

Abstract
The latent membrane protein 1 (LMP1) oncogene carried by Epstein-Barr virus (EBV) is essential for transformation and maintenance of EBV-immortalized B cells in vitro, and it is expressed in most EBV-associated tumor types. The activation of the NF-κB pathway by LMP1 plays a critical role in the upregulation of antiapoptotic proteins. The EBV-encoded EBNA2 transactivator is required for LMP1 activation in latency III, while LMP1 itself appears to be critical for its activation in the latency II gene expression program. In both cases, additional viral and cellular transcription factors are required in mediating transcription activation of the LMP1 promoter. Using DNA affinity purification and chromatin immunoprecipitation assay, we showed here that members of the NF-κB transcription factor family bound to the LMP1 promoter in vitro and in vivo. Electrophoretic mobility shift assay analyses indicated the binding of the p50-p50 homodimer and the p65-p50 heterodimer to an NF-κB site in the LMP1 promoter. Transient transfections and reporter assays showed that the LMP1 promoter is activated by exogenous expression of NF-κB factors in both B cells and epithelial cells. Exogenous expression of NF-κB factors in the EBNA2-deficient P3HR1 cell line induced LMP1 protein expression. Overall, our data are consistent with the presence of a positive regulatory circuit between NF-κB activation and LMP1 expression.
Epstein-Barr virus (EBV) is a ubiquitous human herpesvirus with transforming ability. It has been associated with several malignancies, including Burkitt's lymphoma, nasal NK/T cell lymphoma, nasopharyngeal carcinoma (NPC), and Hodgkin's lymphoma, as well as lymphoproliferative disorders in immunocompromised individuals (53). The latent membrane protein 1 (LMP1) gene is the main EBV oncogene and has the ability to transform human and rodent fibroblasts in vitro (10, 52). LMP1 functions as a constitutively active tumor necrosis factor receptor that induces the activation of several signaling pathways, including those of the nuclear factor-κB (NF-κB) family. LMP1 signaling leads to upregulation of antiapoptotic proteins and provide growth signals in latently infected cells (4).
In EBV-associated tumors, different patterns of latent gene expression programs are observed and have been grouped under three latency types, I, II, and III. LMP1 is expressed in both latency II and III, which include most EBV-related malignancies (37). In latency III cells, LMP1 expression is activated by the viral EBNA2 protein through its proximal ED-L1 promoter. EBNA2 lacks direct DNA binding ability but relies on other cellular and viral transcription factors for its interaction with the promoter region (54). While the RBP-Jκ binding to the LMP1 promoter has been the most established mediator of EBNA2 activation (26), other factors such as PU.1 (23), POU (44), and an AP-2 site binding factor (22) are also involved in EBNA2 activation of LMP1. In latency II cells, EBNA2 is not expressed and LMP1 expression has to occur via alternative mechanisms. In epithelial cells, LMP1 expression is activated through both the ED-L1 promoter and a distal promoter referred to as TR-L1 or ED-L1E. STAT3 (5) and Sp1 and Sp3 transcription factors (51) have been shown to activate the TR-L1 promoter in epithelial cells. Whether the same factors are involved in LMP1 regulation in latency II type tumors not of epithelial origin is unclear.
Recent data have indicated that activation of LMP1 is critically dependent on its own expression in latency II cells mediated by the activation of the JNK signaling pathway (13). This study also presents evidence for an LMP1 autoregulatory loop. In disagreement with Goormachtigh et al. (13), however, we show here that the NF-κB pathway is involved in the activation of the LMP1 promoter and not in its inhibition. The NF-κB family members were shown to bind to the LMP1 promoter in vitro and in vivo. Electrophoretic mobility shift assay (EMSA) analysis indicated that the p50-p50 homodimer and the p65 (RelA)-p50 heterodimer bind to an NF-κB site at positions −79 to −89 of the LMP1 promoter. A mutation in this site led to a decrease in LMP1 promoter activity in reporter assays. Overexpression of NF-κB factors in B cells and epithelial cells activated the LMP1 promoter markedly in the absence of EBNA2. Finally, NF-κB expression in the P3HR1 cell line led to increased LMP1 expression. Overall, our results show that the NF-κB factors upregulate LMP1 expression independently of EBNA2.
MATERIALS AND METHODS
Cell lines and cell culture conditions.
DG75 is an EBV-negative Burkitt's lymphoma cell line (3). CBC-Rael (9), P3HR1 (28), and WW1-LCL (15) are EBV-positive B-cell lines. The cells were maintained in RPMI 1640 medium supplemented with 10% fetal calf serum, 100 U penicillin ml−1, and 100 μg streptomycin ml−1 (Sigma-Aldrich). HEK293 is human embryonic kidney cell line of neuroendothelial origin (43), and MCF7 is a human breast cancer cell line (46). These cell lines were maintained in Dulbecco's modified Eagle's medium (Invitrogen) also supplemented with fetal calf serum, penicillin, and streptomycin.
Plasmids, transfections, and reporter assays.
The expression vectors for NF-κB p50 and p65 (7, 14) and the pIL2RCAT reporter plasmid (38) were obtained from the AIDS Research and Reference Reagent program. The coding sequences for the NF-κB p50 and NF-κB p65 were then cloned into the multiple-cloning site of the pcDNA3 plasmid (Invitrogen).
The LMP1 regulatory sequence (LRS) is defined as positions 169019 to 169692 of B95-8 EBV DNA (GenBank accession no. AJ507799), which corresponds to positions −634 to +40 relative to the LMP1 transcription initiation site (+1). The pgLRS −634CAT and pgLRS −106CAT reporter plasmids have been described previously (44). The pgLRS −634 NF-κBmutCAT and pgLRS −106 NF-κBmutCAT reporter plasmids were constructed by inserting double-stranded oligonucleotides encompassing the mutation in the NF-κB site into the MluI-RsaI-opened LRS reporter plasmids.
Transient transfections of B cells were carried out by electroporation using 5 × 106 cells as described previously (44). Cotransfections in DG75 cells were carried out with 10 μg of reporter plasmids and 2 μg of NF-κB expression vectors or the empty vector. In P3HR1 cells, 10 μg of reporter plasmids and 5 μg of NF-κB expression plasmids were used. For the HEK293 and MCF7 cell lines, approximately 2 × 106 cells were transfected in 25-cm2 cell culture bottles in antibiotic-free medium with a FuGENE6 kit according to the manufacturer's instructions. Cotransfections were carried out using 0.5 μg of the NF-κB expression vectors and 3.5 μg of the reporter plasmids. The cells were harvested after 72 h and assayed for chloramphenicol acetyltransferase (CAT) activity as described previously (40). The cell extracts prepared for reporter assay were also used for immunoblot analysis.
