

Abstract
Human metapneumovirus (HMPV) is a significant respiratory pathogen classified in the Pneumovirinae subfamily of the paramyxovirus family. Recently, we demonstrated that HMPV F protein-promoted cell-cell fusion is stimulated by exposure to low pH, in contrast to what is observed for other paramyxovirus F proteins. In the present study, we examined the potential role of histidine protonation in HMPV F fusion and investigated the role of low pH in HMPV viral entry. Mutagenesis of the three ectodomain histidine residues of the HMPV F protein demonstrated that the mutation of a histidine in the heptad repeat B linker domain (H435) ablated fusion activity without altering cell surface expression or proteolytic processing significantly. Modeling of the HMPV F protein revealed several basic residues surrounding this histidine residue, and the mutation of these residues also reduced fusion activity. These results suggest that electrostatic repulsion in the heptad repeat B linker region may contribute to the triggering of HMPV F. In addition, we examined the effect of inhibitors of endosomal acidification or endocytosis on the entry of a recombinant green fluorescent protein-expressing HMPV. Interestingly, chemicals that raise the pH of endocytic vesicles resulted in a 30 to 50% decrease in HMPV infection, while the inhibitors of endocytosis reduced infection by as much as 90%. These data suggest that HMPV utilizes an endocytic entry mechanism, in contrast to what has been hypothesized for most paramyxoviruses. In addition, our results indicate that HMPV uses the low pH of the endocytic pathway to enhance infectivity, though the role of low pH likely differs from classically described mechanisms.
Since the discovery of human metapneumovirus (HMPV) by van den Hoogen et al. in 2001 (61), numerous reports have confirmed its importance as a human pathogen with worldwide significance (reviewed in references 26 and 31). Infants are most likely to suffer from disease caused by HMPV infection, although adults may also experience symptoms of HMPV infection, particularly those with compromised immune systems (31, 65). HMPV infection can result in respiratory tract disease of various severities, and it is likely the second most common cause of bronchiolitis and lower respiratory tract infection resulting in the hospitalization of very young children (reviewed in references 17 and 31).
The most common cause of respiratory tract disease in infants is respiratory syncytial virus (RSV), a virus closely related to HMPV (64). Both RSV and HMPV are classified in the subfamily Pneumovirinae of the family Paramyxoviridae (21, 60). One clear distinction between viruses of the Pneumovirinae subfamily and the Paramyxovirinae subfamily with regard to the viral entry mechanism has become apparent in recent years. While the homotypic attachment protein (G, H, or HN) of viruses within the Paramyxovirinae subfamily is required for virus attachment and membrane fusion promotion by the viral F protein, the “attachment” proteins (G) of multiple Pneumovirinae subfamily members were shown to be dispensable for entry in cultured cells and also in vivo in the case of HMPV (7, 9, 41, 48, 56). In fact, HMPV with G deleted was infectious in primates (7). Furthermore, the G protein of HMPV did not enhance cell-cell fusion promoted by F in transfected Vero cells (49), suggesting that the HMPV F protein alone is capable of performing both the critical attachment step and efficient membrane fusion.
All viruses are generally classified in either a neutral pH/plasma membrane entry category or a low pH/endocytic entry category (reviewed in references 18 and 38). The trigger of fusion between the viral membrane and the plasma membrane in a neutral pH environment is thought to involve receptor binding (35, 36), while fusion with endosomal membranes is generally triggered by the increased concentration of hydrogen ions within the endosomal pathway (52). Paramyxoviruses are believed to promote fusion under neutral pH conditions at the plasma membrane, and any exceptions to this rule remain controversial (35). Fusion promoted by the rubulavirus SER was first shown to be stimulated by low pH (50), but a subsequent analysis revealed extensive conflicting data (10). Recently, the small interfering RNA knockdown of several proteins involved in endocytosis and vesicular trafficking was shown to inhibit infection by RSV (33). However, inhibitors of endosomal acidification did not have a significant effect on infection, suggesting that RSV uses an endocytic route of entry into cells, but the low pH of endosomes is not an essential trigger of fusion. Conversely, we have shown that cell-cell fusion promoted by HMPV F is stimulated by a low pH (49), but the necessity of low pH in virus entry has not been examined.
The paramyxovirus F protein is a type I fusion protein, indicating that the formation of a six-helix bundle by two individual heptad repeat regions in the trimeric protein is directly linked to the merger of membranes that results in virus entry or cell-cell fusion (35). There must be a specific trigger of fusion because the F protein conformational change is irreversible and premature activation prevents infection (15, 16). The triggering of paramyxovirus F proteins is thought to occur following receptor binding by the attachment protein at the plasma membrane (2, 36). However, the ability of HMPV to infect cells and promote fusion in the absence of an attachment protein suggests that the trigger of fusion for this viral F protein must be different. It is likely that the F protein itself can bind a receptor and that this may play a role in the trigger of fusion. However, the observation of low-pH-stimulated cell-cell fusion by HMPV F raises the possibility that the low pH of the endosomal pathway may be an important physiological trigger of the HMPV F conformational change that results in entry.
Influenza virus HA is also a type I fusion protein, and fusion promoted by this viral protein is known to be triggered by low pH (52). The mechanism of low-pH triggering is hypothesized to involve electrostatic repulsion between residues that become protonated in a low-pH environment and neighboring residues (14, 28). The prefusion conformation of both influenza virus HA and paramyxovirus F proteins is metastable (15, 36); thus, it is possible that minor yet specific changes in protein stability induced by changes in side chain ionization state lead to the extensive reorganization in protein structure that accompanies membrane fusion. Histidine residues, with a side chain pKa of 6.04 in solution, are the amino acids most likely to ionize within the physiological pH range (63). Indeed, histidine residues have been shown to modulate the pH at which fusion by influenza virus HA is achieved (57), and low-pH-induced conformational changes of VSV G were inhibited by the chemical modification of histidine residues (14), observations consistent with the electrostatic repulsion hypothesis of triggering. A specific histidine residue at the domain I to III interface has also been recently shown to be critical for the triggering of the type II tick-borne encephalitis E fusion protein (22). Specific regions in paramyxovirus F proteins have also been implicated in the modulation of fusion activity under neutral pH conditions (23, 46), as the replacement of residues in these regions can increase or decrease fusion depending on the side chain present. One such region, referred to as the heptad repeat B (HRB) linker, is located N terminal to the C-terminal heptad repeat region (46).
We have examined histidine residues in the HMPV F ectodomain for a potential role in low-pH triggering of fusion. In transient expression experiments, we discovered a residue in the HRB linker domain that is important for fusion promotion. Interestingly, this residue is surrounded by several basic residues, the mutation of which also resulted in defective low-pH triggering of fusion. The biological significance of low-pH fusion in virus entry was also examined. Inhibitors of endosomal acidification were found to reduce but not abolish the infectivity of HMPV, while inhibitors of endocytosis resulted in even greater reductions in HMPV infectivity. These results suggest that the low-pH environment encountered after the endocytosis of HMPV may stimulate F protein triggering and subsequent membrane fusion.
MATERIALS AND METHODS
Cell lines.
Vero cells and BSR cells (provided by Karl-Klaus Conzelmann, Max Pettenkofer Institut) were grown in Dulbecco's modified Eagle's medium (DMEM; Gibco Invitrogen, Carlsbad, CA), supplemented with 10% fetal bovine serum (FBS) and 1% penicillin and streptomycin (P/S). The medium of the BSR cells was supplemented with 0.5 mg/ml G-418 sulfate (Gibco Invitrogen, Carlsbad, CA) every third passage to select for T7 polymerase-expressing cells. A549 cells (provided by Hsin-Hsiung Tai, University of Kentucky) were grown in RPMI medium supplemented with 10% FBS and 1% P/S.
Plasmids and antibodies.
The HMPV F gene was subcloned into the pCAGGS mammalian expression vector as described previously (49). All HMPV F protein mutants were created with the gene in pGEM-3Zf(+) using QuikChange site-directed mutagenesis (Stratagene) and subcloned into pCAGGS. All mutants were sequenced in their entirety. Antipeptide antibodies (Genemed Synthesis, San Francisco, CA) were generated using amino acids 524 to 538 of HMPV F.
Viruses.
Recombinant, green fluorescent protein (GFP)-expressing HMPV (clade A2, CAN 97-83 strain) containing a codon-stabilized SH gene was synthesized as described previously (6). HMPV (at a multiplicity of infection [MOI] of 0.01 to 0.03) was propagated in Vero cells incubated at 32°C with Opti-MEM, 2 mM l-glutamine, and 0.3 μg/ml TPCK (N-tosyl-l-phenylalanine chloromethyl ketone)-trypsin replenished every other day. On the 10th day, the cells and medium were collected, SPG (218 mM sucrose, 0.0049 M l-glutamic acid, 0.0038 M KH2PO4, 0.0072 M K2HPO4) from a 10× stock was added, and aliquots were frozen in dry ice/methanol and thawed twice prior to storage at −80°C. Titers of approximately 1 × 107 were achieved with this method. Recombinant GFP-expressing PIV5 (formerly called SV5) was kindly provided by Robert Lamb (Howard Hughes Medical Institute, Northwestern University). GFP was inserted between the P/V and M genes with a duplicate of the EIS sequence that precedes M. PIV5 was propagated in MDBK cells as described previously (43), but PIV5 was also stored in the SPG stabilizing agent. Recombinant VSV, kindly provided by Michael A. Whitt (University of Tennessee Health Science Center, Memphis, Tennessee) in which the G envelope gene was replaced with the GFP gene and pseudotyped with the VSV G glycoprotein was propagated as previously described (55).
