

Abstract
The hyperpermeability of tumor vessels to macromolecules, compared with normal vessels, is presumably due to vascular endothelial growth factor/vascular permeability factor (VEGF/VPF) released by neoplastic and/or host cells. In addition, VEGF/VPF is a potent angiogenic factor. Removal of this growth factor may reduce the permeability and inhibit tumor angiogenesis. To test these hypotheses, we transplanted a human glioblastoma (U87), a human colon adenocarcinoma (LS174T), and a human melanoma (P-MEL) into two locations in immunodeficient mice: the cranial window and the dorsal skinfold chamber. The mice bearing vascularized tumors were treated with a bolus (0.2 ml) of either a neutralizing antibody (A4.6.1) (492 μg/ml) against VEGF/VPF or PBS (control). We found that tumor vascular permeability to albumin in antibody-treated groups was lower than in the matched controls and that the effect of the antibody was time-dependent and influenced by the mode of injection. Tumor vascular permeability did not respond to i.p. injection of the antibody until 4 days posttreatment. However, the permeability was reduced within 6 h after i.v. injection of the same amount of antibody. In addition to the reduction in vascular permeability, the tumor vessels became smaller in diameter and less tortuous after antibody injections and eventually disappeared from the surface after four consecutive treatments in U87 tumors. These results demonstrate that tumor vascular permeability can be reduced by neutralization of endogenous VEGF/VPF and suggest that angiogenesis and the maintenance of integrity of tumor vessels require the presence of VEGF/VPF in the tissue microenvironment. The latter finding reveals a new mechanism of tumor vessel regression—i.e., blocking the interactions between VEGF/VPF and endothelial cells or inhibiting VEGF/VPF synthesis in solid tumors causes dramatic reduction in vessel diameter, which may block the passage of blood elements and thus lead to vascular regression.
Keywords: angiogenesis, vascular obstruction
The microvasculature of solid tumors is, in general, hyperpermeable to macromolecules compared with normal vessels (1–5). This is presumably due to interactions between vascular endothelial cells and the vascular endothelial growth factor/vascular permeability factor (VEGF/VPF) released by neoplastic and/or host cells (6, 7). This hypothesis has been indirectly supported by much experimental evidence. Roberts and Hasan (8) demonstrated that there was a correlation between the in vivo photosensitizer accumulation and the amount of VEGF/VPF secretion in cell culture of three experimental tumor lines. A problem with this study is that the VEGF/VPF expression in vivo may be different from that in vitro because of different local microenvironments. One of the alternative approaches to elucidating the effect of VEGF/VPF on tumor vasculature is to control the expression of the VEGF/VPF in vivo (9, 10). Pötgens et al. (10) demonstrated that the vascular permeability of tumors induced by VEGF/VPF-transfected melanoma cells was higher than that of the controls, which were known to have lower expression of VEGF/VPF. Similarly, local treatment with exogenous VEGF/VPF has increased the permeability of postcapillary venules as well as capillaries of normal tissues (11–13). Mixing VEGF/VPF with an anti-VEGF/VPF antibody before application has abolished the effect of VEGF/VPF on vascular permeability (11, 12). Up to now, there has been no direct evidence in the literature showing that endogenous VEGF/VPF is responsible for the hyperpermeability of tumor vessels. To this end, we designed an experiment to provide information on the role of VEGF/VPF in the regulation of tumor vascular permeability, in which a neutralizing antibody (A4.6.1) against VEGF/VPF was administered systemically, and tumor vascular permeability to bovine serum albumin (BSA) in both treated and control animals was measured afterward.
VEGF/VPF is also a potent vasculogenic and angiogenic factor (6, 7, 14). Loss of a single VEGF/VPF allele results in abnormal formation of blood vessels and thus is embryonic-lethal (15, 16). Furthermore, neutralization of the growth factor via an antibody has led to the inhibition of angiogenesis and tumor growth (17, 18), as well as tumor metastasis (19, 20). In the present study, we report a new finding on the VEGF/VPF-tumor vessel interactions: neutralization of endogenous VEGF/VPF dramatically changes morphology of tumor vessels.