For selection of P3HR1 cells transfected with NF-κB factors, the cells were cotransfected with 20 μg of green fluorescent protein (GFP) expression vector. After 24 h, the transfected cells were sorted in a BD FACSAria cell-sorting system based on their GFP fluorescence. The cells were then incubated for an additional 24 or 48 h before harvest.
EMSAs.
Nuclear extracts were prepared as described by Dignam et al. (6). EMSA binding reactions were carried out as described by Sjoblom et al. (44). All oligonucleotides were purchased from Invitrogen. EMSAs were carried out using double-stranded synthetic 32P-labeled oligonucleotides corresponding to the B95-8 and P3HR1 DNA sequences. For the sequences of the probe and the competitors, see Fig. 2. In supershift experiments, 2 μg of each antibody was used. NF-κB p50 (sc-7178 X) and NF-κB p65 (sc-7151 X) were purchased from Santa Cruz Biotechnology, and the normal rabbit immunoglobulin G (IgG) was purchased from Upstate. The samples were separated by electrophoresis in 5% polyacrylamide gels (acrylamide-bisacrylamide, 29:1) in 0.5× Tris-borate-EDTA for 3 h at 300 V. Alternatively, DNA retardation gels (Invitrogen) were used in 0.5× Tris-borate-EDTA for 2 h at 150 V. The bands were visualized by exposure to a phosphorimager screen and scanned with a Typhoon 9200 scanner (Amersham Biosciences).
FIG. 2.

The NF-κB p50-p50 homodimer and p65-p50 heterodimer bind to the LRS NF-κB site. (A) A 32P-labeled double-stranded synthetic oligonucleotide corresponding to the LRS region from position −74 to −94 of the B95-8 sequence was incubated with nuclear extracts from WW1-LCL cells and subjected to EMSA. Competition reactions were carried out as indicated above the gel. The first lane shows the binding pattern obtained with the nuclear extract. The black arrows indicate specific complexes and the broken arrows nonspecific complexes. Nucleotide sequences of the double-stranded oligonucleotides used in the competition experiment are shown below the gel. The NF-κB binding site in LRS and the NF-κB consensus sequence are in uppercase letters, and the mutated nucleotides are in lowercase letters and underlined. Antibody supershift experiments were carried out as indicated above the gel. The supershifted complexes are indicated by arrowheads on the left. (B) A 32P-labeled double-stranded synthetic oligonucleotide corresponding to the LRS region from position −74 to −94 of the P3HR1 sequence was incubated with nuclear extracts from WW1-LCL cells and subjected to EMSA. Supershift analysis was carried out with the antibodies as indicated above the gel. The first lane shows the binding pattern obtained with the nuclear extract. The black arrows indicate specific complexes and the broken arrows nonspecific complexes.
DNA affinity purification.
DNA affinity purification was performed as described by Atanasiu et al. (1) with some modifications. Biotinylated DNA fragments of EBV were obtained by PCR amplification of the LMP1 promoter. The PCR primers amplified a region of the promoter corresponding to positions −202 to −1 relative to transcription initiation site (+1). Control biotinylated DNA fragments were obtained by PCR of the ampicillin-coding region. The eluted proteins were separated on a 4 to 12% Novex bis-Tris gel (Invitrogen) using sodium dodecyl sulfate-polyacrylamide gel electrophoresis. All protein bands were cut, digested with trypsin, and analyzed by liquid chromatography-tandem mass spectrometry using linear trap quadropole-Fourier transform ion cyclotron resonance (FTICR) mass spectrometer. Immunoblot analysis was also carried out on the eluted proteins.
ChIP assay.
Chromatin immunoprecipitation (ChIP) was carried out according to the protocol provided by Upstate Biotechnology, Inc., with minor modifications. Two EBV-positive cell lines, WW1-LCL and CBC-Rael, were used for ChIP. Briefly, the ChIP extract was sonicated to between 200- and 350-bp DNA fragments on a Diagenode Bioruptor according to manufacturer's protocol (Diagenode, Liège, Belgium). The level of immunoprecipitated DNA was determined by quantitative PCR using primer pairs that amplified the region encompassing the LRS NF-κB site of B95-8 and the Rael origin (B95-8 primers, ACGTCAGAGTAACGCGTGTTTC and GCAGACCCCGCAAATCC; Rael primers, AGGCAGTACGGGTGCAGATT and TTTTTACGCTTACATGCACACA). A primer pair covering another region of EBV (dyad symmetry) (GGGAGATATCGCTGTTCCTTAGG and TGTTACCCAACGGGAAGCAT) was used as a negative control.
Quantitative RT-PCR.
Total RNA was prepared from 1 × 105 cells, using the Qiagen RNeasy Plus minikit as instructed by the manufacturer. Quantitative reverse transcription-PCR (RT-PCR) of LMP1 was carried out as described by Bell et al. (2) with a few modifications. The SuperScript III RTS first-strand cDNA synthesis kit (Invitrogen) was used according to the manufacturer's protocol. An EBV-specific cDNA primer mix (2 μM) (2) was added to each reaction mixture. For quantitative PCR of LMP1, the fluorescent power Sybr green kit was used according to the manufacturer's protocol (Applied Biosystems) with a primer pair amplifying LMP1 of B95-8 origin (2) in combination with melting curve analyses to ensure the specificity of the PCR primers. The amount of each transcript was normalized against the GAPDH (glyceraldehyde-3-phosphate dehydrogenase) gene transcript, quantified with the commercially available predeveloped assay reagent from Applied Biosystems in an ABI HT7900 instrument (Applied Biosystems).
Immunoblot analysis.
Immunoblot analysis was carried out according to standard protocols. The membranes were stained with 0.1% Ponceau S (Sigma) in 5% acetic acid to confirm equal loading and transfer of proteins. The membranes were probed with anti-NF-κB p50, anti-NF-κB p65, anti-IκBα (Cell Signaling), anti-GAPDH (Santa Cruz), and anti-LMP1 (Dako) antibodies and horseradish peroxidase-conjugated secondary antibodies (Cell Signaling). The immunoreactive protein bands were visualized with chemiluminescence reagents (Pierce) and detected using a ChemiDoc instrument (Bio-Rad). The intensity of LMP1 protein bands was measured using the Quantity One software (Bio-Rad).
RESULTS
NF-κB factors bind to an NF-κB site at the proximal LMP1 promoter in vitro and in vivo.