Reporter gene fusion assay.
Vero cells in 6-cm dishes were transfected using Lipofectamine Plus reagents (Invitrogen) with 1.5 μg pCAGGS-HMPV wild-type or mutant F protein and 1.5 μg T7 control plasmid (Promega) containing luciferase cDNA under the control of the T7 promoter. The following day (see Fig. 2 and 3) or 2 days later (incubated at 32°C the second night; Fig. 4), Vero cells in one 6-cm dish were lifted from the plate surface with trypsin, a process which also efficiently cleaved the HMPV F protein. The cells were resuspended in DMEM plus 10% FBS and overlaid onto two 35-mm dishes of confluent BSR cells, which constitutively express the T7 polymerase. The combined cells were incubated at 32°C for 60 min. The cells were then rinsed once with PBS (#14287; Gibco Invitrogen, Carlsbad, CA) (pH 7.2) before adding pH 5, 7, or 4.5 PBS (as indicated in Fig. 2 to 4) buffered with 10 mM HEPES and 5 mM MES (morpholineethanesulfonic acid; the pH 4.5 PBS contains 10 mM MES). The cells were incubated for 4 min at 37°C under the indicated pH conditions, and then Opti-MEM with 0.3 μg/ml TPCK-trypsin was added. The cells were again incubated at 32°C for 1 h, and then the cells were treated with pH 5, 7, or 4.5 PBS as before. DMEM with FBS was added after this treatment, and the cells were incubated at 37°C for 4 h. Finally, the cell lysates were analyzed for luciferase activity using a luciferase assay system (Promega) according to manufacturer's protocol. Light emission was measured using an Lmax luminometer (Molecular Devices, Sunnyvale, CA).
FIG. 2.

Cell surface expression and fusion activity of histidine mutants. (A) Surface expression in Vero cells of transiently expressed, metabolically labeled (5 h, in the presence of 0.3 μg/ml TPCK-trypsin) wild-type F protein (Fwt) and histidine mutants in pCAGGS vectors. Biotinylation of the surface prior to cell lysis, immunoprecipitation, and separation of the total and surface populations by streptavidin pull-down was performed. Proteins were resolved by 10% SDS-PAGE and visualized by autoradiography. (B) Luciferase reporter gene assay of fusion between Vero cells transiently transfected with T7-luciferase and pCAGGS-wild-type F protein (Fwt) or -histidine mutant and BSR cells constitutively expressing the T7 polymerase. Mixed cell populations were exposed to pH 7 or pH 5 buffered PBS as described in Materials and Methods and then lysed and analyzed for luminosity. Data are presented as a percentage of the wild-type F protein luminosity at pH 5, the average value of which from four separate experiments was 6.4. Error bars represent 95% confidence intervals.
FIG. 3.

Stimulation of histidine mutant fusion by decreasing pH. Luciferase reporter gene assay of fusion between Vero cells transiently transfected with T7-luciferase and wild-type F protein (Fwt) or a histidine mutant in the pCAGGS vector or vector alone and BSR cells constitutively expressing the T7 polymerase. Mixed cell populations were exposed to pH 5 or pH 4.5 buffered PBS as described in Materials and Methods and then lysed and analyzed for luminosity. Data are presented as a percentage of the wild-type F protein luminosity at pH 5 (A) and the percent luminosity at pH 4.5 divided by the luminosity at pH 5 from panel A (B). The average luminosity from three separate experiments was 16.5. Error bars represent standard deviations.
FIG. 4.
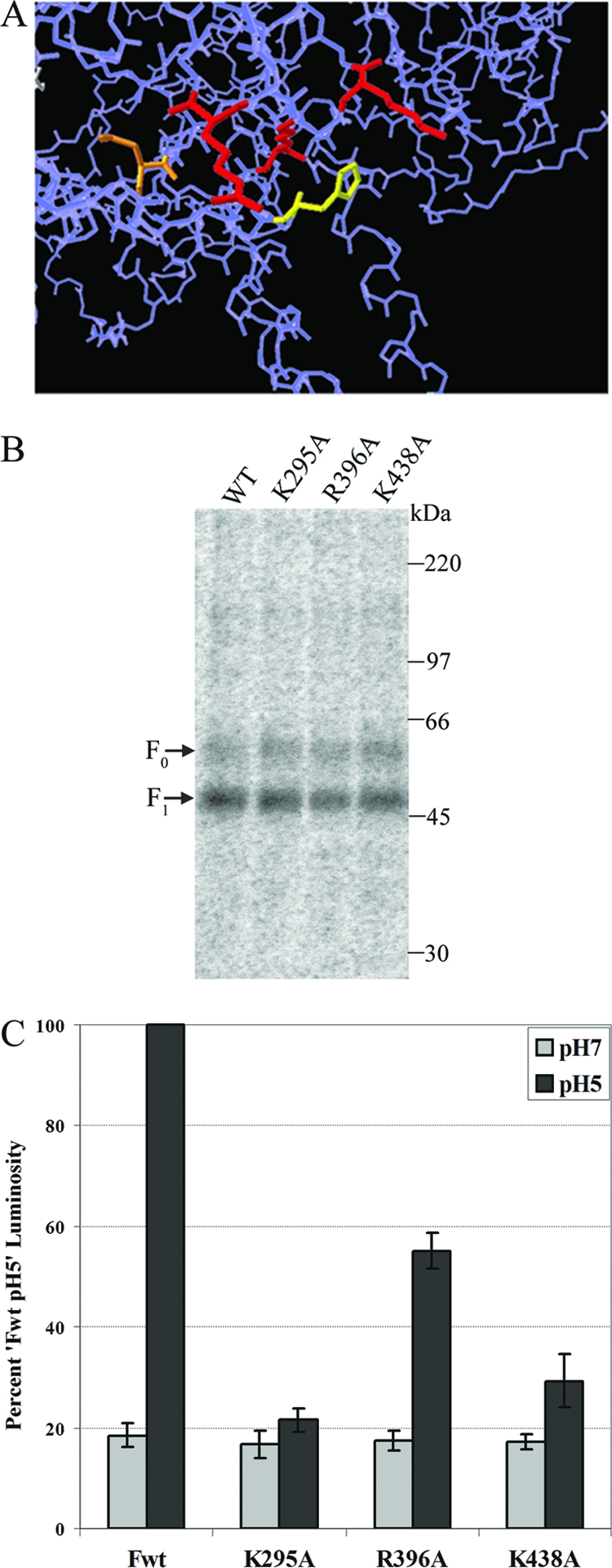
Model, surface expression and fusion activity of basic residue mutants surrounding H435. (A) Image from a homology model of HMPV F based on the prefusion PIV5 F crystal structure. Orange, N353 (glycosylated residue); yellow, H435; red, basic residues (K295, R396, K438). Only the side chains of highlighted residues are shown. (B) Surface expression in Vero cells of transiently expressed, metabolically labeled (5 h, in the presence of 0.3 μg/ml TPCK-trypsin) wild-type F protein and basic residue mutants in pCAGGS vectors. Biotinylation of the surface prior to cell lysis, immunoprecipitation, and separation of the total and surface populations by streptavidin pulldown was performed. Proteins were resolved by 10% SDS-PAGE and visualized by autoradiography. (C) Luciferase reporter gene assay of the fusion between Vero cells transiently transfected with T7- luciferase and pCAGGS-wild-type F protein (Fwt) or -basic residue mutant and BSR cells constitutively expressing the T7 polymerase. Mixed cell populations were exposed to pH 7 or pH 5 buffered PBS as described in Materials and Methods and then lysed and analyzed for luminosity. Data are presented as a percentage of the wild-type F protein luminosity at pH 5, of which the average value from four separate experiments was 12.1. Error bars represent 95% confidence intervals.
Biotinylation.
