs. Importantly, the increase in MHV68 reactivation/replication led to an increase in the frequency of MHV68 latently infected splenocytes 14 days poststimulation.
MATERIALS AND METHODS
Cells, virus, and virus culture.
MHV68 strain WUMS (ATCC VR1465) was used for all virus infections unless indicated. Virus passage, maintenance, and titers were performed as previously described (11). NIH 3T12 cells and mouse embryonic fibroblast (MEF) cells were maintained in Dulbecco's modified Eagle's medium supplemented with 100 U penicillin/ml, 100 mg streptomycin/ml, 10% fetal calf serum (FCS), and 2 mM l-glutamate (cMEM). A20-HE1 and A20-HE2 (16) and A20 were maintained in RPMI medium supplemented with 100 U penicillin/ml, 100 mg streptomycin/ml, 10% FCS, and 2 mM l-glutamate; the A20-HE cells were also maintained with 20 ng/ml hygromycin and 300 μg/ml B sulfate in the medium. The cells were maintained at 37°C in a 5% CO2 environment. MEF cells were prepared from C57BL/6 mice as previously described (43).
Mice, infections, and organ harvests.
Six- to eight-week-old C57BL/6 mice (Jackson Laboratory, MD) were housed and bred in the Whitehead Building vivarium at the Emory University School of Medicine in accordance with all federal, university, and facility regulations. The mice were placed under isoflurane anesthesia prior to intranasal inoculation with 1,000 PFU of virus in 20 μl of cMEM. The mice were anesthetized with isoflurane prior to sacrifice by cervical dislocation. Spleens were homogenized in cMEM and filtered through a 100-μm pore size nylon cell strainer (Becton Dickinson). Erythrocytes were removed with red-blood-cell lysis buffer (Sigma, St. Louis, MO). Splenocytes from three to five mice were used in all experiments. Lungs were harvested intact into cMEM and stored at −80°C.
A20-HE cell stimulation.
A20-HE cells were resuspended at 1 × 106/ml of RPMI complete medium and stimulated for 24 h with either 20 ng/ml phorbol myristate acetate (PMA), 0.5 μg/ml Pam3CSK4, 10 μg/ml poly I:C, 1 μg/ml LPS, 600 μg/ml flagellin, 0.5 μg/ml FLS-1, 0.5 μg/ml ssRNA, or 1 μM CpG (Invivogen). After 24 h, the supernatants were harvested and freeze-thawed three times prior to the plaque assay.
Plaque assays.
The plaque assays were performed as previously described (11), with minor modifications. NIH 3T12 cells were plated onto six-well plates 1 day prior to infection at 2.5 × 105 cells/well. Serial 10-fold dilutions of the supernatants were plated into NIH 3T12 monolayers in a 200-μl volume and allowed to absorb for 1 h at 37°C with rocking every 15 min. Immediately after infection, the plates were overlaid with 2% methylcellulose in cMEM. After 7 days, the plates were stained with a neutral red overlay, and the plaques were scored the next day. The limit of detection for this assay was 50 PFU/ml.
Flow cytometry.
For the flow cytometry analysis, cells were resuspended at 1 × 106 cells/ml in phosphate-buffered saline (PBS) containing 2% FCS, and prior to staining, they were Fc blocked with rat anti-mouse CD16/CD32 for 10 min and then stained with a combination of the following antibodies: fluorescein isothiocyanate-conjugated antibodies to CD69; phycoerythrin (PE)-conjugated antibodies to CD19, CD44, and CD62L; peridinin chlorophyll protein-conjugated antibodies to CD8 and CD4; PE-Cy7-conjugated antibodies to CD69 and CD11a; PacificBlue-conjugated antibodies to CD8, all purchased from BD Pharmingen; and allophycocyanin-conjugated antibody to CD8 purchased from eBioscience. Tetramers to ORF6487 to 495 (H-2Db) were synthesized at the NIH Tetramer Core Facility at Emory University and conjugated to streptavidin-allophycocyanin (Molecular Probes) according to the Core protocol. Antibodies were used at a 1:100 dilution except for the PE- and PE-Cy7-conjugated antibodies, which were used at 1:200, for 20 min on ice in the dark. The cells were then washed with PBS/2% FCS and resuspended in 200 μl. Data were collected on an LSR II flow cytometer (BD Biosciences) and analyzed using FloJo software (TreeStar).
Limiting-dilution ex vivo reactivation analyses.
Limiting-dilution analyses to determine the frequency of cells spontaneously reactivating from latency upon explant was performed as described previously (58, 60). Briefly, bulk splenocytes were resuspended in cMEM and plated in serial twofold dilutions (starting with 106 cells) onto MEF monolayers in 96-well tissue culture plates. Twelve dilutions were plated per sample, and 24 wells were plated per dilution. The wells were scored for cytopathic effect at day 21 postplating. To detect preformed infectious virus, parallel samples of mechanically disrupted cells were plated onto MEF monolayers. This process kills more than 99% of live cells, which allows preformed infectious virus to be discerned from virus reactivating from latently infected cells (58-60). The level of sensitivity of this assay is 0.2 PFU (59). Unless otherwise indicated, significant levels of preformed virus were not detected in these assays.
Limiting-dilution nested-PCR detection of MHV68 genome-positive cells.
The frequency of cells harboring the MHV68 genome was determined using a limiting-dilution analysis coupled with a single-copy-sensitivity nested PCR assay to detect the MHV68 orf50 gene sequence, as previously described (59, 60). Briefly, bulk splenocytes were thawed, counted, and resuspended in isotonic buffer. A series of six threefold serial dilutions, starting with 104 cells/well, were plated in a background of 104 uninfected NIH 3T12 cells in 96-well PCR plates. The cells were lysed prior to the nested PCR by 6 h of treatment at 56°C in the presence of detergent and proteinase K. Then, 10 μl of round 1 PCR mix was added to each well. Following the first-round PCR, 10 μl of round 2 PCR buffer was added to each well and samples were subjected to a second round of PCR. All cell lysis and PCRs were performed on a PrimusHT thermal cycler (MWG Biotech). The products were resolved by ethidium bromide staining on 2% agarose gels. Twelve PCRs were performed for each sample dilution, and a total of six dilutions were performed for each sample. Every PCR plate contained control reactions (uninfected cells and 10 copies, 1 copy, and 0.1 copy of plasmid DNA in a background of 104 cells). All of the assays demonstrated approximately single-copy sensitivity with no false positives.
Ex vivo stimulation of latently infected splenocytes.
Splenocytes were harvested from day 42 infected mice. A total of 106 cells were stimulated with medium alone, 20 ng/ml TPA, 0.5 μg/ml Pam3CSK4, 10 μg/ml poly(I:C), 1 μg/ml LPS, 600 μg/ml flagellin, 0.5 μg/ml FLS-1, 0.5 μg/ml ssRNA, or 1 μM CpG DNA. The stimulated splenocytes were labeled with 1 μM carboxyfluorescein diacetate succinimidyl ester (CFSE) and harvested at different times to assay cellular proliferation or were plated into the limiting-dilution ex vivo reactivation assay, in the presence or absence of TLR ligands.