A TRANSFAC database search of the LMP1 ED-L1 promoter (LRS) was previously used to identify candidate regulatory elements in the promoter (Fig. 1A) (22). In the present study, a DNA affinity purification method (1) was utilized to identify proteins binding to the LMP1 promoter. The LMP1 promoter corresponding to positions −1 to −202 relative to the transcription initiation site (+1) was used as bait to purify proteins from WW1-LCL nuclear extracts. The purified proteins were then identified by mass spectrometry. The p50 and p65 proteins of the NF-κB transcription family were pulled down with the LMP1 promoter sequence-containing DNA. To verify the data, similar DNA affinity purifications were also carried out with a DNA sequence carrying a mutation in the TRANSFAC-identified NF-κB site (at positions −79 to −89) as well as with an unrelated sequence of the same size. The purified proteins were analyzed by immunoblotting using antibodies against the p50 and p65 NF-κB factors. Both proteins were eluted from the wild-type LRS bait, and much lower levels of the factors were eluted from LRS containing the mutated NF-κB site or the unrelated sequence (labeled nonspecific in Fig. 1B).
FIG. 1.

An NF-κB element in the LMP1 promoter binds NF-κB factors p50 and p65. (A) Illustration of the promoter-proximal part of the LRS. The double-stranded DNA sequence is of B95-8 EBV DNA origin. The numbers indicate the nucleotide positions relative to the transcription initiation site (+1). Potential binding sites for different transcription factors identified in a database search (TRANSFAC) as well as previously identified elements are underlined. (B) Immunoblots using NF-κB p50 and p65 antibodies, indicating binding to biotinylated DNA fragments from DNA affinity purification with WW1-LCL nuclear extract. LRS is a biotinylated DNA fragment corresponding to positions −2 to −202 (relative to the transcription initiation site) of the LMP1 promoter, and NF-κB MUT is the same DNA fragment with a mutation in the NF-κB site. Nonspecific indicates an unrelated biotinylated DNA fragment encompassing 200 bp of the ampicillin-coding sequence. (C) ChIP of NF-κB p50 and p65 at the LMP1 promoter and the dyad symmetry (DS) region of EBV in two EBV-positive cell lines. Results of real-time PCR analysis of ChIP assays with antibodies specific to p65, p50, or control IgG are shown. The results are expressed as fold IgG where the IgG control was set to 1. Samples were analyzed in triplicate, and the results are the averages and standard errors of the means (SEM) from three independent experiments.
The status of NF-κB p50 and p65 binding to LRS in vivo was investigated by the ChIP assay using antibodies against p50 and p65 in two latency III cell lines (WW1-LCL and CBC-Rael). The anti-p50 antibody enriched LRS DNA in the precipitates approximately fivefold above IgG in both cell lines used. The p65 antibody failed to coprecipitate the LMP1 promoter. The specificity of the antibody pull-down was verified by checking the enrichment of another EBV region (dyad symmetry), which is not reported to bind these factors. Our results suggest that the p50 protein binds to the LMP1 promoter both in vitro and in vivo, while p65 bound only in vitro under our experimental conditions.
NF-κB binds as a p50-p50 homodimer and as a p50-p65 heterodimer to the LRS NF-κB site.
NF-κB binding to the NF-κB site at positions −89 to −79 of the promoter was further investigated by EMSA. A labeled probe encompassing the LRS NF-κB site [LRS (−94/−74)] was used in EMSA with the WW1-LCL nuclear extract. Four DNA-protein complexes were observed after the binding reaction (Fig. 2A, lane 1). Competition with an excess of unlabeled probe competed away the slower-migrating complexes, indicating that they were specific (Fig. 2A, lane 2). The remaining two complexes were not competed out and were assumed to be nonspecific. Competition with oligonucleotides containing mutations in the NF-κB site failed to eliminate or reduce the specific complexes (Fig. 2A, lanes 3 and 4). Competition with an NF-κB consensus site in an LRS context or an oligonucleotide with an alternative flanking sequence competed away both of the specific complexes (Fig. 2A, lanes 5 and 6). Thus, the specific DNA-protein complexes obtained with this probe contained NF-κB factors that bound to the NF-κB site in LRS. Supershift experiments with an anti-p50 antibody shifted both specific bands, indicating the presence of p50 in both complexes (Fig. 2A, lane 7). The anti-p65 antibody shifted the top specific complex only (Fig. 2A, lane 8), indicating the presence of p65 in this complex. The rabbit IgG did not give rise to any supershifted complex, which supported the specificity of the supershift reactions (Fig. 2A, lane 9). Similar results were obtained with nuclear extracts from the CBC-Rael cell line (data not shown). Supershift analysis with antibodies against other members of the NF-κB family did not result in any shifted complexes (data not shown). Hence, the results suggest that the top slower-moving complex represents a p65-p50 heterodimer and the lower complex a p50-p50 homodimer. The weak intensity of the p65-p50 heterodimer may explain why an enrichment of the LRS precipitate was not observed in the ChIP experiments with the p65 antibody. The weaker intensity might reflect the presence of only low levels of p65 or active p65 or a lower affinity of this complex for the LRS probe.
The LRS NF-κB site at positions −79 to −89 in the P3HR1 virus contains a 1-base-pair mutation relative to the prototype B95-8 sequence (Fig. 2A, legend). EMSA with a P3HR1-NF-κB site probe using supershift analysis indicated the same pattern of factor binding to the site (Fig. 2B). The overall binding affinity of these factors to the P3HR1 probe was less than that observed for the B95-8 probe as determined by specific competition experiments (data not shown).
The conserved NF-κB site in LRS is involved in promoter activation.
To determine the role of the NF-κB site in the regulation of the LMP1 promoter, mutation analysis of LMP1-carrying reporter plasmids was performed. An LRS CAT reporter encompassing the ED-L1 sequence or a shorter sequence including the NF-κB site was used with and without a mutation in the NF-κB site (pgLRS −634CAT, pgLRS −634 NF-κBmutCAT, pgLRS −106CAT, and pgLRS −106 NF-κBmutCAT). This mutation corresponds to the second mutation used in EMSA analysis in the previous competition experiment (Fig. 2A, lane 4) and was shown to abolish NF-κB factor binding to the promoter. Since NF-κB factors are constitutively active in LMP1-expressing EBV-positive cells, the latency III cell line WW1-LCL was used for transfections. The NF-κB mutated reporter in the −634 context showed a 60% decrease in activity relative to the wild-type LMP1 promoter (Fig. 3A). Therefore, the NF-κB site and the factors binding to it have a role in the regulation of the LMP1 promoter. Interestingly, the mutation in the NF-κB site of LMP1 reporter plasmids including only a shorter part of the promoter (LRS −106) did not display a large difference in promoter activity relative to the wild-type promoter. Consequently, it appears that the NF-κB site is involved in the regulation of the promoter but requires the full promoter context (up to position −634) for the activation of the promoter.
FIG. 3.

The NF-κB site mutation leads to reduced LMP1 promoter activity. (A) LRS CAT reporter plasmids encompassing the LMP1 promoter (positions +40 to −634 or +40 to −106), with and without a mutation in the NF-κB site, were transfected in the WW1-LCL cell line. The cells were harvested after 72 h and assayed for CAT activity. The relative CAT activity was obtained by measuring the percentage of chloramphenicol acetylation in the linear range of the CAT assay. The values are the means and SEM from four independent transfections. (B) Alignment of the NF-κB sequence in the LMP1 promoter in different EBV strains. The NF-κB binding site in LRS and the NF-κB consensus sequence are in uppercase letters, and the mutated nucleotides are in lowercase letters and underlined.