Cells in 6-cm dishes were transfected with 3 μg pCAGGS-HMPV F wild-type or mutant protein using Lipofectamine Plus reagents (Invitrogen). At 18 to 24 h posttransfection, the cells were starved in methionine- and cysteine-deficient DMEM for 45 min and then metabolically labeled with Tran[35S] label (100 μCi/ml; MP Biomedicals) with 0.3 μg/ml TPCK-trypsin for 4 to 5 h. The cells were washed three times with cold pH 8 PBS and incubated for 30 min with rocking at 4°C and then 20 min at room temperature with 1 mg/ml EZ-Link Sulfo-NHS-Biotin (Pierce, Rockford, IL) diluted in pH 8 PBS. The cells were washed again three times with pH 8 PBS and then lysed in radioimmunoprecipitation assay buffer containing 100 mM Tris-HCl (pH 7.4), 150 mM NaCl, 0.1% sodium dodecyl sulfate (SDS), 1% Triton X-100, 1% deoxycholic acid, protease inhibitors (1 kallikrein inhibitory unit of aprotinin [Calbiochem, San Diego, CA], 1 mM phenylmethylsulfonyl fluoride [Sigma, St. Louis, MO], a Complete protease inhibitor tablet [Roche Molecular Biochemicals, Indianapolis, IN]), and 25 mM iodoacetamide (Sigma). The lysates were centrifuged at 136,500 × g for 10 min at 4°C, and the supernatants were collected. Antipeptide sera and protein A-conjugated Sepharose beads (Amersham, Piscataway, NJ) were used to immunoprecipitate the F proteins as previously described (43). Immunoprecipitated protein was boiled away from the beads in 10% SDS. Fifteen percent of the total protein was removed for analysis, and the remaining 85% was diluted in biotinylation dilution buffer (20 mM Tris [pH 8], 150 mM NaCl, 5 mM EDTA, 1% Triton X-100, 0.2% bovine serum albumin) and incubated with immobilized streptavidin (Pierce) for 1 h at 4°C. Samples were washed and analyzed via SDS-10% polyacrylamide gel electrophoresis (SDS-PAGE) and visualized using the Typhoon imaging system.
Syncytia.
Vero cells in 35-mm dishes at ∼70% confluence were infected with HMPV (MOI = 0.5) and incubated overnight at 32°C. The next day, infected cells were incubated in Opti-MEM with or without 0.3 μg/ml TPCK-trypsin for 1.5 h at 32°C. The cells were then rinsed once with PBS (pH 7.2) before adding pH 5 or 7 PBS buffered with 10 mM HEPES and 5 mM MES. The cells were incubated for 4 min at 37°C under the indicated pH conditions and washed, and Opti-MEM ± 0.3 μg/ml TPCK-trypsin was added. The cells were again incubated at 32°C for 1 to 2 h and then treated with pH 5 or 7 PBS as before. The trypsin and pH treatment was repeated four times total. DMEM with 2% FBS was added after the last treatment and incubated overnight at 32°C. Photographs were taken the next morning with a Spot Insight Firewire digital camera mounted on a Zeiss Axiovert 100 inverted microscope.
Drug treatments.
Bafilomycin A1 (Calbiochem), concanamycin A (Calbiochem), monensin (Sigma), and dynasore (Tocris Bioscience) stock solutions were made in dimethyl sulfoxide (DMSO). Ammonium chloride (Sigma) and chlorpromazine (Alexis Biochemicals) were dissolved in PBS. Chlorpromazine stock solutions were made fresh for each experiment. Confluent monolayers of cells in 24-well plates were pretreated for 1.5 h for lysosomotropic agents, 1 h for chlorpromazine, or 30 min for dynasore, with the drug or vehicle control (control volume equal to the maximum volume of the drug) at the concentrations indicated. The drugs were diluted (1:100 dilution or less) into DMEM (Vero cells) or RPMI (A549 cells) medium plus FBS and P/S (minus FBS for dynasore). For experiments with ammonium chloride, 20 mM HEPES-buffered DMEM plus FBS and P/S was used during the drug treatments. Infections were carried out in the presence of a drug or vehicle control, such that 30% of the cells on average would be infected in the control wells. After 4 h at 37°C (2 h for the dynasore experiments), the cells were washed and incubated in DMEM or RPMI medium for an additional 6 h (VSV) or 12 to 16 h (HMPV and PIV5) at 37°C. For an analysis of the dynasore effects postinfection, the drug or vehicle control (DMSO) was added 2 h postinfection and then removed after 2 h. The cells were then washed twice with PBS minus calcium or magnesium, and 110 μl/well 0.05% trypsin plus EDTA (Gibco Invitrogen) was added. When the cells had detached from the dish, 110 μl/well 2% formaldehyde was added and the cells were collected for analysis by flow cytometry where the GFP fluorescent intensity of at least 10,000 cells was determined. Data shown in the graphs in Fig. 6 through 9 represent the percentage of GFP-expressing cells in the total population as a percentage of the control treated cells (set to 100%).
FIG. 6.

Virus entry in Vero cells treated with inhibitors of endosomal acidification. Monolayers of Vero cells in 24-well plates were pretreated with drugs: bafilomycin A1 (A), concanamycin A (B), ammonium chloride (C), and monensin (D) or vehicle control diluted in DMEM plus FBS at the concentrations indicated for 1.5 h. The DMEM was buffered with 20 mM HEPES for the ammonium chloride treatments. Infections were carried out in the presence of the drugs or vehicle control such that 30 to 40% of the cells would be infected on average in control wells, and the cells were incubated for 4 h at 37°C. The cells were then washed and incubated in DMEM plus FBS for an additional 6 h (VSV) or 12 to 16 h (HMPV and PIV5) at 37°C. The cells were then detached with trypsin, fixed in 1% formaldehyde, and analyzed for GFP expression by flow cytometry. Data shown represent the number of GFP-expressing cells as a percentage of that of the control, set to 100% for each virus, from at least four separate experiments. Error bars represent 95% confidence intervals.
FIG. 9.

Effect of endocytosis inhibitors on HMPV infection. Virus entry in Vero cells treated with chlorpromazine (A) or dynasore (B). Monolayers of Vero cells in 24-well plates were pretreated with drug (chlorpromazine for 1 h; dynasore for 30 min) or vehicle control diluted at the concentrations shown. Infections were carried out in the presence of the drug or vehicle control such that 30 to 40% of the cells would be infected on average in the control wells, and the cells were incubated at 37°C for either 4 h (chlorpromazine) or 2 h (dynasore). The cells were then washed and incubated in DMEM plus FBS for an additional 6 h (VSV) or 12 to 16 h (HMPV and PIV5) at 37°C. For analysis of the dynasore effects postinfection, drug or vehicle control (DMSO) was added 2 h postinfection and then removed after 2 h. For all samples, the cells were detached with trypsin, fixed in 1% formaldehyde, and analyzed for GFP expression by flow cytometry. Data shown represent the number of GFP-expressing cells as a percentage of that of the control, set to 100% for each virus. Error bars represent 95% confidence intervals.
pH pretreatment of virus.
Twenty microliters of HMPV stock (resulting in an ∼20% infection after pH 7 treatment), 1.3 μl PIV5 stock diluted to 20 μl in Opti-MEM plus 1× SPG which is the medium used for HMPV propagation and freezing, or 1.4 μl of VSV diluted 1:100 in Opti-MEM and brought to a 20-μl volume in Opti-MEM plus 1× SPG were aliquoted into empty wells of a 24-well plate. Eighty microliters of 15 mM citric acid-150 mM NaCl buffer with a pH of either 4.1 or 7 was added to each virus in individual wells and then incubated at 37°C for 10 min. Next, 450 μl of Opti-MEM was added to each well. Confluent monolayers of Vero cells in a 24-well plate were washed twice, and 500 μl of each virus solution was distributed onto the cells. Ten hours (VSV) or 16 to 20 h postinfection, the cells were harvested and analyzed for GFP expression as described above. The quantities of low-pH buffer required to lower the pH of the original virus solution to pH 5 and then neutralize that solution with Opti-MEM were determined on a larger scale prior to the experiment.
Homology modeling.
DeepView/Swiss-PdbViewer v3.7 (www.expasy.org/spdbv/) was used to generate a model of the HMPV F protein from the molecular coordinates (mmdbId:37132) determined from the crystal structure of the prefusion form of PIV5 F (66). The HMPV F sequence was loaded to model on each monomer of PIV5 F and then fitted onto the structure. The three modeled HMPV F monomers were merged, and the merged structure was energy minimized with 100 steps of steepest descent.
RESULTS
We have previously demonstrated that cell-cell fusion promoted by HMPV F is strongly stimulated by brief exposure to low pH and that there is a steady increase in fusion as the pH is decreased from pH 7.0 to pH 4.5 (49). The pKa of the histidine side chain in solution is 6.04, and no other amino acid side chain has a pKa (in solution) that can ionize within the physiological pH range (63). Since exposure of the HMPV F protein to low pH appears to trigger the HMPV F protein conformational change that results in membrane fusion, we hypothesized that the protonation of histidine side chains that takes place in a low pH environment might be important for F protein triggering. To determine the potential importance of the F protein stability and function of the three histidine residues in the HMPV F ectodomain, we created a model of HMPV F from the molecular coordinates of the prefusion crystal structure of PIV5 F (66). Interestingly, when we examined the location of the histidine residues in our homology model of the HMPV F protein, we noticed that each histidine residue was present in a potentially important location (Fig. 1). Histidine 332 appears to be buried but sits at the interface of two monomers. Histidine 368 lies adjacent to the fusion peptide, and it is predicted to form hydrogen bonds with amino acids in the fusion peptide. Lastly, histidine 435 sits at the base of the head of the molecule, where it is surrounded by several basic residues and is also in close proximity to a site of glycosylation. Histidine 435 is just N terminal of HRB, in a region which has been termed the HRB linker domain, and residues in this region have been shown to modulate fusion promoted by PIV5 F (46).