In vivo administration of TLR ligands.
C57BL/6 mice were infected with 1,000 PFU intranasally and, at 42 days postinfection, were injected intraperitoneally (i.p.) with either 200 μl PBS, 15 μg LPS, or 20 μg CpG DNA in 200 μl PBS. The splenocytes and lungs were then harvested at days 1, 3, 7, and 14 poststimulation and processed as described above.
Measurement of viral persistence in the lung.
Persistent MHV68 replication in the lungs was measured using a modified form of the limiting-dilution ex vivo reactivation assay described above. Briefly, the left lung from each animal was mechanically disrupted as described previously (56). This disrupts >99% of the cells present but has less than a twofold effect on the titer of the preformed infectious virus (58). The homogenate was plated in a twofold-dilution series onto MEFs in 96-well tissue culture plates—12 dilutions of 16 replicate wells were plated. The appearance of a cytopathic effect (CPE) was monitored microscopically and read 21 days postplating. The individual lungs from three to five mice were used in all experiments.
Statistical analyses.
All data were analyzed by using GraphPad Prism software (GraphPad Prism). Viral titer data were statistically analyzed using the Mann-Whitney nonparametric two-tailed t test. Based on Poisson distribution, the frequencies of reactivation and viral genome-positive cells were obtained from a nonlinear regression fit of the data where the regression line intersects 63.2%. The frequencies of reactivation and viral genome-positive cells were statistically analyzed by an unpaired two-tailed t test of the log 63.2% effective concentration.
RESULTS
B-cell stimulation by TLR ligands drives MHV68 reactivation from latently infected B-cell lines.
To determine whether TLR ligands can trigger MHV68 reactivation, we used murine A20 B cells latently infected with MHV68 (A20-HE cell lines), which we have previously shown are responsive to a number of reactivation stimuli (16). Two independently derived A20-HE cell lines were treated with different TLR ligands or PMA as a positive control. The supernatants from the untreated and treated A20-HE cells were recovered at 24 h postinduction and assayed for changes in virus production by plaque assay on murine NIH 3T12 fibroblasts (Fig. 1A). To control for the possible toxic effects of these treatments on NIH 3T12 fibroblasts, a parallel series of treatments of the MHV68-negative parental murine A20 B-cell line was carried out. Importantly, supernatants from the treated A20 cells were also plated onto monolayers of NIH 3T12 cells, and no cytopathic effect was detected under any of the treatment conditions (data not shown). The stimulation of A20-HE cells with PMA resulted in a strong induction of virus reactivation (Fig. 1A). In addition, stimulation with the TLR ligands poly(I:C) (TLR3), LPS (TLR4), flagellin (TLR5), and CpG DNA (TLR9) also elicited substantial increases in virus reactivation/replication (Fig. 1A). It should be noted that we performed reverse transcriptase PCR using intron-spanning, gene-specific primers for mouse TLRs 1 to 9 and found that the A20-HE cells express all of these TLRs (data not shown). Thus, we conclude that these data demonstrate that stimulation through a subset of TLRs can promote MHV68 reactivation from latently infected B-cell lines.
FIG. 1.

Stimulation with TLR ligands enhances the reactivation of MHV68 latently infected B-cell lines in vitro. Two independently derived MHV68 latently infected cell lines (A20-HE1 and -HE2 cell lines) were treated with the indicated reagents. Tissue culture supernatants were harvested at 24 h posttreatment and virus titers determined by plaque assay. The data shown are representative of the results from three independent experiments. (A) The following ligands were used to stimulate specific TLRs as follows: Pam3CSK4 (TLR1/2), HKLM (heat-killed Listeria monocytogenes) (TLR2), poly(I:C) (TLR3), LPS (TLR4), flagellin (TLR5), FSL1 (TLR6/2), ssRNA (TLR7), and CpG DNA (TLR9). (B) Dose response of A20-HE1 and -HE2 cells to ssRNA (TLR7).
HIV infection can induce KSHV reactivation (36, 57). Furthermore, it has also been shown that TLR7 can recognize HIV ssRNA (4, 21, 35). Thus, we wanted to further investigate the role of TLR7 signaling and MHV68 reactivation since the previous failure of MHV68 to reactivate in response to ssRNA could reflect a true lack of response or alternatively may be due to the experimental setup. To ensure that the concentration of ssRNA used to stimulate the A20-HE cells would be sufficient to induce MHV68 reactivation, we performed a dose-response with ssRNA (Fig. 1B). Notably, we observed that varying the concentration of ssRNA from 0.25 μg to 10 μg made no difference in the inability of TLR7 stimulation to induce MHV68 reactivation (Fig. 1B).
TLR stimulation leads to early B-cell activation and proliferation, which correlates with an increase in virus reactivation from primary splenocytes.
The B-cell receptor-dependent stimulation of MHV68 latently infected splenocytes leads to an increase in B-cell activation and proliferation that correlates with viral reactivation (39). To investigate the impact of TLR stimulation, we harvested splenocytes from mice 42 dpi after intranasal infection. Notably, by 42 dpi, there is barely detectable spontaneous virus reactivation from splenocytes detected in our limiting-dilution ex vivo reactivation assay (54). We focused our analysis on those TLR ligands that generated the largest stimulation of virus reactivation from the A20-HE cell lines (Fig. 1). The TLR ligands poly(I:C), LPS, flagellin, and CpG DNA were added to bulk splenocytes and B-cell activation and proliferation examined over several days poststimulation. Within 24 h poststimulation, a significant number of the B cells were activated, as evidenced by an upregulation of CD69 compared to unstimulated control cells (Fig. 2A). We also assessed proliferation by CFSE labeling cells. CFSE is a dye that labels cells by incorporation into the cell and with each cell division leads to dilution of the dye in daughter cells. The CFSE-labeled cells were stimulated with TLR ligands and analyzed by flow cytometry for the loss of CFSE staining at several time points poststimulation. At 24 h poststimulation, there was very little evidence of B-cell proliferation (Fig. 2B, open histogram). However, by 120 h poststimulation, all the B cells present in the cultures had proliferated to various extents, with those cultures treated with TLR ligands exhibiting greater proliferation than the unstimulated culture (Fig. 2B, shaded histogram). These data indicate that TLR stimulation is effective in activating B cells and driving them to proliferate.
FIG. 2.

The ex vivo TLR stimulation of splenic B cells induces B-cell activation and proliferation. (A) Bulk splenocytes were harvested 42 dpi from mice and were either stimulated with the indicated TLR ligand or left unstimulated in medium. Cells were harvested at the indicated times poststimulation, labeled with anti-CD69 and anti-CD19 antibodies, and analyzed by flow cytometry. Error bars represent standard deviations between the results from individual mice. (B) Mice were intranasally infected with 1,000 PFU wild-type MHV68, and at 42 dpi, bulk splenocytes were labeled with CFSE and stimulated with the indicated treatments. At several time points poststimulation, the cells were stained with anti-CD19 antibody and analyzed by flow cytometry for the loss of CFSE. The flow cytometry histograms shown were gated on CD19+ cells. The unfilled histograms represent CFSE-staining levels at 24 h poststimulation, while the shaded histograms represent CFSE staining at 120 h poststimulation. The results shown are representative of the results obtained from three independent experiments with three to four mice per group.