A sequence alignment of the LMP1 promoter sequences from a number of EBV isolates and cell lines representing type 1, type 2, and epithelial isolates indicated that this NF-κB site is highly conserved (Fig. 3B). In fact the only virus with a sequence variation in this site reported so far is the P3HR1 virus. The mutation in this site has most probably occurred in cell culture, as it is not present in the P3HR1 parental cell line, Jijoy. The conservation of the NF-κB site in the LMP1 promoter supports its importance in expression regulation.
Expression of p50 and p65 activates the LMP1 promoter in B cells and epithelial cells.
Cotransfection of p50 and p65 expression plasmids with LRS reporter plasmids was utilized to monitor the effect of active p50 and p65 on the LMP1 promoter in several cell lines. NF-κB activation usually occurs as a result of extracellular stimuli. In the absence of an inducer, NF-κB factors are sequestered in the cytoplasm by their interactions with the inhibitory IκB proteins (12). Expression of p50 and p65 is used here to obtain an excess of the dimers relative to the cellular IκB inhibitors that would result in an increased fraction of free and active dimers. Notably, the activities of the p50-p50 homodimer and the p50-p65 heterodimer are regulated by different members of the IκB family (12, 18) and by the levels of p50 and p65 available to form dimers with. Thus, the level of activity of the exogenously expressed NF-κB factors is dependent on the context of the cellular inhibitory IκB as well as the cellular NF-κB factors present in each cell line. As a control for NF-κB activity, an NF-κB-responsive reporter plasmid (pIL2RCAT) that contains the NF-κB-responsive site of the interleukin-2 receptor (IL-R) promoter (38) was included. The expression of p50 and p65 in transfected cells was confirmed by immunoblot analysis using specific antibodies (Fig. 4). Overexpression of p50 in the EBV-negative DG75 cells led to a large increase in LMP1 promoter activity relative to its basal level (approximately 50-fold) (Fig. 4A). The LRS reporter with the NF-κB site mutation was also activated in the presence of p50 expression but three times less than the wild-type promoter, indicating that most of the p50-induced activation is through this NF-κB site. The IL-2R promoter was also markedly activated by p50 expression. Overexpression of p65 also activated both the LMP1 promoter (approximately 20-fold) and the IL-2R promoter in reporter plasmids. Activation of the LMP1 promoter by p65 occurred strictly through the NF-κB site, as the pgLRS −634NF-κBmut reporter was not activated by p65. Since p50 and p65 bind the LMP1 promoter as a heterodimer, their expression plasmids were cotransfected together with the reporter plasmids. The IL-2R reporter was activated to about the same level as it was by p50 alone. The LMP1 promoter was also activated by the combination of p50 and p65, but only to a one-third of the activation seen with p50 expression alone (approximately a 20-fold increase relative to basal LRS −634 activity). An almost identical pattern of reporter activity was observed in the EBV-positive P3HR1 cell line (Fig. 4B). P3HR1 contains a deletion in the EBNA2 gene and as such exhibits only background LMP1 expression and is referred to as a latency II-III cell line.
FIG. 4.

NF-κB p50 and p65 expression induces LRS reporter plasmid activation in different cell lines. (A) The p50 and p65 expression vectors, compensated with empty vector (pcDNA3.1), were transfected separately or together into DG75 cells, together with the pgLRS −634CAT, pgLRS-634 NF-κBmutCAT, or pIL2RCAT reporter plasmid as shown. The relative CAT activity was obtained by measuring the percentage of chloramphenicol acetylation in the linear range of the CAT assay. The values shown are the means and standard deviations from three independent transfections. The expression of p50 and p65 was detected by immunoblotting using specific antibodies. The exogenously expressed p50 contains an additional five amino acids from the p105 precursor and therefore migrates slightly above the endogenous p50 on the gel. (B) Transfection and immunoblot analysis were carried out in P3HR1 cells as described for panel A. (C) Transfection and immunoblot analysis were carried out in MCF7 cells as described for panel A. (D) Transfection and immunoblot analysis were carried out in HEK293 cells as described for panel A. The values are the means and SEM from four to six independent transfections in each cell line.
The facts that LMP1 expression in epithelial cells is EBNA2 independent and that the NF-κB p50 and p65 proteins activated the LMP1 promoter in the absence of EBNA2 raised questions about the effect of these factors on LMP1 activation in epithelial cells. The epithelial cell line MCF7 was cotransfected with LMP1 reporters and NF-κB factors in a manner similar to that for DG75 and P3HR1 transfections (Fig. 4C). In the MCF7 cell line, the basal LRS activity was higher than the background level. Interestingly, in the absence of exogenous expression of NF-κB factors, the LRS −634NF-κBmut reporter exhibited levels three times higher than the activity of the wild-type reporter. It is possible that the mutated vector bound factors that mediated activation in this cell line. The expression of p50, alone or together with p65, activated the wild-type LRS sixfold and threefold, respectively. Exogenous expression of p65 alone failed to activate the LRS or the control pIL2R reporter plasmid, indicating that p65 overexpression alone did not lead to increased p50-p65 activity in this cell line. Expression of p50 alone activated the NF-κB mutated LRS reporter twofold relative to the basal NF-κB mutated reporter, which was three times less than the wild-type induction. Overall, both the p50-p50 homodimer and the p50-p65 heterodimer activated the LMP1 promoter through the NF-κB site in the MCF7 cell line.
The neuroendothelial HEK293 cell line contains low levels of NF-κB factors, providing an environment with a low background level of NF-κB activity. For this reason, the induction of the LMP1 reporter by NF-κB factors was also investigated in this cell line (Fig. 4D). As with the epithelial MCF7 cell line, in the absence of NF-κB factors, the LRS reporter with a mutated NF-κB site showed slightly higher activity than the wild-type reporter. Overexpression of p50 activated the promoter 6-fold, while p65 gave rise to a 10-fold increase in promoter activity relative to the basal LRS activity (Fig. 4C). The combination of p50 and p65 expression had an additive effect, resulting in a 20-fold increase in activity. The effect of p50 and p65 on the LMP1 promoter was mediated exclusively by the LRS NF-κB site, as no significance increase in the NF-κB mutated reporter was observed. The IL-2R reporter showed a similar pattern of activation, while the overall increase in activity was less than that for the LMP1 promoter in all cases. Overall, NF-κB factors seem to be potent activators of the LMP1 promoter in HEK293 cells.