FIG. 1.

Homology model of HMPV F based on the prefusion PIV5 F crystal structure. For all images, except the top right (H332), cyan indicates HRB, green indicates HRA, white indicates a fusion peptide, red indicates the P1 residue of the cleavage site, orange indicates N-linked glycosylation sites, and yellow indicates histidine residues. For the H332 image, red indicates monomer one, white indicates monomer two, and yellow indicates H332. In the middle right H368 image, dotted lines indicate predicted hydrogen bonds. For clarity, the side chains of all the residues, except single highlighted residues, have been removed.
Each of the three HMPV F protein histidine residues was mutated to alanine, as well as to asparagine, an amino acid that is similar in size to histidine and polar but that cannot be protonated. To examine the cell surface expression and proteolytic processing of wild-type and mutant F proteins, Vero cells were transiently transfected with wild-type or mutant F protein genes under the control of a chicken actin promoter in the pCAGGS vector. The cells were metabolically labeled in the presence of TPCK-trypsin, and biotinylation of the surface protein was performed. Following immunoprecipitation, the proteins were analyzed by SDS-PAGE. The proteolytic processing of HMPV F from the precursor protein, F0, into the fusion-competent disulfide-linked subunits, F1 and F2, requires the addition of exogenous protease in cell culture (8, 49). Thus, trypsin was added to the labeling medium in order to examine the efficiency of the cleavage for each mutant, as a lack of processing could indicate defects in folding and would likely affect the activity of the protein. As shown in Fig. 2A, the mutation of H332 to either alanine or asparagine resulted in reduced surface expression, with no visible cleaved protein. Both of these mutants appeared as a single tight band with the same mobility as the faster-migrating portion of wild-type F0 (Fig. 2A), indicating a loss of complex carbohydrate branching. This suggests that the side chain of the residue at position 332 is important for the proper folding of the F protein, a finding that is consistent with our model predicting that this residue is buried and sits at the interface of two monomers. Interestingly, the alanine and asparagine mutants of H368 differed in their expression and cleavage patterns with respect to the wild type (Fig. 2A). While the surface expression and cleavage of the H368A mutant were similar to or slightly less than those of the wild type on average, the levels of the H368N mutant on the surface of cells were significantly greater than those of the wild type. These data suggest that side-chain packing near the fusion peptide may modulate the folding efficiency or stability of the F protein. Finally, the mutation of H435 to both alanine and asparagine resulted in levels of surface expression and cleavage that were similar to the wild-type F protein, suggesting that the mutation of H435 does not significantly affect the folding, transport, or proteolytic processing of the F protein.
To examine the fusion activity of the histidine mutants, we performed a luciferase reporter gene assay (Fig. 2B). Vero cells were transfected with a wild-type or mutant F protein gene in a pCAGGS vector as well as a plasmid containing luciferase cDNA under the control of a T7 promoter. The Vero cells were then treated with trypsin to both detach them from the plates and cleave the HMPV F protein and were then overlaid onto BSR cells, which constitutively express the T7 polymerase. The mixed cell populations were briefly exposed to buffered pH 7 or pH 5 PBS twice to trigger fusion. Four hours later, luciferase activity, indicative of fusion between Vero and BSR cells, was measured. Interestingly, the only histidine mutant capable of promoting substantial levels of fusion when exposed to low pH was H368N (Fig. 2B). It is not surprising that H332A and H332N were unable to promote fusion under both neutral and low pH conditions, as these mutants did not appear to fold properly. Fusion promoted by the H368A mutant when exposed to low pH was only slightly above background levels, which could represent a defect in low-pH triggering of fusion considering that the surface expression of this mutant was similar to that of the wild type. On the other hand, the H368N mutant promoted fusion at levels similar to or slightly less than those of the wild type (Fig. 2B), suggesting that the protonation of H368 is not required for fusion promotion. However, this mutant displayed increased levels of cleaved F protein on the surface, which suggests that either this further increase in F1 surface expression does not correlate with increased fusion activity or that there is also a slight defect in fusion promotion by this mutant. Finally, both the H435A and H435N mutants demonstrated severely compromised fusion activity (Fig. 2B), which is interesting considering that the expression and cleavage of these mutants were very similar to the expression and cleavage of wild-type F (Fig. 2A). Fusion promoted by each of the histidine mutants was also examined in syncytium assays, in which polykaryon formation is observed microscopically following trypsin and low-pH treatment of transfected cell monolayers. The extent of the syncytium formation by each of the mutants was consistent with the reporter gene results (data not shown).
The lack of fusion promoted by mutants of H435, despite normal expression levels, suggests this HRB linker residue may be essential for the triggering of fusion by low pH. Our earlier results had demonstrated the increased stimulation of HMPV F-promoted fusion by pH levels below 5 (49). To test whether further decreases in pH would allow fusion promotion in the absence of an ionizable residue at position 435, fusions of the wild-type F protein and histidine mutants were compared at pH 5 and pH 4.5 (Fig. 3A and B). Fusions by the H435N and H368N mutants were stimulated approximately 50% by further reducing the pH to 4.5, a similar level of stimulation to that seen with the wild-type F protein. In contrast, fusion promoted by the H332N mutant was not stimulated by lowering the pH, as expected of a mutant whose fusion defect results from folding and surface expression defects (Fig. 2), thus confirming that the lowering of the pH has a specific effect on fusion. These results indicate that, while H435 plays an important role in the promotion of HMPV F fusion, residues in addition to H435 can participate in low-pH triggering of fusion.
The fusion defective phenotype of the H435 alanine and asparagine mutants, in light of their relatively normal appearance on a polyacrylamide gel, prompted us to examine this region of the protein further. As mentioned previously, the homology model of HMPV F indicates that H435 is surrounded by at least three basic residues, K295, R396, and K438 (Fig. 4A). We hypothesized that the protonation of H435 leads to electrostatic repulsion within this group of amino acids, which is at least partially responsible for triggering of the F protein conformational change. We therefore examined the cell surface expression and fusion of alanine mutants of each of these basic residues (K295A, R396A, and K438A). Our results indicate that the cell surface expression and proteolytic processing of each of the basic residue mutants are similar to those of the wild type (Fig. 4B), suggesting that the mutation of each of these basic residues individually does not significantly impact protein folding or transport. However, fusion promotion by each of these mutants is strongly reduced, with the K295A mutant showing background fusion levels and the K438A mutant promoting fusion just above that. The level of fusion promoted by the R396A mutant is approximately 50% of the wild-type levels (Fig. 4C), which is consistent with our structural model indicating that this basic residue is the furthest of the three from H435. These results suggest that each of these positive charges in the HRB linker region is important for the low-pH triggering of fusion.
The data presented here and in our previous publication strongly support a role for low pH in HMPV F protein-promoted fusion (49). However, as a paramyxovirus, it would be unusual for this virus to require low pH for cell entry. Thus, we chose to examine the importance of low pH in viral entry using a recombinant GFP-expressing HMPV virus with a codon-stabilized SH gene (6). Western blot analysis suggested that the vast majority of the F protein in the HMPV preparations was proteolytically processed during propagation; thus, infections were not carried out in the presence of trypsin. We first wished to determine if syncytium formation in virus-infected cells was stimulated by exposure to low pH. A lack of pronounced syncytium formation in HMPV-infected cells has been noted in several publications (1, 8, 12, 26, 34). The cells infected with HMPV at an MOI of 0.5 were therefore treated with or without TPCK-trypsin, followed by exposure to buffered pH 7 or pH 5 PBS, and examined for syncytium formation under a fluorescence microscope. As shown in Fig. 5, neither trypsin treatment nor low-pH exposure alone was sufficient to induce obvious syncytium formation. However, when infected cells were treated with trypsin to proteolytically process the HMPV F protein on the cell surface and then briefly incubated in low-pH buffer, there was extensive syncytium formation visible (Fig. 5). This shows that the presence of other viral proteins does not alter the low-pH stimulation of F protein-promoted cell-cell fusion and that the low-pH stimulation of fusion is not an artifact of our transient expression system.
FIG. 5.

Syncytium formation in HMPV-infected cells. A recombinant GFP-expressing HMPV was used to infect Vero cells (MOI = 0.5). Infected cells were incubated in Opti-MEM ± 0.3 μg/ml TPCK-trypsin at 32°C the day after infection, except when being treated with pH 5 or 7 PBS. Cells were treated with buffered pH 5 or 7 PBS four times throughout the day for 4 min at 37°C. Photographs were taken the next morning.