To assess whether TLR-dependent B-cell activation and proliferation had any impact on MHV68 reactivation from splenocytes, we recovered splenocytes 42 dpi and plated them in a limiting-dilution reactivation assay either with medium alone or with the addition of either PMA, poly(I:C), LPS, flagellin, or CpG DNA. Notably, ex vivo stimulation with PMA and/or the TLR ligands resulted in a substantial increase in the frequency of splenocytes reactivating from latency (Fig. 3). Since the readout of the ex vivo reactivation assay is the presence of CPE on monolayers of MEFs, we also assessed whether any of these treatments alone caused the appearance of CPE on monolayers of MEFs. Notably, none of the treatment conditions used led to detectable CPE on the MEF monolayers (data not shown). Furthermore, a quantitative PCR analysis of the MHV68 genomes demonstrated an increase in viral genomes at 48 and 72 h following LPS or CpG DNA stimulation (data not shown). Taken together, these findings demonstrate that ex vivo TLR stimulation induces B-cell activation, proliferation, and enhanced MHV68 reactivation from latency.
FIG. 3.

Stimulation of MHV68 reactivation from explanted splenocytes with TLR ligands. Forty-two days following a 1,000 PFU wild-type MHV68 intranasal inoculation, bulk splenocytes were obtained and were analyzed for the frequency of cells reactivating virus with and without TLR stimulation upon explant. The data shown represent the results from three independent experiments, with cells pooled from three to four mice per experimental group. Based on these limiting-dilution analyses, the frequency of cells reactivating virus under each condition was as follows: no treatment (medium), ∼1 in 500,000; PMA, 1 in 30,000; poly(I:C), 1 in 62,000; LPS, 1 in 30,000; flagellin, 1 in 30,000; and CpG DNA, 1 in 19,000.
TLR4 and TLR9 stimulation in vivo drives B-cell activation and increased virus reactivation/replication.
We next tested the capacity of TLR agonists to induce B-cell activation in vivo. For these analyses, we used LPS and CpG DNA, the two most potent ligands for driving reactivation ex vivo. Also, LPS is a known B-cell mitogen, and CpG DNA directly triggers B-cell activation (27). The mice were intranasally infected with 1,000 PFU intranasally, and at 42 dpi, the mice received i.p. administration of either PBS, 15 μg LPS, or 20 μg CpG DNA. Splenocytes were harvested at different times poststimulation and analyzed by flow cytometry for B-cell activation by an upregulation of CD69 expression. What we observed was similar to that for ex vivo stimulation: the peak of the B-cell activation occurred 1 day after both LPS and CpG DNA were administered (Fig. 4). We observed a ca. threefold increase in B-cell activation in the LPS- and CpG DNA-treated mice compared to that for the PBS-treated control mice (Fig. 4B). Importantly, these data demonstrate that TLR ligand-induced B-cell activation occurs in mice latently infected with MHV68.
FIG. 4.
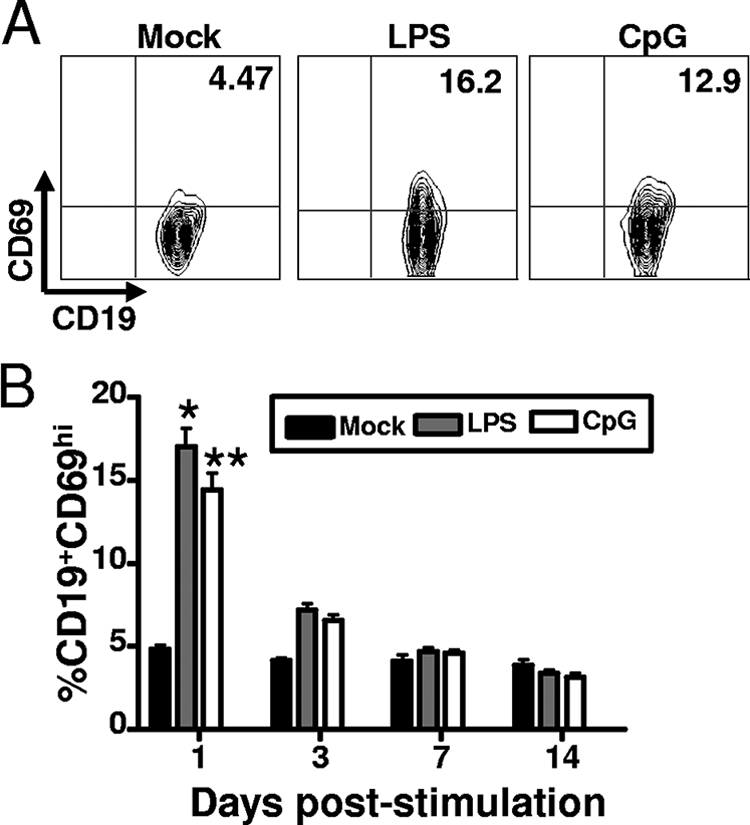
LPS and CpG DNA treatment in vivo leads to B-cell activation. Forty-two days after the mice were intranasally infected with 1,000 PFU of wild-type MHV68, the mice were administered PBS, 15 μg LPS, or 20 μg CpG DNA via i.p. injection. Splenocytes were harvested at different times poststimulation, stained with anti-CD19 and anti-CD69 antibodies, and analyzed by flow cytometry. (A) Representative flow cytometry plots of CD69 expression levels in purified B-cell fractions (splenocytes were pooled from four to five mice per experimental group). The values shown in the upper right quadrants are the percentages of CD19+ cells that express CD69 activation marker. (B) Percentage of CD19+ CD69hi cells at the indicated days poststimulation. Error bars represent the standard deviations between the results for individual mice. The percentages of CD19+ CD69hi cells in the spleens of mice treated with LPS (P < 0.0001) or CpG DNA (P < 0.0001) at day 1 posttreatment were significantly different from that of the PBS-treated mice. *, LPS stimulation induced a statistically significant increase in the percentage of activated B cells compared to those treated with PBS. **, CpG stimulation induced a statistically significant increase in the percentage of activated B cells compared to those treated with PBS.