Taken together, these results show that the LMP1 reporter is activated by both p50-p50 and p50-p65 dimers, irrespective of the host cell line. The levels of induction, however, and the relative levels of activation of the two dimers vary according the cell line used rather than cell type. Since it is known that LMP1 itself activates the p50-p50 and p50-p65 dimers, our results suggest that the LMP1 promoter is most likely activated by these NF-κB dimers in LMP1-expressing cells.
Expression of p50 and p65 induces LMP1 expression.
To study the effect of p50 and p65 on the endogenous LMP1 expression, exogenous expression of these factors in P3HR1 was carried out. Due to low transfection efficiency in P3HR1 cells, the cells were also cotransfected with a GFP expression vector and the transfected cells were selected before analyses, based on their GFP fluorescence. The effect of p50 and p65 overexpression on LMP1 RNA and protein levels was then investigated by quantitative RT-PCR (Fig. 5A) and immunoblotting (Fig. 5B), respectively. The LMP1 RNA level (normalized against GAPDH) increased by 1.5-fold in the presence of p50 expression and only slightly in the presence of p65 expression alone (Fig. 5A). Expression of both p50 and p65, however, gave rise to a 2.5-fold increase in LMP1 RNA (Fig. 5A). The increase in LMP1 expression was also observed at the protein level by immunoblot analysis of the LMP1 protein (Fig. 5B). Immunoblotting against GAPDH confirmed equal protein loading. Immunoblotting against IκBα, a known target of the p50-p65 dimer, confirmed the activity of these factors in transfected cells. The relative LMP1 protein levels were estimated from the measured intensity of the bands. Generally, the upregulation at the protein level appeared to be more than that observed at the RNA level. This is probably due to the longer half-life of the LMP1 protein relative to its RNA. The increase in LMP1 expression observed here was not as strong as that seen in the reporter system. This may be explained by our observation that the binding affinity of NF-κB to the LMP1 promoter in the B95.8 probe was stronger than that in the P3HR1 probe in EMSA. Our results here support the involvement of NF-κB p50 and p65 proteins in the positive regulation of LMP1 expression. The data also suggest that p50-p65 is the more potent activating dimer of the endogenous LMP1 promoter in this case. It is not unusual that endogenous promoters behave differently than their corresponding reporter plasmids due to differences in their location in the genome and epigenetic status. The apparent differences in sensitivity to the different NF-κB factors in the LMP1 promoter in P3HR1 cells may also be due to the 1-base-pair mutation in the P3HR1 NF-κB site relative to the B95.8 sequence that the reporter plasmids contain.
FIG. 5.
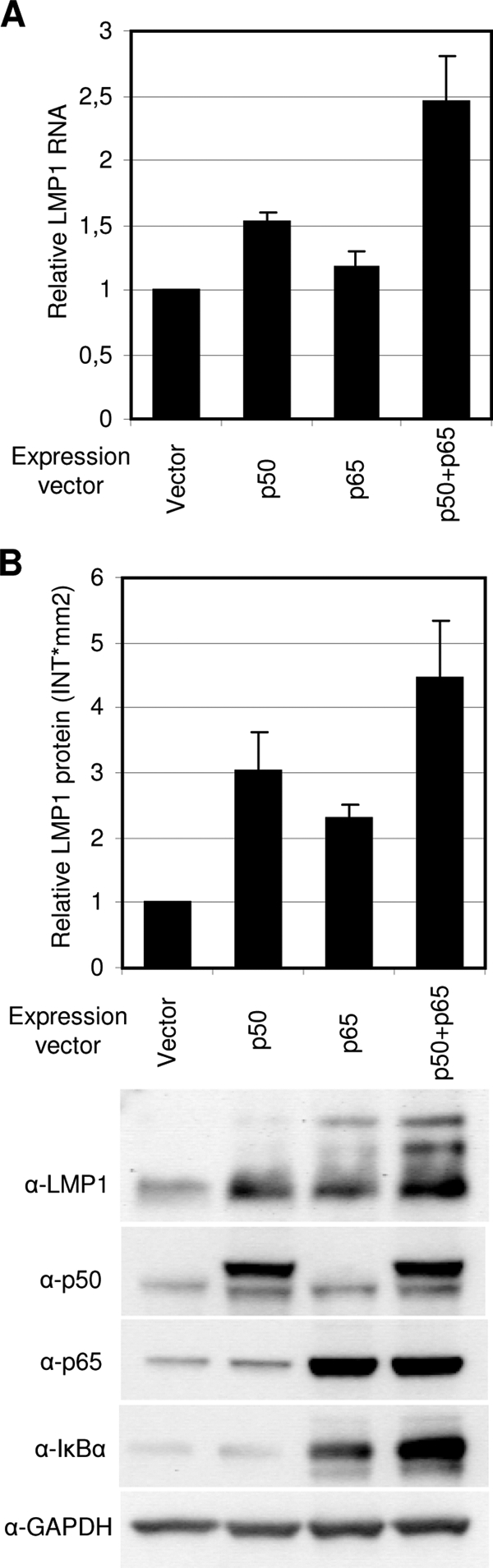
NF-κB expression induces endogenous LMP1 expression in P3HR1 cells. P3HR1 cells were transfected with p50, p65, p50, and p65 or control vector together with a GFP expression vector. Transfected cells were selected based on GFP fluorescence in a flow cytometer after 24 h and then incubated for an additional 24 h or 48 h before harvest. (A) LMP1 RNA levels in transfected cells were analyzed using quantitative RT-PCR. The LMP1 RNA level was normalized against GAPDH, and the change in LMP1 RNA in response to NF-κB factors was measured relative to the vector (set to 1). The values are the means and SEM from five independent transfections. (B) Immunoblot analysis was used to detect LMP1, p65, p50, IκBα, and GAPDH protein levels in transfected cells. The intensity of LMP1 protein bands was measured using the Quantity One software, and the change in LMP1 RNA in response to NF-κB factors was measured relative to the vector (set to 1).
DISCUSSION
The regulation of LMP1 gene expression appears to be governed by the cell type, cellular context of transcription factors, viral pattern of gene expression, and extracellular stimuli. Furthermore, several recent studies have reported different LMP1 autoregulatory mechanisms (13, 29, 30, 33). We have previously reported several regulatory elements at the LMP1 ED-L1 promoter that are involved in its regulation together with or independently of EBNA2 (54). In the present study, we demonstrated the binding of NF-κB transcription factors p50-p65 and p50-p50 to the LMP1 promoter. This occurred through a conserved NF-κB site in LRS at positions −79 to −89 relative to the transcription initiation site. Activation of the NF-κB factors led to a large increase in promoter activity and an increase in endogenous LMP1 expression.
Our data are of particular interest since LMP1 is a well-established activator of the NF-κB pathway itself (45). Activation of this pathway is required for LMP1 transformation of rodent fibroblasts (17). Therefore, our data suggest the presence of an autoregulatory loop, where LMP1 activation of the NF-κB signaling pathway leads to upregulation of its own expression. Further, the study presents an alternative mechanism for the activation of the LMP1 ED-L1 promoter in the absence of EBNA2 expression.