In nature, a virus is most likely to encounter low pH following endocytosis and trafficking through the endosomal pathway. To determine if endosomal pH plays a role in the entry of HMPV, we utilized several known inhibitors of endosomal acidification. Bafilomycin A1 and concanamycin A are specific inhibitors of the vacuolar type H+-ATPase (V-ATPase), ammonium chloride is a weak base that buffers cellular pH, and monensin is an ionophore which facilitates the transport of H+ ions across membranes. As a control for pH-dependent entry, we used recombinant VSV in which the G envelope gene was replaced with the GFP gene and pseudotyped with the VSV G glycoprotein. For a pH-independent entry control, recombinant GFP-expressing PIV5 was used (10, 11, 55). Vero cells were pretreated with each drug for 1.5 h and then infected with each virus in the presence of the drug. After 4 h, the cells were washed and fresh medium was added. The number of GFP-positive cells was determined by flow cytometry. This time course of drug treatment was chosen because the drugs appeared to have nonspecific effects on infection when they were left on the cells for longer periods of time. Drug treatment beyond the time used in our experiments often impacted GFP expression in the negative control, and there appeared to be a significant inhibition of HMPV infection when the drug was added 6 h postinfection and left overnight.
Under the conditions described above, 100 nM and 200 nM doses of bafilomycin A1 resulted in a nearly complete inhibition of VSV infection and had no effect on the efficiency of PIV5 infection (Fig. 6A). Interestingly, bafilomycin A1 treatment of Vero cells resulted in an approximately 40% reduction in HMPV infection. Concanamycin A is a macrolide antibiotic similar in structure to bafilomycin A1, but concanamycin A is more specific and is as effective as bafilomycin A1 at lower concentrations (19). The effect of 5 nM and 10 nM doses of concanamycin A on infections of VSV and PIV5 was the same as that of bafilomycin A1, and concanamycin A resulted in an approximately 50% reduction in HMPV infection (Fig. 6B). Thus, two specific inhibitors of vacuolar acidification significantly impact HMPV infection, but the inhibitors do not completely block infection as they do VSV infection, suggesting that there are differences in the entry mechanisms of these viruses. Ammonium chloride, on the other hand, appeared to have only minor effects on the entry of HMPV in Vero cells, and doses that inhibited VSV infection affected PIV5 and HMPV infection to similar levels (Fig. 6C). However, the ionophore monensin inhibited HMPV infection while not significantly inhibiting PIV5 infection (Fig. 6D). Again, the effects of monensin were greater on VSV infection than on HMPV infection, with HMPV infection reduced by approximately 30% at the highest dose tested. Higher concentrations affected PIV5 infection as measured by GFP expression, thus likely impacting cell health.
Vero cells, which are monkey kidney cells, are a common cell type used in studies of viruses. We used Vero cells for the majority of our experiments because these cells are not significantly affected by the trypsin conditions needed to cleave the F protein. However, we wanted to examine the effect of endosomal acidification inhibitors in a more physiologically relevant cell type. Thus, we examined the effect of endosomal acidification inhibitors on HMPV infection in A549 cells, a human alveolar type II-like epithelial cell line, which others have characterized as a model to study lower airway epithelial cell responses to HMPV infection (3). Bafilomycin A1 and concanamycin A had the same effect on infection by all viruses in A549 cells that they had in Vero cells, except bafilomycin A1 had a somewhat larger effect on HMPV infection, reducing infection by approximately 50% in these cells (Fig. 7A and B). Interestingly, ammonium chloride also had a larger negative impact on HMPV infection in A549 cells, and in these cells, ammonium chloride did not inhibit PIV5 infection at any of the doses tested (Fig. 7C). Thus, optimal HMPV infection of human lower airway cells is significantly impacted by conditions that alter low pH in cellular vesicles.
FIG. 7.

Virus entry in A549 cells treated with inhibitors of endosomal acidification. Monolayers of A549 cells in 24-well plates were pretreated with a drug, bafilomycin A1 (A), concanamycin A (B), or ammonium chloride (C), or a vehicle control diluted in RPMI medium plus FBS at the concentrations indicated for 1.5 h. DMEM buffered with 20 mM HEPES was used for the ammonium chloride treatments. Infections were carried out in the presence of the drugs or the vehicle control such that 20 to 30% of the cells would be infected on average in control wells, and the cells were incubated for 4 h at 37°C. The cells were then washed and incubated in RPMI medium plus FBS for an additional 6 h (VSV) or 12 to 16 h (HMPV and PIV5) at 37°C. The cells were then detached with trypsin, fixed in 1% formaldehyde, and analyzed for GFP expression by flow cytometry. Data shown represent the number of GFP-expressing cells as a percentage of that of the control, set to 100% for each virus, from at least three separate experiments. Error bars represent 95% confidence intervals.
Some viral proteins, such as influenza virus HA, that are activated by low pH may be inactivated by exposure to low pH when the virus is in solution and a target membrane is absent (13). Other viral proteins, however, must bind to a receptor or be internalized before low pH can cause a conformational change (40, 53, 58). VSV G is a unique viral protein that may actually undergo reversible conformational changes (24). Thus, low pH will inactivate the viral entry glycoprotein, but neutralization of the pH prior to its addition to cells will reactivate the virus. We exposed GFP-expressing HMPV, PIV5, and VSV to a pH 5 or pH 7 citric acid solution for 10 min at 37°C and then neutralized the solution with Opti-MEM prior to their addition to the cells. We then determined the effect of this pretreatment on the infection efficiency of each virus in Vero cells. The low-pH pretreatment of HMPV resulted in more than a 50% reduction in the number of cells infected (Fig. 8). This low-pH pretreatment resulted in a reduction in the number of cells infected by PIV5 but to a much smaller extent than that seen with HMPV, suggesting that low-pH pretreatment more specifically affects HMPV. Interestingly, low-pH pretreatment increased VSV infection, though the reason for this is unknown.
FIG. 8.

Low versus neutral pH pretreatment effect on virus infection. Eighty microliters of pH 4.1 or 7 citric acid buffer was added to each virus brought to 20 μl total volume in Opti-MEM plus SPG and incubated at 37°C for 10 min. The quantity of virus used resulted in 20 to 40% of the cells on average being infected. Next, 450 μl of Opti-MEM was added to neutralize the virus solution, and 500 μl of the final solution was used to infect monolayers of Vero cells in a 24-well plate. Ten hours (VSV) or 16 to 20 h (HMPV and PIV5) postinfection, the cells were detached with trypsin, fixed in 1% formaldehyde, and analyzed for GFP expression by flow cytometry. Data shown represent the number of GFP-expressing cells as a percentage of that of the pH 7 treatment, set to 100% for each virus, from four separate experiments. Error bars represent 95% confidence intervals.
A viral entry mechanism incorporating low pH suggests a requirement for the endocytosis of viral particles. To examine a potential role for clathrin-mediated endocytosis in HMPV entry, we treated cells with the clathrin inhibitor chlorpromazine prior to and during the initial infection. VSV and PIV5 were again examined in parallel, as VSV has been shown to require clathrin-mediated endocytosis and PIV5 is hypothesized to enter cells at the plasma membrane (10, 54). Interestingly, at low doses of chlorpromazine, there was an increase in the number of cells infected with VSV (Fig. 9A). Chlorpromazine is reported to have a number of effects on cellular processes unrelated to endocytosis, which may account for the apparent increase in VSV infection at low concentrations of the drug. Furthermore, very high concentrations of chlorpromazine at neutral pH have been shown to promote fusion by inducing positive membrane curvature (39). Nevertheless, at higher doses of 10 μg/ml, there was a large decrease in VSV infection (Fig. 9). At this dose, there was a minor decrease in PIV5 infection, which may be due to the visible cytopathic effect of the drug at this concentration. Still, the number of HMPV-infected cells was reduced in the presence of 10 μg/ml chlorpromazine to an extent much greater than that of PIV5-infected cells and even more so than that of VSV-infected cells, with a 70% reduction in the number of cells infected compared to the control (Fig. 9).
To further explore the potential role of endocytosis in HMPV entry, we examined the effect of dynasore, a small molecule inhibitor of dynamin (32), on HMPV entry. The addition of dynasore inhibited both VSV and HMPV entry when present 30 min prior to infection and during the first 2 h of infection (Fig. 9B; pre/during samples), indicating that dynamin is important for the entry of both of these pathogens. In contrast, the addition of dynasore for the same total length of time starting 2 h postinfection (Fig. 9B; post samples) resulted in only minor decreases in infection, demonstrating that the major role of dynamin is during the initial stage of infection. These results strongly support a role for endocytosis in HMPV entry, in contrast to the route of entry for the majority of paramyxoviruses.
DISCUSSION
Since the discovery of human metapneumovirus just 7 years ago (61), it has proven to be unique among paramyxoviruses. The F protein is not cleaved by endogenous proteases, unlike what is observed for most F proteins (8, 49), and the attachment protein is not essential for entry and does not appear to enhance fusion (9, 49). Our work presented here indicates that HMPV entry is dependent on endocytosis and that low pH within endosomes affects the efficiency of infection of host cells. RSV, a closely related virus in the subfamily Pneumovirinae of the paramyxovirus family, displays a number of important similarities to HMPV: the F protein of RSV is also capable of promoting fusion in the absence of an attachment protein (56), and RSV entry was recently shown to occur via clathrin-mediated endocytosis (33). These observations suggest that endocytic entry may be a characteristic of members of the Pneumovirinae subfamily, in contrast to other paramyxoviruses. For both RSV and HMPV, endosomal entry may provide control of F protein triggering in the absence of the tight coupling between attachment protein binding and fusion that is observed for members of the Paramyxoviridae family.