We next assessed whether LPS or CpG DNA treatment of mice in vivo impacted MHV68 reactivation. After i.p. administration of LPS or CpG DNA, splenocytes were harvested at different times poststimulation and plated for reactivation. Notably, no further stimulation was given upon the explant of splenocytes. At day 1 poststimulation, we did not detect any stimulation-induced change in MHV68 reactivation (Fig. 5) or any increase in the presence of preformed infectious virus in the spleen (the latter was assessed by mechanically disrupting splenocytes) (Fig. 5). In contrast, by 3 days poststimulation, we were able to detect a more than twofold increase in MHV68 reactivating from latency in mice treated with LPS or CpG DNA (Fig. 5). This modest increase in virus reactivation correlated with a substantial increase in the presence of preformed infectious virus in the spleens of mice that received LPS or CPG DNA treatment (Fig. 5). The induction of virus reactivation and the presence of preformed infection virus were apparent at both days 3 and 7 poststimulation and appeared to largely subside by 2 weeks posttreatment (Fig. 5). These data provide compelling evidence that in vivo TLR stimulation can trigger virus reactivation in vivo.
FIG. 5.
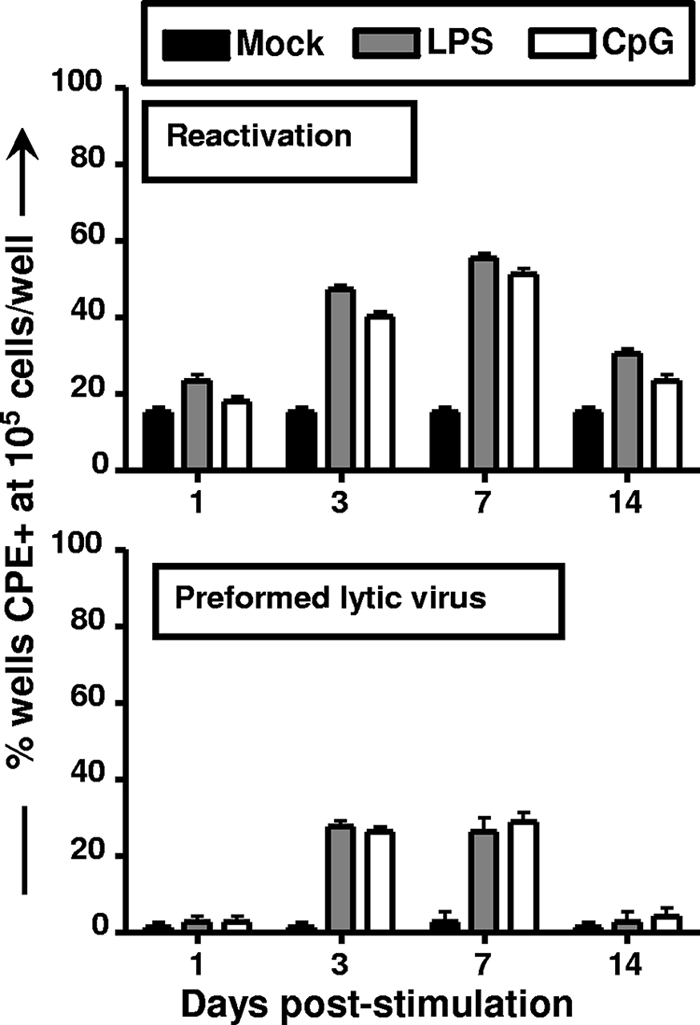
LPS and CpG DNA treatment of latently infected mice stimulate the reactivation of MHV68. Mice were infected intranasally with 1,000 PFU of wild-type MHV68. At 42 dpi, the mice were treated with PBS, LPS, or CpG DNA via i.p. injection. Limiting-dilution analyses assessed the impact of in vivo TLR stimulation on virus reactivation upon explant at the indicated days poststimulation. Shown in the top panel are the percentages of wells in which viral CPE were detected when 105 cells were plated. Similarly, plating mechanically disrupted cells assessed the presence of preformed infection virus. The frequency of wells exhibiting CPE when 105 cell equivalents were plated is shown in the bottom panel. The data shown represent the results compiled from three independent experiments, with cells pooled from four to five mice per experimental group.
To extend these analyses, we assessed whether TLR stimulation led to the recrudescence of virus in the lungs by performing a limiting-dilution assay on lung homogenate. Following an intranasal infection, the lungs are the primary site of acute lytic replication, which is cleared by ca. 12 days postinfection (54). At day 42 postinfection, the PBS-treated mice had very low levels of replicating virus in the lungs (Fig. 6). As observed in the spleen, there was a significant increase in virus replication in the lungs of mice that received either LPS or CpG DNA which could be detected at both days 3 and 7 posttreatment and was largely cleared by 2 weeks posttreatment (Fig. 6).
FIG. 6.

Replication of MHV68 in the lungs following in vivo TLR stimulation. Mice infected with 1,000 PFU wild-type MHV68 intranasally were treated with PBS, LPS, or CpG DNA at 42 dpi, followed by analysis of virus replication in the lungs at the indicated days posttreatment. Serial dilutions of lung homogenates were plated onto MEF indicator monolayers and scored for viral CPE 21 days postplanting with 16 replicates plated per dilution. Each point on the scatter plot represents the analysis of lung homogenate from an individual mouse, and the data represent analyses of the results from three independent experiments containing four to five mice per condition per experiment. Statistically significant differences were observed at both day 3 posttreatment (mock versus LPS treated, P < 0.0001; mock versus CpG DNA treated, P < 0.0001) and day 7 posttreatment (mock versus LPS treated, P < 0.0001; mock versus CpG DNA treated, P < 0.0001). Nonlinear regression analyses were performed to determine the 50% tissue culture infective dose.
MHV68 reactivation in vivo correlates with an increase in virus-specific, activated, and effector CD8+ T cells.
If LPS and CpG DNA induce MHV68 reactivation and subsequent virus replication in vivo, we reasoned that a CD8+ T-cell response to the reactivating virus would ensue. To test this hypothesis, we assessed the frequency of CD8+ T cells by following viral-specific, activated, and effector CD8+ T cells. One day poststimulation, there was little difference in the MHV68-specific CD8+ T-cell response between PBS-treated and LPS- or CpG-treated mice, as examined by tetramer staining of MHV68 ORF6487 to 495 (data not shown). However, by day 7 poststimulation, there was a statistically significant increase in the MHV68-specific CD8+ T-cell response (Fig. 7A). This increase in the MHV68-specific response corresponded with a global increase in CD8+ T-cell activation in LPS- and CpG-treated mice, as seen by a downregulation of CD62L (Fig. 7B). Finally, we observed an increase in effector CD44hi CD62Llo CD8+ T cells in mice that received LPS or CpG DNA (Fig. 7C). These data indicate that an increase in MHV68 reactivation/replication corresponds to an increase in the CD8+ T-cell response, suggesting the reengagement of an adaptive immune response to MHV68 reactivation/replication.
FIG. 7.