Two hypotheses were tested in the study: (i) endogenous VEGF/VPF increases tumor microvascular permeability to macromolecules, and (ii) the integrity of tumor vessels is maintained through constant stimulation with VEGF/VPF present in the tissue microenvironment, so the removal of the stimulant will lead to vessel regression.§ To test these hypotheses, we transplanted human tumors with different VEGF/VPF expression levels into severe combined immunodeficient (SCID) mice at two locations: cranial window and dorsal skinfold chamber (4, 21). The tumor-bearing animals were treated systemically with either the anti-VEGF/VPF antibody or the vehicle saline after the tumors were fully vascularized. The permeability, diameter, density, and length of tumor vessels were then quantified at different time points posttreatment to elucidate the time dependence of the response to the anti-VEGF/VPF treatment. The methods of the quantification were similar to those used in our previous studies (3, 4, 21), which were based on intravital fluorescence microscopy and video image analysis.
MATERIALS AND METHODS
Animal and Tumor Models.
The dorsal skinfold chamber and cranial window preparations have been described in detail in previous studies (4, 21, 22). The chunks of four tumor lines were transplanted into cranial windows in SCID mice: LS174T (a human colon adenocarcinoma) (21), MCaIV (a murine mammary adenocarcinoma) (4), U87 (a human glioblastoma) (4), and P-MEL (a human melanoma kindly provided by D. L. Fraker, National Institutes of Health). Only the first two tumor lines could grow in the dorsal skinfold chambers.
Clonogenic Assay.
The procedure was similar to that described by Gerweck et al. (23). In brief, U87 cells were cultured in T75 flasks with DMEM (Sigma) containing 10% (vol/vol) heat-inactivated fetal bovine serum (Sigma), 1% penicillin and streptomycin (P-0781, Sigma), and 2% (vol/vol) HCl (1 M, Fisher). Single cell suspensions were obtained by trypsinization, and the cells were plated into T30 flasks at various densities (number of cells/flask): 40, 80, and 160. Duplicates were prepared at each cell density. The cells were further cultured in the fresh medium for 24 h and then treated with either PBS (0.6 ml) or the anti-VEGF/VPF antibody (0.6 ml, 492 μg/ml) in 3 ml of medium for 6 h. Then, the medium was removed, and the cells were continuously cultured with 6 ml of fresh medium. Two weeks later, each flask was washed with 6 ml of saline, and the cells were fixed with 5 ml of methanol for 5 min and stained with crystal violet (5 g dissolved in 100 ml of methanol and 900 ml of distilled water) for 5 min. Finally, the number of colonies per flask was counted.
Northern Blot Analysis. Isolation of total RNA from cultured tumor cells.
Tumor cells (≈107) were treated with 1 ml of Ultraspec RNA and homogenized, and the total cellular RNA was extracted (100–200 μg) (Biotecx, Laboratories, Houston).
Isolation of total RNA from tumor tissues.
The procedure was similar to that described by Gramza et al. (24). In brief, human tumors (≈100 mg) grown in immunodeficient mice were removed, put in the liquid nitrogen immediately, and stored in the −70°C freezer for later analysis. The frozen tissue was contained in a small plastic bag (Kapak, Minneapolis), kept frozen via repeated immersion into the liquid nitrogen, and continuously smashed with a hammer. The tissue powder was placed in 1 ml of RNA isolation reagent, TRIzol (GIBCO) at room temperature and homogenized for 15 sec with a Polytron (Brinkmann). The homogenate was then mixed with 0.2 ml of chloroform and centrifuged for 15 min at 12,000 × g (4°C). The aqueous phase was transferred to a fresh tube, and the total RNA was extracted.
VEGF/VPF mRNA measurement.
Thirty micrograms of the total RNA was separated by electrophoresis via 1% agarose gel containing 1.7% (vol/vol) formaldehyde, transferred to a GeneScreenPlus membrane (Biotechnology System, NEN), and hybridized with a 32P-labeled VEGF/VPF cDNA probe synthesized by PCR with the following forward and reverse oligonucleotide primers: 5′-GGA ATT CAA GCT TGC CAC CAT GAA CTT CTC GCT GTC TTG-3′ and 5′-GGG ATC CGC GGC CGC TCA CCG CCT CGG CTT GTC ACA TCT-3′, respectively (kindly provided by Brian Seed, Massachusetts General Hospital, Boston). The hybridized filter was autoradiographed using Kodak XAR film at −80°C for 16–18 h or exposed to a Storage Phosphor Screen (Molecular Dynamics) for 5 h. The radioactivity, which was proportional to the amount of VEGF/VPF mRNA, was quantified using the PhosphorImager (model 410A; Molecular Dynamics) and was normalized by the amount of rRNA in each tumor line.