RSV and HMPV do differ in their mechanisms of F protein proteolytic activation. RSV F protein activation requires cleavage at two distinct furin sites (25), in contrast to the single furin site present in most F proteins. The HMPV F protein from the majority of strains lacks the ability to be efficiently processed by cellular proteases (47) and must undergo cleavage by extracellular proteases prior to fusion triggering. The absence of HMPV F protein proteolytic processing by intracellular proteases would provide additional control of triggering, preventing the premature activation of F protein in the lower pH environment of the secretory pathway prior to viral assembly. The subsequent cleavage of F protein in the virion by proteases such as the serine protease TMPRSS2 (51) would then be required prior to F protein triggering and function.
One important difference between RSV and HMPV is the effect of low pH on the efficiency of viral entry. RSV entry, though dependent on endocytosis, was not significantly affected by lysosomotropic agents (33). Our results indicate a 30 to 50% reduction in HMPV entry when intracellular pH is raised, suggesting that the low pH environment of the endosomal pathway can stimulate fusion and entry. The two major subtypes of HMPV, clade A and clade B, both contain multiple strains currently circulating in the human population, with clade A viruses often seen at a more prevalent rate (29). A recent report (27) found differences in the effect of pH on fusion promoted by HMPV F proteins from different strains. The F proteins from two clade A viruses, including the CAN 97-83 strain used in our study, showed strong stimulation by low pH, while one clade A F protein did not display fusion under any of the tested conditions. In contrast, the F proteins from two clade B viruses promoted equivalent levels of fusion at neutral and low pH (27). An analysis of the role of endocytosis for clade B viruses has not been performed to date, but these results suggest that the level of low-pH stimulation may differ between various HMPV strains.
Our experiments using specific inhibitors of the V-ATPase strongly indicate that exposure to low pH provides a potent stimulation of F protein fusion, resulting in more-efficient entry. However, we observed two thought-provoking results when various inhibitors of endosomal acidification were used to examine HMPV entry in parallel with pH-dependent and -independent control viruses: (i) ammonium chloride and monensin did not inhibit entry to the same extent as bafilomycin A1 and concanamycin A, and (ii) there was about a 50% maximum inhibition of HMPV entry with any drug, as opposed to the nearly complete inhibition of VSV entry.
A recent examination of Jaagsiekte sheep retrovirus entry also noted strong inhibition by bafilomycin A1 but weak inhibition by ammonium chloride (5). The authors of this recent study suggested that the difference might be due to the rapid recovery of endosomal pH following removal of the ammonium chloride (44), allowing for viral particles that were trapped in the endocytic pathway to then enter. However, bafilomycin A1 inhibition is also reversible (67). It is possible that viral particles may persist in vesicles for long periods (40), especially under high pH conditions when the activity of proteases in the lysosome is decreased. We did not detect a difference in HMPV inhibition by acidification inhibitors when the incubation time with the drug was extended to 6 h postinfection. However, we were unable to determine the effect of inhibitors on infection without removing the inhibitor at a time prior to analysis due to nonspecific effects on infection, as measured by decreased GFP expression in our negative control virus-infected cells. The initiation of GFP expression resulting from HMPV and PIV5 infection was not evident until more than 12 h postinfection, and it is likely that drug treatments for these extended periods had cytotoxic effects.
It is possible that the difference in the inhibitions of bafilomycin A1 and ammonium chloride is a consequence of their relative effectiveness in neutralizing the endosomal pH. Experiments measuring the pH in cellular vesicles during treatment with acidification inhibitors have found that neither type of inhibitor raises the pH of endosomes to neutrality unless excessive concentrations are used, and the effectiveness of bafilomycin A1 and ammonium chloride in raising the pH was often not equivalent and can depend on cell type (37, 42, 67). We have shown that HMPV F protein-promoted fusion increases steadily as the pH to which the protein is exposed decreases (49). This observation suggests that the probability of F protein triggering is inversely proportional to pH, and there is not a specific pH that must be reached in order for all F proteins to trigger. It is also likely that more than one F protein must undergo a conformational change in order for membrane fusion to occur (20). Therefore, some proteins may trigger in a neutral or near-neutral environment, perhaps following receptor binding, but low pH may facilitate fusion by increasing the number of F proteins to trigger. Thus, by raising the pH slightly, the inhibitors of endosomal acidification simply decrease the probability that enough F proteins will trigger to cause fusion before the virus is degraded. If the inhibitors raised the pH to different extents, the number of entry events would reflect the probability of F protein triggering under those conditions. It is important to note, however, that VSV entry, with a pH threshold of approximately 6.2 (45), was efficiently inhibited by all drugs tested. Thus, the endosomal pH was raised to at least this level with each of the tested agents. Since the triggering of HMPV fusion in cell-cell fusion systems was not significantly stimulated by pH treatments above 6, there are either differences in the pH requirements between cell-cell and virus-cell fusion or additional factors are at work. Further experiments will be needed to clarify this important point.
Alternatively, chemicals that alter the pH of endocytic vesicles have in some cases been shown to block the trafficking of certain cargo (4, 30, 59). It is possible that the specific V-ATPase inhibitors bafilomycin A1 and concanamycin A alter or block the trafficking of endocytosed HMPV such that the entry or postentry steps are disturbed and the effect of these drugs on infection is unrelated to pH. Further studies are necessary to determine if inhibition of the V-ATPase affects the trafficking of viral particles, and this information would be particularly important due to the widespread use of these inhibitors in studies of viral entry. However, RSV entry was shown to be no more affected by 10 nM bafilomycin A1 treatment than a control virus, and RSV appears to be a unique virus that requires endocytosis but not low pH for entry (33). Unless post-endocytic trafficking of RSV is unimportant, the lack of inhibition by bafilomycin A1 would suggest that this inhibitor does not affect trafficking in the system used.
The mutagenesis of histidine residues in the ectodomain of HMPV F revealed the importance of a residue in the HRB linker domain to low pH-induced fusion. Furthermore, several basic residues, which are structurally in close proximity to this histidine residue, also appear to facilitate the low pH-induced F protein conformational changes that are responsible for membrane fusion. Histidine residues are important in many biological reactions due to their propensity to ionize within the physiological pH range (63). We hypothesize that H435 of the HMPV F protein becomes protonated under low-pH conditions and that this protonation results in electrostatic repulsion between the histidine and neighboring lysine and arginine residues, thus destabilizing the prefusion conformation of the molecule. However, it is likely that other F protein residues contribute to low-pH triggering as well. The pKa of charged amino acid side chains is influenced by the local environment of the side chains, and the average number of side chain-associated protons can increase when the pH is lowered (28). Our analysis of histidine mutant fusion at pH 4.5 suggests that fusion can still be stimulated when the residue at position 435 is not capable of accepting a proton. Thus, H435, while important for fusion, is likely not the only residue that contributes to low-pH triggering of fusion.
Each of the F protein histidine residues is conserved in the subtypes of HMPV (62). The basic residues, with the exception of R396, which tolerated mutation better than the others, are also conserved. It is also interesting to note that modeling suggests there is a glycosylation site (N353), which we have verified is utilized, near H435 in the prefusion structure. The mutation of N353 resulted in defective proteolytic processing and a loss of fusion activity (49), and it is possible that there are interactions between the negative charges on this carbohydrate modification and the multiple positive charges in the vicinity that are necessary for protein stability. Finally, a change from a glycine to a glutamic acid residue at position 294 in clade B viruses has been correlated with the loss of low-pH stimulation of HMPV F fusion (27). Though the authors of this report suggest the proximity of residue G294 to histidine 368, our modeled structure indicates that G294 lies within 6 Å of H435 (but over 25 Å from H368). While it is unclear why the models differ, the close proximity of residue 294 to H435 in our model and its presence adjacent to the critical basic residue K295 strongly suggest that the effect of changes at position 294 on low-pH triggering of fusion relates to the alteration of the electrostatic interactions in the HRB linker region.
While further experiments are needed to more carefully define the endocytic route utilized by HMPV and the role of endocytosis in the entry of different strains of HMPV, our results and those of Kolokoltsov et al. (33) indicate that endocytic entry may be a common feature of members of the Pneumovirinae subfamily, in contrast to what has been reported for the Paramyxovirinae subfamily. The partial reduction in HMPV infection by the inhibitors of endosomal acidification is a novel observation and suggests that at least some strains of HMPV may have evolved to use the low pH of the endocytic pathway to enhance infectivity, although the role of low pH likely differs from classically described mechanisms.
Acknowledgments
We are grateful to Peter Collins (Laboratory of Infectious Diseases, NIAID, Bethesda, MD) for providing the recombinant GFP-HMPV, Robert Lamb (HHMI, Northwestern University) for providing GFP-PIV5, and Michael Whitt (University of Tennessee Health Science Center, Memphis, Tennessee) for providing VSV pseudotypes. We thank Guy Boivin (Centre Hospitalier Universitaire de Québec, Quebec City, Quebec, Canada) for his permission to obtain HMPV. We also thank Cara Pager for preparing the VSV pseudotypes used in these studies and Jennifer Strange and Greg Bauman from the University of Kentucky's Flow Cytometry Core Facility for their assistance in processing samples.