Increase in CD8+ T-cell responses following the in vivo stimulation of MHV68 reactivation. Mice were infected intranasally with 1,000 PFU of wild-type MHV68 and at 42 dpi were stimulated with PBS, LPS, or CpG DNA, and splenocytes were harvested at day 7 poststimulation. (A) Cells were stained with anti-CD8, a major histocompatibility complex I tetramer to an epitope in the MHV68 p56 antigen (ORF6487 to 495), as an indicator of the MHV68-specific CD8+ T-cell response. Each point on the scatter plot represents the percentage of CD8+ p56+ CD8+ T cells from the spleen of an individual mouse. The percentages of tetramer-positive CD8+ T cells in the spleens of mice treated with LPS (P < 0.0001) or CpG DNA (P < 0.0001) were significantly different from that of the control (PBS-treated) mice. (B) Splenocytes recovered from the control and treated mice were stained with anti-CD8 and anti-CD62L. Each data point on the scatter plot represents the percentage of CD8+ CD62Llo T cells, as a marker of activated CD8+ T cells, from the spleen of a single mouse. The percentages of activated CD8+ T cells in the spleens of mice treated with LPS (P < 0.0001) or CpG DNA (P < 0.0001) were significantly different from that of the PBS-treated mice. (C) To examine effector CD8+ T cells, splenocytes were stained with anti-CD8, anti-CD62L, and anti-CD44. Each point on the scatter plot represents the percentage of CD8+ CD62Llo CD44hi T cells in the spleen of a single mouse. The percentages of effector CD8+ T cells in the spleens of mice treated with LPS (P < 0.0001) or CpG DNA (P < 0.0001) were significantly different from that of the PBS-treated mice. The horizontal bars represent the mean percentages. The data shown represent the results of three individual experiments with four to five mice per group, including the naïve mice.
The TLR-mediated induction of MHV68 reactivation/replication leads to an increase in MHV68 latency in vivo.
The homeostatic maintenance of latency during chronic infection might involve the periodic reactivation and reseeding of latency reservoirs. To test whether TLR-induced reactivation leads to changes in the levels of virus latency in the spleen, we analyzed changes in the frequency of splenocytes harboring MHV68 between 1 and 14 days poststimulation. We found the frequencies of viral genome-positive cells were similar at day 1 following PBS (1/3,600), LPS (1/4,000) and CpG (1/4,500) treatment (Fig. 8). Remarkably, while the PBS-treated controls exhibited the same frequencies of viral genome-positive splenocytes at day 14 (1/4,000), mice treated with LPS (1/550) or CpG (1/650) had a substantial increase in the frequency of viral genome-positive cells at day 14 poststimulation (Fig. 8). Therefore, we conclude that the TLR-stimulated induction of MHV68 reactivation appears to seed latency in vivo. These data suggest that periodic heterologous infections may contribute to the homeostatic maintenance of latent gammaherpesvirus infection through TLR-mediated virus reactivation/replication.
FIG. 8.

Stimulation of MHV68 reactivation in vivo leads to an increase in the frequency of latently infected splenocytes. Mice were infected with 1,000 PFU wild-type MHV68 intranasally and at 42 dpi were treated i.p. with PBS, LPS, or CpG DNA. Bulk splenocytes from day 1 and day 14 posttreatment were assayed for the frequency of viral genome-positive cells by limiting-dilution PCR. The data shown represent the results of three independent experiments using four to five mice for each experimental group per experiment.
DISCUSSION
The stimuli for herpesvirus reactivation in vivo during natural infections are largely unknown. Here we report that signaling through specific TLRs can induce MHV68 reactivation, both in vitro and in vivo, raising the question of whether heterologous infections play a significant role in episodic gammaherpesvirus reactivation/replication in the chronically infected host. Notably, we observed that 14 days post-TLR stimulation there was an increase in the frequency of MHV68 latently infected splenocytes, suggesting that TLR-driven virus reactivation/replication may be one mechanism by which chronic MHV68 infection is maintained/renewed. Virus reactivation is clearly important for the horizontal transmission of herpesviruses and may for some herpesviruses play an important role in maintaining reservoirs of “functional” latently infected cells in the chronically infected host (8, 19, 30, 52, 53). The latter issue may be particularly important for gammaherpesviruses, whose genomes are known to be heavily methylated in the long-term latency reservoirs (10, 41). Methylated cytosines are prone to oxidative deamination, leading to the production of thymine which, if not repaired prior to cellular proliferation, can become fixed mutations in the viral genome (5, 6). Thus, episodic virus reactivation may ensure the persistence and propagation of “functional” viral genomes (and perhaps reflects an ongoing mechanism by which viral fitness is maintained).
Is there any evidence indicating a role for heterologous infections triggering herpesvirus reactivation? In support of this idea, latently infected EBV cell lines can be induced to reactivate upon superinfection with CMV (3). Similarly, HIV-1 infection of KSHV-infected PEL cells causes KSHV reactivation (36, 57). Many infections, both bacterial and viral, trigger an immune response by their PAMPs. TLRs are expressed on a wide variety of cells, including antigen-presenting cells. Signaling through TLRs can trigger HIV-1 replication in latently infected mast cells (46). KSHV-infected PEL cells can be induced to reactivate in the presence of LPS (38), while the in vivo administration of LPS also stimulates murine CMV reactivation (13). Finally, we previously demonstrated that following i.p. inoculation, MyD88−/− mice exhibited a defect in MHV68 reactivation but not the establishment of latency (17). The latter suggested a potential role for TLR signaling in MHV68 reactivation. As a surrogate for heterologous infections, we assessed whether TLR ligands could induce virus reactivation in vivo at 42 dpi. Day 42 is a time point at which very little spontaneous MHV68 reactivation is observed (62). We focused our analyses on B-cell latency since by day 42 the vast majority of MHV68 latently infected splenocytes are B cells (62). LPS and CpG DNA treatment increased the frequency of activated splenic B cells compared to that for the PBS-treated mice 1 day after administration. This was consistent with the early kinetics of B-cell activation upon TLR stimulation ex vivo. In vivo TLR stimulation significantly increased MHV68 reactivation from latency, with both reactivation and the presence of preformed infectious virus in the spleen peaking at 7 days poststimulation (cleared by day 14). As discussed above, by day 14, we observed a significant increase in the frequency of viral genome-positive splenocytes in mice that received LPS or CpG DNA treatment compared to that for the PBS-treated mice. This increase implies either the reseeding of latency following virus reactivation and subsequent virus replication and/or the expansion of the pool of latently infected cells through TLR-driven cell proliferation. Either mechanism may facilitate the maintenance of chronic gammaherpesvirus infection.
We have previously demonstrated that anti-Ig/anti-CD40 stimulation ex vivo can induce both the proliferation of B cells and MHV68 reactivation/replication (39). In the present study, we show similar results using TLR agonists. The immediate downstream pathway activated following the TLR stimulation of a B cell is distinct from that following anti-CD40/anti-Ig stimulation; however, both pathways eventually lead to the activation of similar downstream transcription factors, notably NF-κB and AP-1, as well as interferon regulatory factors (47). Since multiple stimulation pathways lead to the activation of similar downstream transcription factors, one cannot clearly decipher what cellular factors and their expression patterns are necessary to drive the reactivation of MHV68. Transcription Element Search software (TESS) analyses have revealed putative NF-κB and interferon regulatory factor binding sites in the promoter of orf50, whose gene product RTA is the master lytic activator of MHV68 replication, being required for both acute virus replication and reactivation from latency (32, 42). The activation of one or more of these transcription factors may not only induce B-cell activation and proliferation but also bind to the orf50 promoter and cause the production of RTA, leading to the initiation of the lytic cycle. Interestingly, the overexpression of the NF-κB subunit p65 has been shown to inhibit MHV68 replication in tissue culture (7). One interpretation is that the overexpression of a single NF-κB subunit may alter the dynamic complex formation needed for reactivation. Notably, however, we have previously shown that NF-κB is not essential for reactivation in vivo (29). The Raf/MEK/extracellular signal-regulated kinase pathway is known to trigger KSHV reactivation (15). Also, growth factors or cell cycle regulatory proteins induced by NF-κB may also play a combinatorial role in MHV68 reactivation.