While the induction of the LMP1 promoter in reporter assays by the p50-p50 homodimer and p50-p65 heterodimer varied in different cell lines, both complexes seem to play a role in LMP1 expression. Even though the p50 homodimer is generally thought of as a repressor of transcription activity (39), several investigations indicate a positive regulatory role for this complex in promoter activation via interaction with the Bcl-3 coactivator (12, 16, 47). The reported activation of a p50 homodimer/Bcl-3 complex in NPCs and its function in transcription activation (49, 50), together with our results, suggest the possibility that LMP1 may be regulated by this complex in NPCs.
The NF-κB transcription factors are induced by many stimuli and regulate genes involved in growth, differentiation, and immune response (35). Aberrant regulation of the NF-κB signaling pathway results in an array of different human diseases, including cancer (8). Not surprisingly, this pathway has been a target of many viral pathogens, including EBV (20). Furthermore, many viruses that induce NF-κB activity also contain NF-κB binding sites in their own promoters (35). A positive autoregulatory circuit between LMP1 and NF-κB is therefore a conceivable mechanism to ensure sustained viral gene expression. A positive autoregulatory loop in LMP1 expression may seem disadvantageous since high levels of LMP1 are known to induce cytostasis and inhibit the activity of viral and cellular promoters (11, 24, 32, 42). However, several mechanisms of LMP1 downregulation that ensure appropriate LMP1 expression levels have also been reported (29, 31, 34).
Several lines of evidence suggest that NF-κB factors may be involved in LMP1 regulation in both latency types II and III and in both B cells and epithelial cells. First, the heterodimer p50-p65 and the homodimer p50-p50 are activated by LMP1 in both B cells and epithelial cells (19, 36). Second, the LMP1 promoter was activated by the p50-p50 and p50-p65 factors in our reporter assays in both B cells and epithelial cells. Third, our results showed that the activation of LMP1 reporters by NF-κB factors was independent of EBNA2. Finally, the ED-L1 promoter is used in both latency types, even if it is not the only promoter used in latency II (41). It is noteworthy that overexpression of the NF-κB factors in continuously growing lymphoblastoid cell lines (LCLs) did not significantly affect the level of LMP1 promoter activity in reporter assays (data not shown). This is most likely due to the presence of active NF-κB factors even in the absence of exogenous NF-κB expression in these cells.
While our data are consistent with the notion of LMP1 autoregulation proposed by Goormachtigh et al. (13), our results are in disagreement regarding the role of the NF-κB pathway in LMP1 regulation. Notably, the NF-κB p50 homodimer was not investigated by this study. We did not observe an inhibitory effect of NF-κB on LMP1 in our B cells or epithelial cells corresponding to that seen by Goormachtigh and colleagues.
To date, it seems that EBNA2 in latency III cells (25) and LMP1 itself in latency II cells (13) are the main factors for LMP1 regulation. Considering the fact that both mechanisms require additional cellular and viral factors, it is interesting that none of the identified candidates so far appear to be critical for the promoter activation. NF-κB is a particularly good candidate for LMP1 upregulation, as it is activated by EBV infection prior to LMP1 expression (47) and can be involved in LMP1 gene regulation even at the early stages of infection in the absence of EBNA2. It is likely that the NF-κB factors require the cooperation of other factors at the promoter for transactivation, as indicated by our finding that reporter plasmids containing the NF-κB site but lacking the upstream promoter sequences were not activated. In fact, it has been shown that promoter activation by NF-κB factors is regulated not only by interactions with coactivators but also by protein-protein interactions with other factors binding in the promoter region (21, 27, 48). Since EBNA2 is a very efficient activator of LMP1 gene expression in latency III cells in culture, the relative contribution of NF-κB factors was not determined in these cells. In our LCLs, NF-κB inhibition using small interfering RNA did not affect LMP1 expression levels. However, our results suggest that these factors may play an important role in LMP1 expression in the absence of EBNA2, as in latency II cells, or at different stages of the EBV life cycle in its host where EBNA2 is not expressed.
Acknowledgments
We thank Katarina Junevik (Sahlgrenska University Hospital, Gothenburg) for her help with fluorescence-activated cell sorting. We also thank P. Lieberman and his group members and C. Chau (Wistar Institute, PA) for their valuable contributions with regard to the DNA affinity purification and ChIP methods. We acknowledge A. Sjöblom-Hallen for her contributions to the early stages of this project. Gel band cut-out, in-gel trypsin digestion, and FTICR-liquid chromatography-tandem mass spectrometry peptide sequencing for protein identification were carried out by the Proteomics Core Facility at the University of Gothenburg, Gothenburg. The following reagents were obtained through the AIDS Research and Reference Reagent Program, Division of AIDS, NIAID, HIH: pBluescript-NF-κB 1 (p50) and pRSV-RelA (p65) from Gary Nabel and Niel Perkins and pIL2R from Michael Leonardo.
This study was supported by grants from the Swedish Medical Research Council (project 5667), the Swedish Cancer Society, the Swedish Society for Medical Research, Assar Gabrielsson's Foundation, IngaBritt and Arne Lundberg's Foundation, and the Sahlgrenska University Hospital Foundation.
Footnotes
Published ahead of print on 19 November 2008.
REFERENCES
- 1.Atanasiu, C., L. Lezina, and P. M. Lieberman. 2005. DNA affinity purification of Epstein-Barr virus OriP-binding proteins. Methods Mol. Biol. 292267-276. [DOI] [PubMed] [Google Scholar]
- 2.Bell, A. I., K. Groves, G. L. Kelly, D. Croom-Carter, E. Hui, A. T. Chan, and A. B. Rickinson. 2006. Analysis of Epstein-Barr virus latent gene expression in endemic Burkitt's lymphoma and nasopharyngeal carcinoma tumour cells by using quantitative real-time PCR assays. J. Gen. Virol. 872885-2890. [DOI] [PubMed] [Google Scholar]
- 3.Ben-Bassat, H., N. Goldblum, S. Mitrani, T. Goldblum, J. M. Yoffey, M. M. Cohen, Z. Bentwich, B. Ramot, E. Klein, and G. Klein. 1977. Establishment in continuous culture of a new type of lymphocyte from a “Burkitt-like” malignant lymphoma (line D.G.-75). Int. J. Cancer 1927-33. [DOI] [PubMed] [Google Scholar]
- 4.Cahir-McFarland, E., and E. Kieff. 2005. Epstein-Barr virus latent infection membrane protein 1, p. 553-570. In E. S. Robertson (ed.), Epstein-Barr virus. Caister Academic Press, Cambridge, England.