Experimental Procedures.
The experimental protocol was similar to our previous studies (3, 4). In brief, the animals were anesthetized with ketamine/xylazine and fixed on a polycarbonate plate. The plate was placed on the stage of an intravital fluorescence microscope (Axioplan, Zeiss) equipped with the fluorescence filter set for rhodamine (Omega Optical, Brattleboro, VT), an intensified charge-coupled device video camera (C2400-88, Hamamatsu Photonics, Hamamatsu, Japan), a videocassette recorder (SVO-9500MD, Sony), and a photomultiplier (9203B, EMI, Rockaway, NJ). The anti-VEGF/VPF treatment was performed 7–23 days posttumor transplantation, depending on the tumor vascularization and the treatment protocols. The number of animals (N) used in each group was between three and seven, as indicated in Results and in the figure and table legends.
In each group, the animals were treated systemically via either the tail vein or i.p. injections of an anti-VEGF/VPF antibody, A4.6.1 (492 μg/ml) (18), or PBS. A fixed dose of 0.2 ml was administered for each systemic injection (i.p. or i.v.) of either the antibody or PBS. The vascular permeability and morphology measurements were performed 6 h to 11 days posttreatment. The details of the permeability measurement have been described previously (3, 4). In brief, a bolus of rhodamine labeled BSA (0.15 ml) (A847, Molecular Probes) was injected into the tail veins of the anesthetized animals, and the tumor vascular permeability was calculated based on the intensity measurement of the fluorescence from the tumor tissue. The vessel diameter, length, and density were measured via the image analysis of the tumor vasculature in five different areas (≈0.5 mm in length) per tumor. The vascular or blood volume per unit area was calculated based on the vessel diameter (D) and length (L).
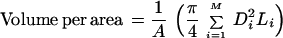 |
where M is the total number of vessels in the region with the area A.
Statistics.
The Mann–Whitney U test was used to compare the differences in permeability or vascular morphology between treated and control groups. The difference was considered significant if the P value was less than 0.05.
RESULTS
The tumor vascular permeability and morphology were quantified in both antibody-treated groups and in the matched controls. The results are reported as the median and range of the measurements unless specified below. The letters N and n represent the numbers of animals and total vessels, respectively.
Tumor Vascular Permeability.
Tumor vascular permeability to rhodamine-labeled BSA was lower in anti-VEGF/VPF antibody-treated animals compared with the permeability in matched controls. The level of reduction was time-dependent as shown in Tables 1–3. When the antibody was administered i.p., the vascular permeability of U87 tumors did not change until 4 days after i.p. injection (Table 1) even though the dose of injection in the first group (i.e., 8 h postinjection) was twice as high as that in the second and third groups. However, the vascular permeability of U87 tumors was reduced within 6 h post-i.v. injection of the same amount of antibody (Table 3). The time-dependent change in the vascular permeability was also examined in LS174T tumors, and the maximum decrease in permeability was reached at 3 days post-i.v. injection of the antibody (Table 2). At day 5, the difference in permeability between antibody-treated and control animals became insignificant (Table 2), presumably because of the shifted balance between antibody clearance from the tumors and the constant production of VEGF/VPF by tumor as well as infiltrated host cells.
Table 1.