TLR signaling likely represents one of many pathways that lead to MHV68 reactivation. Signals that lead to the terminal differentiation of latently infected memory B cells are also a likely trigger (31), as has been shown for EBV and KSHV (25, 31, 64). Apoptotic signaling pathways triggered by activation through TLR-mediated pathways may be another (2). TLR signaling leads to an array of proinflammatory cytokine production (interleukin-12, tumor necrosis factor alpha, alpha/beta interferon, and/or interleukin-2), all of which have autocrine and paracrine effects that may contribute to MHV68 reactivation. TLRs induce the expression of selectins, chemokines, and chemokine receptor genes that regulate cell migration to the sites of inflammation (24). Chemokines that cause B-cell migration into secondary lymphoid organs during an immune response, such as secondary lymphoid tissue chemokines and B-lymphocyte chemokines, as well as MIP-3α and MIP-3β, may also play a role in triggering virus reactivation in vivo. Furthermore, the latter may be a mechanism that could bring the reactivating B cell in close proximity to proliferating B cells, facilitating de novo latency establishment.
Coincident with the peak of in vivo reactivation following TLR stimulation, we also observed an increase in the CD8+ T-cell response. Both an increase in MHV68-specific CD8+ T cells and a general increase in activated and effector CD8+ T-cell responses were observed. This indicates that the host immune system responds to reactivating virus in vivo, likely in an attempt to control virus replication. Thus, periodic virus reactivation in vivo may, in turn, keep the MHV68-specific immune response primed to control chronic MHV68 infection.
Thus, in conclusion, we have demonstrated that TLR signaling can trigger MHV68 reactivation and subsequent virus replication in vivo. To our knowledge, this is the first report demonstrating the in vivo stimulation of gammaherpesvirus reactivation. We hypothesize that MHV68 may not only take advantage of this innate immune response to trigger reactivation but also utilize the expansion of latently infected B cells and/or the reactivation of MHV68 from B cells reseeding latency reservoirs to facilitate the homeostatic maintenance of MHV68 latency.
Footnotes
Published ahead of print on 19 November 2008.
REFERENCES
- 1.Alexopoulou, L., A. C. Holt, R. Medzhitov, and R. A. Flavell. 2001. Recognition of double-stranded RNA and activation of NF-kappaB by Toll-like receptor 3. Nature 413732-738. [DOI] [PubMed] [Google Scholar]
- 2.Aliprantis, A. O., R. B. Yang, D. S. Weiss, P. Godowski, and A. Zychlinsky. 2000. The apoptotic signaling pathway activated by Toll-like receptor-2. EMBO J. 193325-3336. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Arcenas, R., and R. H. Widen. 2002. Epstein-Barr virus reactivation after superinfection of the BJAB-B1 and P3HR-1 cell lines with cytomegalovirus. BMC Microbiol. 220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Beignon, A. S., K. McKenna, M. Skoberne, O. Manches, I. DaSilva, D. G. Kavanagh, M. Larsson, R. J. Gorelick, J. D. Lifson, and N. Bhardwaj. 2005. Endocytosis of HIV-1 activates plasmacytoid dendritic cells via Toll-like receptor-viral RNA interactions. J. Clin. Investig. 1153265-3275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Bird, A. 1992. The essentials of DNA methylation. Cell 705-8. [DOI] [PubMed] [Google Scholar]
- 6.Bird, A. P. 1986. CpG-rich islands and the function of DNA methylation. Nature 321209-213. [DOI] [PubMed] [Google Scholar]
- 7.Brown, H. J., M. J. Song, H. Deng, T. T. Wu, G. Cheng, and R. Sun. 2003. NF-κB inhibits gammaherpesvirus lytic replication. J. Virol. 778532-8540. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Cesarman, E. 2002. The role of Kaposi's sarcoma-associated herpesvirus (KSHV/HHV-8) in lymphoproliferative diseases. Recent Results Cancer Res. 15927-37. [DOI] [PubMed] [Google Scholar]
- 9.Cesarman, E., P. S. Moore, P. H. Rao, G. Inghirami, D. M. Knowles, and Y. Chang. 1995. In vitro establishment and characterization of two acquired immunodeficiency syndrome-related lymphoma cell lines (BC-1 and BC-2) containing Kaposi's sarcoma-associated herpesvirus-like (KSHV) DNA sequences. Blood 862708-2714. [PubMed] [Google Scholar]
- 10.Chen, J., K. Ueda, S. Sakakibara, T. Okuno, C. Parravicini, M. Corbellino, and K. Yamanishi. 2001. Activation of latent Kaposi's sarcoma-associated herpesvirus by demethylation of the promoter of the lytic transactivator. Proc. Natl. Acad. Sci. USA 984119-4124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Clambey, E. T., H. W. T. Virgin, and S. H. Speck. 2002. Characterization of a spontaneous 9.5-kilobase deletion mutant of murine gammaherpesvirus 68 reveals tissue-specific genetic requirements for latency. J. Virol. 766532-6544. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Coban, C., K. J. Ishii, T. Kawai, H. Hemmi, S. Sato, S. Uematsu, M. Yamamoto, O. Takeuchi, S. Itagaki, N. Kumar, T. Horii, and S. Akira. 2005. Toll-like receptor 9 mediates innate immune activation by the malaria pigment hemozoin. J. Exp. Med. 20119-25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Cook, C. H., J. Trgovcich, P. D. Zimmerman, Y. Zhang, and D. D. Sedmak. 2006. Lipopolysaccharide, tumor necrosis factor alpha, or interleukin-1β triggers reactivation of latent cytomegalovirus in immunocompetent mice. J. Virol. 809151-9158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Diebold, S. S., T. Kaisho, H. Hemmi, S. Akira, and C. Reis e Sousa. 2004. Innate antiviral responses by means of TLR7-mediated recognition of single-stranded RNA. Science 3031529-1531. [DOI] [PubMed] [Google Scholar]
- 15.Ford, P. W., B. A. Bryan, O. F. Dyson, D. A. Weidner, V. Chintalgattu, and S. M. Akula. 2006. Raf/MEK/ERK signalling triggers reactivation of Kaposi's sarcoma-associated herpesvirus latency. J. Gen. Virol. 871139-1144. [DOI] [PubMed] [Google Scholar]