- 5.Chen, H., J. M. Lee, Y. Zong, M. Borowitz, M. H. Ng, R. F. Ambinder, and S. D. Hayward. 2001. Linkage between STAT regulation and Epstein-Barr virus gene expression in tumors. J. Virol. 752929-2937. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Dignam, J. D., R. M. Lebovitz, and R. G. Roeder. 1983. Accurate transcription initiation by RNA polymerase II in a soluble extract from isolated mammalian nuclei. Nucleic Acids Res. 111475-1489. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Duckett, C. S., N. D. Perkins, T. F. Kowalik, R. M. Schmid, E. S. Huang, A. S. Baldwin, Jr., and G. J. Nabel. 1993. Dimerization of NF-κB2 with RelA(p65) regulates DNA binding, transcriptional activation, and inhibition by an I κB-α (MAD-3). Mol. Cell. Biol. 131315-1322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Dutta, J., Y. Fan, N. Gupta, G. Fan, and C. Gelinas. 2006. Current insights into the regulation of programmed cell death by NF-kappaB. Oncogene 256800-6816. [DOI] [PubMed] [Google Scholar]
- 9.Ernberg, I., K. Falk, J. Minarovits, P. Busson, T. Tursz, M. Masucci, and G. Klein. 1989. The role of methylation in the phenotype-dependent modulation of Epstein-Barr nuclear antigen 2 and latent membrane protein genes in cells latently infected with Epstein-Barr virus. J. Gen. Virol. 702989-3002. (Erratum, 71:488, 1990.) [DOI] [PubMed] [Google Scholar]
- 10.Fahraeus, R., L. Rymo, J. S. Rhim, and G. Klein. 1990. Morphological transformation of human keratinocytes expressing the LMP gene of Epstein-Barr virus. Nature 345447-449. [DOI] [PubMed] [Google Scholar]
- 11.Floettmann, J. E., K. Ward, A. B. Rickinson, and M. Rowe. 1996. Cytostatic effect of Epstein-Barr virus latent membrane protein-1 analyzed using tetracycline-regulated expression in B cell lines. Virology 22329-40. [DOI] [PubMed] [Google Scholar]
- 12.Gilmore, T. D. 2006. Introduction to NF-kappaB: players, pathways, perspectives. Oncogene 256680-6684. [DOI] [PubMed] [Google Scholar]
- 13.Goormachtigh, G., T. S. Ouk, A. Mougel, D. Tranchand-Bunel, E. Masy, C. Le Clorennec, J. Feuillard, G. W. Bornkamm, C. Auriault, E. Manet, V. Fafeur, E. Adriaenssens, and J. Coll. 2006. Autoactivation of the Epstein-Barr virus oncogenic protein LMP1 during type II latency through opposite roles of the NF-κB and JNK signaling pathways. J. Virol. 807382-7393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Gorman, C., R. Padmanabhan, and B. H. Howard. 1983. High efficiency DNA-mediated transformation of primate cells. Science 221551-553. [DOI] [PubMed] [Google Scholar]
- 15.Gregory, C. D., R. J. Murray, C. F. Edwards, and A. B. Rickinson. 1988. Downregulation of cell adhesion molecules LFA-3 and ICAM-1 in Epstein-Barr virus-positive Burkitt's lymphoma underlies tumor cell escape from virus-specific T cell surveillance. J. Exp. Med. 1671811-1824. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Grimm, T., S. Schneider, E. Naschberger, J. Huber, E. Guenzi, A. Kieser, P. Reitmeir, T. F. Schulz, C. A. Morris, and M. Sturzl. 2005. EBV latent membrane protein-1 protects B cells from apoptosis by inhibition of BAX. Blood 1053263-3269. [DOI] [PubMed] [Google Scholar]
- 17.He, Z., B. Xin, X. Yang, C. Chan, and L. Cao. 2000. Nuclear factor-kappaB activation is involved in LMP1-mediated transformation and tumorigenesis of rat-1 fibroblasts. Cancer Res. 601845-1848. [PubMed] [Google Scholar]
- 18.Heissmeyer, V., D. Krappmann, F. G. Wulczyn, and C. Scheidereit. 1999. NF-kappaB p105 is a target of IkappaB kinases and controls signal induction of Bcl-3-p50 complexes. EMBO J. 184766-4778. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Herrero, J. A., P. Mathew, and C. V. Paya. 1995. LMP-1 activates NF-κB by targeting the inhibitory molecule I kBα. J. Virol. 692168-2174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Hiscott, J., T. L. Nguyen, M. Arguello, P. Nakhaei, and S. Paz. 2006. Manipulation of the nuclear factor-kappaB pathway and the innate immune response by viruses. Oncogene 256844-6867. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Hoffmann, A., G. Natoli, and G. Ghosh. 2006. Transcriptional regulation via the NF-kappaB signaling module. Oncogene 256706-6716. [DOI] [PubMed] [Google Scholar]
- 22.Jansson, A., P. Johansson, W. Yang, L. Palmqvist, A. Sjoblom-Hallen, and L. Rymo. 2007. Role of a consensus AP-2 regulatory sequence within the Epstein-Barr Virus LMP1 promoter in EBNA2 mediated transactivation. Virus Genes 35203-214. [DOI] [PubMed] [Google Scholar]
- 23.Johannsen, E., E. Koh, G. Mosialos, X. Tong, E. Kieff, and S. R. Grossman. 1995. Epstein-Barr virus nuclear protein 2 transactivation of the latent membrane protein 1 promoter is mediated by J κ and PU. 1. J. Virol. 69253-262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Kaykas, A., and B. Sugden. 2000. The amino-terminus and membrane-spanning domains of LMP-1 inhibit cell proliferation. Oncogene 191400-1410. [DOI] [PubMed] [Google Scholar]
- 25.Kempkes, B., M. Pawlita, U. Zimber-Strobl, G. Eissner, G. Laux, and G. W. Bornkamm. 1995. Epstein-Barr virus nuclear antigen 2-estrogen receptor fusion proteins transactivate viral and cellular genes and interact with RBP-J kappa in a conditional fashion. Virology 214675-679. [DOI] [PubMed] [Google Scholar]
- 26.Kieff, E., and A. B. Rickinson. 2001. Epstein-Barr virus and its replication, p. 2511-2574. In D. M. Knipe and P. M. Howley (ed.), Fields virology, 4th ed. Lippincott-Raven Publishers, Philadelphia, PA.