This study was supported by predoctoral fellowship 0715355B from the Great Rivers affiliate of the American Heart Association, a research grant from the March of Dimes, and research grant 1R21AI074783-01 from the National Institutes of Health.
Footnotes
Published ahead of print on 26 November 2008.
REFERENCES
- 1.Alvarez, R., K. S. Harrod, W. J. Shieh, S. Zaki, and R. A. Tripp. 2004. Human metapneumovirus persists in BALB/c mice despite the presence of neutralizing antibodies. J. Virol. 7814003-14011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Bagai, S., and R. A. Lamb. 1995. Quantitative measurement of paramyxovirus fusion: differences in requirements of glycoproteins between simian virus 5 and human parainfluenza virus 3 or Newcastle disease virus. J. Virol. 696712-6719. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Bao, X., T. Liu, L. Spetch, D. Kolli, R. P. Garofalo, and A. Casola. 2007. Airway epithelial cell response to human metapneumovirus infection. Virology 36891-101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Bayer, N., D. Schober, E. Prchla, R. F. Murphy, D. Blaas, and R. Fuchs. 1998. Effect of bafilomycin A1 and nocodazole on endocytic transport in HeLa cells: implications for viral uncoating and infection. J. Virol. 729645-9655. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Bertrand, P., M. Cote, Y. M. Zheng, L. M. Albritton, and S. L. Liu. 2008. Jaagsiekte sheep retrovirus utilizes a pH-dependent endocytosis pathway for entry. J. Virol. 822555-2559. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Biacchesi, S., B. R. Murphy, P. L. Collins, and U. J. Buchholz. 2007. Frequent frameshift and point mutations in the SH gene of human metapneumovirus passaged in vitro. J. Virol. 816057-6067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Biacchesi, S., Q. N. Pham, M. H. Skiadopoulos, B. R. Murphy, P. L. Collins, and U. J. Buchholz. 2006. Modification of the trypsin-dependent cleavage activation site of the human metapneumovirus fusion protein to be trypsin independent does not increase replication or spread in rodents or nonhuman primates. J. Virol. 805798-5806. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Biacchesi, S., M. H. Skiadopoulos, K. C. Tran, B. R. Murphy, P. L. Collins, and U. J. Buchholz. 2004. Recovery of human metapneumovirus from cDNA: optimization of growth in vitro and expression of additional genes. Virology 321247-259. [DOI] [PubMed] [Google Scholar]
- 9.Biacchesi, S., M. H. Skiadopoulos, L. Yang, E. W. Lamirande, K. C. Tran, B. R. Murphy, P. L. Collins, and U. J. Buchholz. 2004. Recombinant human metapneumovirus lacking the small hydrophobic SH and/or attachment G glycoprotein: deletion of G yields a promising vaccine candidate. J. Virol. 7812877-12887. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Bissonnette, M. L., S. A. Connolly, D. F. Young, R. E. Randall, R. G. Paterson, and R. A. Lamb. 2006. Analysis of the pH requirement for membrane fusion of different isolates of the paramyxovirus parainfluenza virus 5. J. Virol. 803071-3077. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Blumenthal, R., A. Bali-Puri, A. Walter, D. Covell, and O. Eidelman. 1987. pH-dependent fusion of vesicular stomatitis virus with Vero cells. Measurement by dequenching of octadecyl rhodamine fluorescence. J. Biol. Chem. 26213614-13619. [PubMed] [Google Scholar]
- 12.Boivin, G., Y. Abed, G. Pelletier, L. Ruel, D. Moisan, S. Cote, T. C. Peret, D. D. Erdman, and L. J. Anderson. 2002. Virological features and clinical manifestations associated with human metapneumovirus: a new paramyxovirus responsible for acute respiratory-tract infections in all age groups. J. Infect. Dis. 1861330-1334. [DOI] [PubMed] [Google Scholar]
- 13.Boulay, F., R. W. Doms, I. Wilson, and A. Helenius. 1987. The influenza hemagglutinin precursor as an acid-sensitive probe of the biosynthetic pathway. EMBO J. 62643-2650. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Carneiro, F. A., F. Stauffer, C. S. Lima, M. A. Juliano, L. Juliano, and A. T. Da Poian. 2003. Membrane fusion induced by vesicular stomatitis virus depends on histidine protonation. J. Biol. Chem. 27813789-13794. [DOI] [PubMed] [Google Scholar]
- 15.Carr, C. M., C. Chaudhry, and P. S. Kim. 1997. Influenza hemagglutinin is spring-loaded by a metastable native conformation. Proc. Natl. Acad. Sci. USA 9414306-14313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Connolly, S. A., and R. A. Lamb. 2006. Paramyxovirus fusion: real-time measurement of parainfluenza virus 5 virus-cell fusion. Virology 355203-212. [DOI] [PubMed] [Google Scholar]
- 17.Deffrasnes, C., M. E. Hamelin, and G. Boivin. 2007. Human metapneumovirus. Semin. Respir. Crit. Care Med. 28213-221. [DOI] [PubMed] [Google Scholar]
- 18.Dimitrov, D. S. 2004. Virus entry: molecular mechanisms and biomedical applications. Nat. Rev. Microbiol. 2109-122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Drose, S., K. U. Bindseil, E. J. Bowman, A. Siebers, A. Zeeck, and K. Altendorf. 1993. Inhibitory effect of modified bafilomycins and concanamycins on P- and V-type adenosinetriphosphatases. Biochemistry 323902-3906. [DOI] [PubMed] [Google Scholar]
- 20.Dutch, R. E., S. B. Joshi, and R. A. Lamb. 1998. Membrane fusion promoted by increasing surface densities of the paramyxovirus F and HN proteins: comparison of fusion reactions mediated by simian virus 5 F, human parainfluenza virus type 3 F, and influenza virus HA. J. Virol. 727745-7753. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Easton, A. J., J. B. Domachowske, and H. F. Rosenberg. 2004. Animal pneumoviruses: molecular genetics and pathogenesis. Clin. Microbiol. Rev. 17390-412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Fritz, R., K. Stiasny, and F. X. Heinz. 2008. Identification of specific histidines as pH sensors in flavivirus membrane fusion. J. Cell Biol. 183353-361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Gardner, A. E., and R. E. Dutch. 2007. A conserved region in the F2 subunit of paramyxovirus fusion proteins is involved in fusion regulation. J. Virol. 818303-8314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Gaudin, Y. 2000. Reversibility in fusion protein conformational changes. The intriguing case of rhabdovirus-induced membrane fusion. Subcell. Biochem. 34379-408. [DOI] [PubMed] [Google Scholar]
- 25.Gonzalez-Reyes, L., M. B. Ruiz-Arguello, B. Garcia-Barreno, L. Calder, J. A. Lopez, J. P. Albar, J. J. Skehel, D. C. Wiley, and J. A. Melero. 2001. Cleavage of the human respiratory syncytial virus fusion protein at two distinct sites is required for activation of membrane fusion. Proc. Natl. Acad. Sci. USA 989859-9864. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Hamelin, M. E., Y. Abed, and G. Boivin. 2004. Human metapneumovirus: a new player among respiratory viruses. Clin. Infect. Dis. 38983-990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Herfst, S., V. Mas, L. S. Ver, R. J. Wierda, A. D. Osterhaus, R. A. Fouchier, and J. A. Melero. 2008. Low-pH-induced membrane fusion mediated by human metapneumovirus F protein is a rare, strain-dependent phenomenon. J. Virol. 828891-8895. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Huang, Q., R. Opitz, E. W. Knapp, and A. Herrmann. 2002. Protonation and stability of the globular domain of influenza virus hemagglutinin. Biophys. J. 821050-1058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Huck, B., G. Scharf, D. Neumann-Haefelin, W. Puppe, J. Weigl, and V. Falcone. 2006. Novel human metapneumovirus sublineage. Emerg. Infect. Dis. 12147-150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Hurtado-Lorenzo, A., M. Skinner, J. El Annan, M. Futai, G. H. Sun-Wada, S. Bourgoin, J. Casanova, A. Wildeman, S. Bechoua, D. A. Ausiello, D. Brown, and V. Marshansky. 2006. V-ATPase interacts with ARNO and Arf6 in early endosomes and regulates the protein degradative pathway. Nat. Cell Biol. 8124-136. [DOI] [PubMed] [Google Scholar]
- 31.Kahn, J. S. 2006. Epidemiology of human metapneumovirus. Clin. Microbiol. Rev. 19546-557. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Kirchhausen, T., E. Macia, and H. E. Pelish. 2008. Use of dynasore, the small molecule inhibitor of dynamin, in the regulation of endocytosis. Methods Enzymol. 43877-93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Kolokoltsov, A. A., D. Deniger, E. H. Fleming, N. J. Roberts, Jr., J. M. Karpilow, and R. A. Davey. 2007. Small interfering RNA profiling reveals key role of clathrin-mediated endocytosis and early endosome formation for infection by respiratory syncytial virus. J. Virol. 817786-7800. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Kuiken, T., B. G. van den Hoogen, D. A. van Riel, J. D. Laman, G. van Amerongen, L. Sprong, R. A. Fouchier, and A. D. Osterhaus. 2004. Experimental human metapneumovirus infection of cynomolgus macaques (Macaca fascicularis) results in virus replication in ciliated epithelial cells and pneumocytes with associated lesions throughout the respiratory tract. Am. J. Pathol. 1641893-1900. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Lamb, R. A., and T. S. Jardetzky. 2007. Structural basis of viral invasion: lessons from paramyxovirus F. Curr. Opin. Struct. Biol. 17427-436. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Lamb, R. A., R. G. Paterson, and T. S. Jardetzky. 2006. Paramyxovirus membrane fusion: lessons from the F and HN atomic structures. Virology 34430-37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Lin, H. J., P. Herman, and J. R. Lakowicz. 2003. Fluorescence lifetime-resolved pH imaging of living cells. Cytometry A 5277-89. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Marsh, M., and A. Helenius. 2006. Virus entry: open sesame. Cell 124729-740. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Melikyan, G. B., S. A. Brener, D. C. Ok, and F. S. Cohen. 1997. Inner but not outer membrane leaflets control the transition from glycosylphosphatidylinositol-anchored influenza hemagglutinin-induced hemifusion to full fusion. J. Cell Biol. 136995-1005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Mothes, W., A. L. Boerger, S. Narayan, J. M. Cunningham, and J. A. Young. 2000. Retroviral entry mediated by receptor priming and low pH triggering of an envelope glycoprotein. Cell 103679-689. [DOI] [PubMed] [Google Scholar]
- 41.Naylor, C. J., P. A. Brown, N. Edworthy, R. Ling, R. C. Jones, C. E. Savage, and A. J. Easton. 2004. Development of a reverse-genetics system for avian pneumovirus demonstrates that the small hydrophobic (SH) and attachment (G) genes are not essential for virus viability. J. Gen. Virol. 853219-3227. [DOI] [PubMed] [Google Scholar]
- 42.Ohkuma, S., S. Shimizu, M. Noto, Y. Sai, K. Kinoshita, and H. Tamura. 1993. Inhibition of cell growth by bafilomycin A1, a selective inhibitor of vacuolar H(+)-ATPase. In Vitro Cell. Dev. Biol. Anim. 29A862-866. [DOI] [PubMed] [Google Scholar]
- 43.Paterson, R. G., and R. A. Lamb. 1993. The molecular biology of influenza viruses and paramyxoviruses, p. 35-73. In A. Davidson and R. M. Elliott (ed.), Molecular virology: a practical approach. IRL Oxford University Press, Oxford, England.