- 16.Forrest, J. C., and S. H. Speck. 2008. Establishment of B-cell lines latently infected with reactivation-competent murine gammaherpesvirus 68 provides evidence for viral alteration of a DNA damage-signaling cascade. J. Virol. 827688-7699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Gargano, L. M., J. M. Moser, and S. H. Speck. 2008. Role for MyD88 signaling in murine gammaherpesvirus 68 latency. J. Virol. 823853-3863. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Gradoville, L., D. Kwa, A. El-Guindy, and G. Miller. 2002. Protein kinase C-independent activation of the Epstein-Barr virus lytic cycle. J. Virol. 765612-5626. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Grundhoff, A., and D. Ganem. 2004. Inefficient establishment of KSHV latency suggests an additional role for continued lytic replication in Kaposi sarcoma pathogenesis. J. Clin. Investig. 113124-136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Hayashi, F., K. D. Smith, A. Ozinsky, T. R. Hawn, E. C. Yi, D. R. Goodlett, J. K. Eng, S. Akira, D. M. Underhill, and A. Aderem. 2001. The innate immune response to bacterial flagellin is mediated by Toll-like receptor 5. Nature 4101099-1103. [DOI] [PubMed] [Google Scholar]
- 21.Heil, F., H. Hemmi, H. Hochrein, F. Ampenberger, C. Kirschning, S. Akira, G. Lipford, H. Wagner, and S. Bauer. 2004. Species-specific recognition of single-stranded RNA via toll-like receptor 7 and 8. Science 3031526-1529. [DOI] [PubMed] [Google Scholar]
- 22.Hemmi, H., T. Kaisho, O. Takeuchi, S. Sato, H. Sanjo, K. Hoshino, T. Horiuchi, H. Tomizawa, K. Takeda, and S. Akira. 2002. Small anti-viral compounds activate immune cells via the TLR7 MyD88-dependent signaling pathway. Nat. Immunol. 3196-200. [DOI] [PubMed] [Google Scholar]
- 23.Hemmi, H., O. Takeuchi, T. Kawai, T. Kaisho, S. Sato, H. Sanjo, M. Matsumoto, K. Hoshino, H. Wagner, K. Takeda, and S. Akira. 2000. A Toll-like receptor recognizes bacterial DNA. Nature 408740-745. [DOI] [PubMed] [Google Scholar]
- 24.Huang, Q., D. Liu, P. Majewski, L. C. Schulte, J. M. Korn, R. A. Young, E. S. Lander, and N. Hacohen. 2001. The plasticity of dendritic cell responses to pathogens and their components. Science 294870-875. [DOI] [PubMed] [Google Scholar]
- 25.Jenner, R. G., K. Maillard, N. Cattini, R. A. Weiss, C. Boshoff, R. Wooster, and P. Kellam. 2003. Kaposi's sarcoma-associated herpesvirus-infected primary effusion lymphoma has a plasma cell gene expression profile. Proc. Natl. Acad. Sci. USA 10010399-10404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Jurk, M., F. Heil, J. Vollmer, C. Schetter, A. M. Krieg, H. Wagner, G. Lipford, and S. Bauer. 2002. Human TLR7 or TLR8 independently confer responsiveness to the antiviral compound R-848. Nat. Immunol. 3499. [DOI] [PubMed] [Google Scholar]
- 27.Krieg, A. M., A. K. Yi, S. Matson, T. J. Waldschmidt, G. A. Bishop, R. Teasdale, G. A. Koretzky, and D. M. Klinman. 1995. CpG motifs in bacterial DNA trigger direct B-cell activation. Nature 374546-549. [DOI] [PubMed] [Google Scholar]
- 28.Krug, A., A. R. French, W. Barchet, J. A. Fischer, A. Dzionek, J. T. Pingel, M. M. Orihuela, S. Akira, W. M. Yokoyama, and M. Colonna. 2004. TLR9-dependent recognition of MCMV by IPC and DC generates coordinated cytokine responses that activate antiviral NK cell function. Immunity 21107-119. [DOI] [PubMed] [Google Scholar]
- 29.Krug, L. T., J. M. Moser, S. M. Dickerson, and S. H. Speck. 2007. Inhibition of NF-kappaB activation in vivo impairs establishment of gammaherpesvirus latency. PLoS Pathog. 3e11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Kuppers, R. 2003. B cells under influence: transformation of B cells by Epstein-Barr virus. Nat. Rev. Immunol. 3801-812. [DOI] [PubMed] [Google Scholar]
- 31.Laichalk, L. L., and D. A. Thorley-Lawson. 2005. Terminal differentiation into plasma cells initiates the replicative cycle of Epstein-Barr virus in vivo. J. Virol. 791296-1307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Liu, S., I. V. Pavlova, H. W. Virgin IV, and S. H. Speck. 2000. Characterization of gammaherpesvirus 68 gene 50 transcription. J. Virol. 742029-2037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Lund, J., A. Sato, S. Akira, R. Medzhitov, and A. Iwasaki. 2003. Toll-like receptor 9-mediated recognition of Herpes simplex virus-2 by plasmacytoid dendritic cells. J. Exp. Med. 198513-520. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Lund, J. M., L. Alexopoulou, A. Sato, M. Karow, N. C. Adams, N. W. Gale, A. Iwasaki, and R. A. Flavell. 2004. Recognition of single-stranded RNA viruses by Toll-like receptor 7. Proc. Natl. Acad. Sci. USA 1015598-5603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Meier, A., G. Alter, N. Frahm, H. Sidhu, B. Li, A. Bagchi, N. Teigen, H. Streeck, H. J. Stellbrink, J. Hellman, J. van Lunzen, and M. Altfeld. 2007. MyD88-dependent immune activation mediated by human immunodeficiency virus type 1-encoded Toll-like receptor ligands. J. Virol. 818180-8191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Merat, R., A. Amara, C. Lebbe, H. de The, P. Morel, and A. Saib. 2002. HIV-1 infection of primary effusion lymphoma cell line triggers Kaposi's sarcoma-associated herpesvirus (KSHV) reactivation. Int. J. Cancer 97791-795. [DOI] [PubMed] [Google Scholar]
- 37.Miller, G., L. Heston, E. Grogan, L. Gradoville, M. Rigsby, R. Sun, D. Shedd, V. M. Kushnaryov, S. Grossberg, and Y. Chang. 1997. Selective switch between latency and lytic replication of Kaposi's sarcoma herpesvirus and Epstein-Barr virus in dually infected body cavity lymphoma cells. J. Virol. 71314-324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Morris, T. L., R. R. Arnold, and J. Webster-Cyriaque. 2007. Signaling cascades triggered by bacterial metabolic end products during reactivation of Kaposi's sarcoma-associated herpesvirus. J. Virol. 816032-6042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Moser, J. M., J. W. Upton, K. S. Gray, and S. H. Speck. 2005. Ex vivo stimulation of B cells latently infected with gammaherpesvirus 68 triggers reactivation from latency. J. Virol. 795227-5231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Ozinsky, A., D. M. Underhill, J. D. Fontenot, A. M. Hajjar, K. D. Smith, C. B. Wilson, L. Schroeder, and A. Aderem. 