- 27.Kim, T. K., and T. Maniatis. 1997. The mechanism of transcriptional synergy of an in vitro assembled interferon-beta enhanceosome. Mol. Cell 1119-129. [DOI] [PubMed] [Google Scholar]
- 28.Klein, G., B. Giovanella, A. Westman, J. S. Stehlin, and D. Mumford. 1975. An EBV-genome-negative cell line established from an American Burkitt lymphoma; receptor characteristics. EBV infectibility and permanent conversion into EBV-positive sublines by in vitro infection. Intervirology 5319-334. [DOI] [PubMed] [Google Scholar]
- 29.Lee, D. Y., and B. Sugden. 2008. The latent membrane protein 1 oncogene modifies B-cell physiology by regulating autophagy. Oncogene 272833-2842. [DOI] [PubMed] [Google Scholar]
- 30.Lee, D. Y., and B. Sugden. 2008. The LMP1 oncogene of EBV activates PERK and the unfolded protein response to drive its own synthesis. Blood 1112280-2289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Lo, A. K., K. F. To, K. W. Lo, R. W. Lung, J. W. Hui, G. Liao, and S. D. Hayward. 2007. Modulation of LMP1 protein expression by EBV-encoded microRNAs. Proc. Natl. Acad. Sci. USA 10416164-16169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Narbonnet, S., and B. Mariame. 2006. The Epstein-Barr virus oncoprotein LMP1 inhibits the activity of viral or cellular promoters without inducing cytostasis. Virology 350381-393. [DOI] [PubMed] [Google Scholar]
- 33.Ning, S., A. M. Hahn, L. E. Huye, and J. S. Pagano. 2003. Interferon regulatory factor 7 regulates expression of Epstein-Barr virus latent membrane protein 1: a regulatory circuit. J. Virol. 779359-9368. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Ning, S., L. E. Huye, and J. S. Pagano. 2005. Interferon regulatory factor 5 represses expression of the Epstein-Barr virus oncoprotein LMP1: braking of the IRF7/LMP1 regulatory circuit. J. Virol. 7911671-11676. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Pahl, H. L. 1999. Activators and target genes of Rel/NF-kappaB transcription factors. Oncogene 186853-6866. [DOI] [PubMed] [Google Scholar]
- 36.Paine, E., R. I. Scheinman, A. S. Baldwin, Jr., and N. Raab-Traub. 1995. Expression of LMP1 in epithelial cells leads to the activation of a select subset of NF-κB/Rel family proteins. J. Virol. 694572-4576. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Pattle, S. B., and P. J. Farrell. 2006. The role of Epstein-Barr virus in cancer. Expert Opin. Biol. Ther. 61193-1205. [DOI] [PubMed] [Google Scholar]
- 38.Pierce, J. W., M. Lenardo, and D. Baltimore. 1988. Oligonucleotide that binds nuclear factor NF-kappa B acts as a lymphoid-specific and inducible enhancer element. Proc. Natl. Acad. Sci. USA 851482-1486. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Plaksin, D., P. A. Baeuerle, and L. Eisenbach. 1993. KBF1 (p50 NF-kappa B homodimer) acts as a repressor of H-2Kb gene expression in metastatic tumor cells. J. Exp. Med. 1771651-1662. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Ricksten, A., A. Olsson, T. Andersson, and L. Rymo. 1988. The 5′ flanking region of the gene for the Epstein-Barr virus-encoded nuclear antigen 2 contains a cell type specific cis-acting regulatory element that activates transcription in transfected B-cells. Nucleic Acids Res. 168391-8410. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Sadler, R. H., and N. Raab-Traub. 1995. The Epstein-Barr virus 3.5-kilobase latent membrane protein 1 mRNA initiates from a TATA-less promoter within the first terminal repeat. J. Virol. 694577-4581. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Sandberg, M. L., A. Kaykas, and B. Sugden. 2000. Latent membrane protein 1 of Epstein-Barr virus inhibits as well as stimulates gene expression. J. Virol. 749755-9761. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Shaw, G., S. Morse, M. Ararat, and F. L. Graham. 2002. Preferential transformation of human neuronal cells by human adenoviruses and the origin of HEK 293 cells. FASEB J. 16869-871. [DOI] [PubMed] [Google Scholar]
- 44.Sjoblom, A., A. Jansson, W. Yang, S. Lain, T. Nilsson, and L. Rymo. 1995. PU box-binding transcription factors and a POU domain protein cooperate in the Epstein-Barr virus (EBV) nuclear antigen 2-induced transactivation of the EBV latent membrane protein 1 promoter. J. Gen. Virol. 762679-2692. [DOI] [PubMed] [Google Scholar]
- 45.Soni, V., E. Cahir-McFarland, and E. Kieff. 2007. LMP1 TRAfficking activates growth and survival pathways. Adv. Exp. Med. Biol. 597173-187. [DOI] [PubMed] [Google Scholar]
- 46.Soule, H. D., J. Vazguez, A. Long, S. Albert, and M. Brennan. 1973. A human cell line from a pleural effusion derived from a breast carcinoma. J. Natl. Cancer Inst. 511409-1416. [DOI] [PubMed] [Google Scholar]
- 47.Sugano, N., W. Chen, M. L. Roberts, and N. R. Cooper. 1997. Epstein-Barr virus binding to CD21 activates the initial viral promoter via NF-kappaB induction. J. Exp. Med. 186731-737. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Ten, R. M., V. Blank, O. Le Bail, P. Kourilsky, and A. Israel. 1993. Two factors, IRF1 and KBF1/NF-kappa B, cooperate during induction of MHC class I gene expression by interferon alpha beta or Newcastle disease virus. C. R. Acad. Sci. III 316496-501. [PubMed] [Google Scholar]
- 49.Thornburg, N. J., R. Pathmanathan, and N. Raab-Traub. 2003. Activation of nuclear factor-kappaB p50 homodimer/Bcl-3 complexes in nasopharyngeal carcinoma. Cancer Res. 638293-8301. [PubMed] [Google Scholar]
- 50.Thornburg, N. J., and N. Raab-Traub. 2007. Induction of epidermal growth factor receptor expression by Epstein-Barr virus latent membrane protein 1 C-terminal-activating region 1 is mediated by NF-κB p50 homodimer/Bcl-3 complexes. J. Virol. 8112954-12961. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Tsai, C. N., C. M. Lee, C. K. Chien, S. C. Kuo, and Y. S. Chang. 1999. Additive effect of Sp1 and Sp3 in regulation of the ED-L1E promoter of the EBV LMP 1 gene in human epithelial cells. Virology 261288-294. [DOI] [PubMed] [Google Scholar]
- 52.Wang, D., D. Liebowitz, and E. Kieff. 1985. An EBV membrane protein expressed in immortalized lymphocytes transforms established rodent cells. Cell 43831-840. [DOI] [PubMed] [Google Scholar]
- 53.Young, L. S., and A. B. Rickinson. 2004. Epstein-Barr virus: 40 years on. Nat. Rev. Cancer 4757-768. [DOI] [PubMed] [Google Scholar]
- 54.Zetterberg, H., and L. Rymo. 2005. EBNA2 transcription regulation in EBV latency, p. 439-462. In E. S. Robertson (ed.), Epstein-Barr virus. Caister Academic Press, Cambridge, England.