- 44.Poole, B., and S. Ohkuma. 1981. Effect of weak bases on the intralysosomal pH in mouse peritoneal macrophages. J. Cell Biol. 90665-669. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Roche, S., and Y. Gaudin. 2002. Characterization of the equilibrium between the native and fusion-inactive conformation of rabies virus glycoprotein indicates that the fusion complex is made of several trimers. Virology 297128-135. [DOI] [PubMed] [Google Scholar]
- 46.Russell, C. J., K. L. Kantor, T. S. Jardetzky, and R. A. Lamb. 2003. A dual-functional paramyxovirus F protein regulatory switch segment: activation and membrane fusion. J. Cell Biol. 163363-374. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Schickli, J. H., J. Kaur, N. Ulbrandt, R. R. Spaete, and R. S. Tang. 2005. An S101P substitution in the putative cleavage motif of the human metapneumovirus fusion protein is a major determinant for trypsin-independent growth in Vero cells and does not alter tissue tropism in hamsters. J. Virol. 7910678-10689. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Schmidt, U., J. Beyer, U. Polster, L. J. Gershwin, and U. J. Buchholz. 2002. Mucosal immunization with live recombinant bovine respiratory syncytial virus (BRSV) and recombinant BRSV lacking the envelope glycoprotein G protects against challenge with wild-type BRSV. J. Virol. 7612355-12359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Schowalter, R. M., S. E. Smith, and R. E. Dutch. 2006. Characterization of human metapneumovirus F protein-promoted membrane fusion: critical roles for proteolytic processing and low pH. J. Virol. 8010931-10941. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Seth, S., A. Vincent, and R. W. Compans. 2003. Activation of fusion by the SER virus F protein: a low-pH-dependent paramyxovirus entry process. J. Virol. 776520-6527. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Shirogane, Y., M. Takeda, M. Iwasaki, N. Ishiguro, H. Takeuchi, Y. Nakatsu, M. Tahara, H. Kikuta, and Y. Yanagi. 2008. Efficient multiplication of human metapneumovirus in Vero cells expressing the transmembrane serine protease TMPRSS2. J. Virol. 828942-8946. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Skehel, J. J., and D. C. Wiley. 2000. Receptor binding and membrane fusion in virus entry: the influenza hemagglutinin. Annu. Rev. Biochem. 69531-569. [DOI] [PubMed] [Google Scholar]
- 53.Smith, J. G., W. Mothes, S. C. Blacklow, and J. M. Cunningham. 2004. The mature avian leukosis virus subgroup A envelope glycoprotein is metastable, and refolding induced by the synergistic effects of receptor binding and low pH is coupled to infection. J. Virol. 781403-1410. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Sun, X., V. K. Yau, B. J. Briggs, and G. R. Whittaker. 2005. Role of clathrin-mediated endocytosis during vesicular stomatitis virus entry into host cells. Virology 33853-60. [DOI] [PubMed] [Google Scholar]
- 55.Takada, A., C. Robison, H. Goto, A. Sanchez, K. G. Murti, M. A. Whitt, and Y. Kawaoka. 1997. A system for functional analysis of Ebola virus glycoprotein. Proc. Natl. Acad. Sci. USA 9414764-14769. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Techaarpornkul, S., N. Barretto, and M. E. Peeples. 2001. Functional analysis of recombinant respiratory syncytial virus deletion mutants lacking the small hydrophobic and/or attachment glycoprotein gene. J. Virol. 756825-6834. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Thoennes, S., Z. N. Li, B. J. Lee, W. A. Langley, J. J. Skehel, R. J. Russell, and D. A. Steinhauer. 2008. Analysis of residues near the fusion peptide in the influenza hemagglutinin structure for roles in triggering membrane fusion. Virology 370403-414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Tscherne, D. M., C. T. Jones, M. J. Evans, B. D. Lindenbach, J. A. McKeating, and C. M. Rice. 2006. Time- and temperature-dependent activation of hepatitis C virus for low-pH-triggered entry. J. Virol. 801734-1741. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Umata, T., Y. Moriyama, M. Futai, and E. Mekada. 1990. The cytotoxic action of diphtheria toxin and its degradation in intact Vero cells are inhibited by bafilomycin A1, a specific inhibitor of vacuolar-type H(+)-ATPase. J. Biol. Chem. 26521940-21945. [PubMed] [Google Scholar]
- 60.van den Hoogen, B. G., T. M. Bestebroer, A. D. Osterhaus, and R. A. Fouchier. 2002. Analysis of the genomic sequence of a human metapneumovirus. Virology 295119-132. [DOI] [PubMed] [Google Scholar]
- 61.van den Hoogen, B. G., J. C. de Jong, J. Groen, T. Kuiken, R. de Groot, R. A. Fouchier, and A. D. Osterhaus. 2001. A newly discovered human pneumovirus isolated from young children with respiratory tract disease. Nat. Med. 7719-724. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.van den Hoogen, B. G., S. Herfst, L. Sprong, P. A. Cane, E. Forleo-Neto, R. L. de Swart, A. D. Osterhaus, and R. A. Fouchier. 2004. Antigenic and genetic variability of human metapneumoviruses. Emerg. Infect. Dis. 10658-666. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Voet, D., and J. G. Voet. 2004. Biochemistry, 3rd ed., vol. 1. John Wiley & Sons, Inc., Hoboken, NJ.
- 64.Welliver, R. C. 2003. Review of epidemiology and clinical risk factors for severe respiratory syncytial virus (RSV) infection. J. Pediatr. 143S112-S117. [DOI] [PubMed] [Google Scholar]
- 65.Williams, J. V., P. A. Harris, S. J. Tollefson, L. L. Halburnt-Rush, J. M. Pingsterhaus, K. M. Edwards, P. F. Wright, and J. E. Crowe, Jr. 2004. Human metapneumovirus and lower respiratory tract disease in otherwise healthy infants and children. N. Engl. J. Med. 350443-450. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Yin, H. S., X. Wen, R. G. Paterson, R. A. Lamb, and T. S. Jardetzky. 2006. Structure of the parainfluenza virus 5 F protein in its metastable, prefusion conformation. Nature 43938-44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Yoshimori, T., A. Yamamoto, Y. Moriyama, M. Futai, and Y. Tashiro. 1991. Bafilomycin A1, a specific inhibitor of vacuolar-type H(+)-ATPase, inhibits acidification and protein degradation in lysosomes of cultured cells. J. Biol. Chem. 26617707-17712. [PubMed] [Google Scholar]