2000. The repertoire for pattern recognition of pathogens by the innate immune system is defined by cooperation between toll-like receptors. Proc. Natl. Acad. Sci. USA 9713766-13771. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Paulson, E. J., and S. H. Speck. 1999. Differential methylation of Epstein-Barr virus latency promoters facilitates viral persistence in healthy seropositive individuals. J. Virol. 739959-9968. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Pavlova, I. V., H. W. Virgin IV, and S. H. Speck. 2003. Disruption of gammaherpesvirus 68 gene 50 demonstrates that Rta is essential for virus replication. J. Virol. 775731-5739. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Pollock, J. L., and H. W. Virgin IV. 1995. Latency, without persistence, of murine cytomegalovirus in the spleen and kidney. J. Virol. 691762-1768. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Poltorak, A., X. He, I. Smirnova, M. Y. Liu, C. Van Huffel, X. Du, D. Birdwell, E. Alejos, M. Silva, C. Galanos, M. Freudenberg, P. Ricciardi-Castagnoli, B. Layton, and B. Beutler. 1998. Defective LPS signaling in C3H/HeJ and C57BL/10ScCr mice: mutations in Tlr4 gene. Science 2822085-2088. [DOI] [PubMed] [Google Scholar]
- 45.Speck, S. H., T. Chatila, and E. Flemington. 1997. Reactivation of Epstein-Barr virus: regulation and function of the BZLF1 gene. Trends Microbiol. 5399-405. [DOI] [PubMed] [Google Scholar]
- 46.Sundstrom, J. B., D. M. Little, F. Villinger, J. E. Ellis, and A. A. Ansari. 2004. Signaling through Toll-like receptors triggers HIV-1 replication in latently infected mast cells. J. Immunol. 1724391-4401. [DOI] [PubMed] [Google Scholar]
- 47.Takeda, K., and S. Akira. 2004. TLR signaling pathways. Semin. Immunol. 163-9. [DOI] [PubMed] [Google Scholar]
- 48.Takeuchi, O., K. Hoshino, T. Kawai, H. Sanjo, H. Takada, T. Ogawa, K. Takeda, and S. Akira. 1999. Differential roles of TLR2 and TLR4 in recognition of gram-negative and gram-positive bacterial cell wall components. Immunity 11443-451. [DOI] [PubMed] [Google Scholar]
- 49.Takeuchi, O., A. Kaufmann, K. Grote, T. Kawai, K. Hoshino, M. Morr, P. F. Muhlradt, and S. Akira. 2000. Cutting edge: preferentially the R-stereoisomer of the mycoplasmal lipopeptide macrophage-activating lipopeptide-2 activates immune cells through a Toll-like receptor 2- and MyD88-dependent signaling pathway. J. Immunol. 164554-557. [DOI] [PubMed] [Google Scholar]
- 50.Takeuchi, O., T. Kawai, P. F. Muhlradt, M. Morr, J. D. Radolf, A. Zychlinsky, K. Takeda, and S. Akira. 2001. Discrimination of bacterial lipoproteins by Toll-like receptor 6. Int. Immunol. 13933-940. [DOI] [PubMed] [Google Scholar]
- 51.Takeuchi, O., S. Sato, T. Horiuchi, K. Hoshino, K. Takeda, Z. Dong, R. L. Modlin, and S. Akira. 2002. Cutting edge: role of Toll-like receptor 1 in mediating immune response to microbial lipoproteins. J. Immunol. 16910-14. [DOI] [PubMed] [Google Scholar]
- 52.Thorley-Lawson, D. A. 2001. Epstein-Barr virus: exploiting the immune system. Nat. Rev. Immunol. 175-82. [DOI] [PubMed] [Google Scholar]
- 53.Thorley-Lawson, D. A., and A. Gross. 2004. Persistence of the Epstein-Barr virus and the origins of associated lymphomas. N. Engl. J. Med. 3501328-1337. [DOI] [PubMed] [Google Scholar]
- 54.Tibbetts, S. A., J. Loh, V. Van Berkel, J. S. McClellan, M. A. Jacoby, S. B. Kapadia, S. H. Speck, and H. W. Virgin IV. 2003. Establishment and maintenance of gammaherpesvirus latency are independent of infective dose and route of infection. J. Virol. 777696-7701. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Usherwood, E. J., J. P. Stewart, and A. A. Nash. 1996. Characterization of tumor cell lines derived from murine gammaherpesvirus-68-infected mice. J. Virol. 706516-6518. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.van Dyk, L. F., H. W. Virgin IV, and S. H. Speck. 2003. Maintenance of gammaherpesvirus latency requires viral cyclin in the absence of B lymphocytes. J. Virol. 775118-5126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Varthakavi, V., P. J. Browning, and P. Spearman. 1999. Human immunodeficiency virus replication in a primary effusion lymphoma cell line stimulates lytic-phase replication of Kaposi's sarcoma-associated herpesvirus. J. Virol. 7310329-10338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Weck, K. E., M. L. Barkon, L. I. Yoo, S. H. Speck, and H. I. Virgin. 1996. Mature B cells are required for acute splenic infection, but not for establishment of latency, by murine gammaherpesvirus 68. J. Virol. 706775-6780. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Weck, K. E., S. S. Kim, H. W. Virgin IV, and S. H. Speck. 1999. B cells regulate murine gammaherpesvirus 68 latency. J. Virol. 734651-4661. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Weck, K. E., S. S. Kim, H. W. Virgin IV, and S. H. Speck. 1999. Macrophages are the major reservoir of latent murine gammaherpesvirus 68 in peritoneal cells. J. Virol. 733273-3283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.West, J. T., and C. Wood. 2003. The role of Kaposi's sarcoma-associated herpesvirus/human herpesvirus-8 regulator of transcription activation (RTA) in control of gene expression. Oncogene 225150-5163. [DOI] [PubMed] [Google Scholar]
- 62.Willer, D. O., and S. H. Speck. 2003. Long-term latent murine gammaherpesvirus 68 infection is preferentially found within the surface immunoglobulin D-negative subset of splenic B cells in vivo. J. Virol. 778310-8321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Wu, T. T., E. J. Usherwood, J. P. Stewart, A. A. Nash, and R. Sun. 2000. Rta of murine gammaherpesvirus 68 reactivates the complete lytic cycle from latency. J. Virol. 743659-3667. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Yu, F., J. N. Harada, H. J. Brown, H. Deng, M. J. Song, T. T. Wu, J. Kato-Stankiewicz, C. G. Nelson, J. Vieira, F. Tamanoi, S. K. Chanda, and R. Sun. 2007. Systematic identification of cellular signals reactivating Kaposi sarcoma-associated herpesvirus. PLoS Pathog. 3e44. [DOI] [PMC free article] [PubMed] [Google Scholar]