Reduction in tumor vascular permeability after anti-VEGF/VPF treatment: U87 tumors in cranial windows
| Control | Antibody-treated | Conditions |
|---|---|---|
| 1.00 (0.61–1.65) | 1.24 (0.58–1.62) | Two boluses of anti-VEGF antibody (N = 6) or PBS (N = 7) were injected i.p. at 0 and 4 h, and the tumor microvascular permeability was measured at 8 h |
| 1.33 (0.92–2.09) | 0.92 (0.40–1.59) | A bolus of anti-VEGF antibody (N = 4) or PBS (N = 4) was injected i.p., and the tumor microvascular permeability was measured at 2 days posttreatment |
| 2.85 (2.30–3.71) | 0.91 (0.51–2.06)* | A bolus of anti-VEGF antibody (N = 4) or PBS (N = 4) was injected i.p., and the tumor microvascular permeability was measured at 4 days posttreatment |
| 1.62 (1.50–2.07) | 0.73 (0.62–1.05)* | The anti-VEGF antibody (N = 4) or PBS (N = 4) was injected i.p. on days 7, 10, 14, and 17 after tumor tissue transplantation, and the tumor microvascular permeability was measured on day 18 |
The permeability data (10−7 cm/s) are shown as median (range). The antibody (492 μg/ml) or PBS was administered 7–23 days after tumor transplantation, depending on growth rate and vascularization of tumors. The dose of each injection was 0.2 ml.
P < 0.05 vs the matched control.
Table 3.
Reduction in tumor vascular permeability after anti-VEGF/VPF treatment: Tumors in cranial windows
| Control | Antibody-treated | Tumor lines |
|---|---|---|
| 1.11 | 0.66 | |
| (0.92–2.82) | (0.58–0.75)* | U87 |
| 3.98 | 1.50 | |
| (3.60–5.06) | (0.79–2.13)* | LS174T |
| 0.98 | 1.29 | |
| (0.89–1.15) | (1.13–1.48) | P-MEL |
The permeability data (10−7 cm/s) are shown as median (range). The antibody (492 μg/ml) or PBS was administered 7–23 days after tumor transplantation, depending on the growth rate and vascularization of tumors. The dose of each injection was 0.2 ml. The measurement was performed at 6 h after a bolus i.v. injection of the anti-VEGF antibody (N = 3) or PBS (N = 3).
P < 0.05 vs the matched control.
Table 2.
Reduction in tumor vascular permeability after anti-VEGF/VPF treatment: LS174T tumors in dorsal skinfold chambers
| Control | Antibody-treated | Time, h |
|---|---|---|
| 2.26 | 1.69 | |
| (0.95–3.69) | (0.83–1.83) | 6 |
| 4.78 | 1.20 | |
| (3.50–5.16) | (1.07–3.22)* | 24 |
| 2.33 | 0.65 | |
| (2.05–5.87) | (0.63–0.75)* | 72 |
| 2.24 | 0.88 | |
| (0.66–8.08) | (0.49–4.07) | 120 |
The permeability data (10−7 cm/s) are shown as median (range). The antibody (492 μg/ml) or PBS was administered 7–23 days after tumor transplantation, depending on the growth rate and vascularization of tumors. The dose of each injection was 0.2 ml. The measurement was performed at various time points after a bolus i.v. injection of the anti-VEGF antibody (N = 3) or PBS (N = 3).
P < 0.05 vs the matched control.
The reduction in vascular permeability of the human melanoma xenograft (P-MEL) transplanted in the cranial window was not significant at 6 h postinjection of the anti-VEGF/VPF antibody (Table 3). At 48 h, the medians of the permeability values were smaller in the antibody-treated animals (2.1 vs 4.0 × 10−7 cm/sec, N = 3 in both groups), but they were not statistically significant. The lesser response of the P-MEL tumors to the antibody treatment could be attributed to lower VEGF/VPF expression by melanoma cells. To test this hypothesis, we performed Northern blot analysis of VEGF/VPF mRNA extracted from both tumor tissues and cultured cells. The relative amount of the mRNA in tumor tissues was 5.2, 3.2, and 1 for U87, LS174T, and P-MEL, respectively. However, the relative amount of the mRNA in cultured cells was 7.0, 1.3, and 1 for U87, LS174T, and P-MEL, respectively, suggesting that the VEGF/VPF expression in vivo might be different from that in vitro. In any case, the VEGF/VPF expression in the P-MEL tumor was low. Thus, these data further supported our hypothesis that the reduction in vascular permeability after antibody injection resulted from neutralization of the endogenous VEGF/VPF.
The response to anti-VEGF/VPF treatment was compared between LS174T tumors transplanted at two locations: the cranial window and the dorsal skinfold chamber (Tables 2 and 3). The results did not show a significant difference between these two groups even though the permeability of the controls was significantly higher for tumors transplanted in the cranial windows than in the dorsal skinfold chambers.
The anti-VEGF/VPF antibody A4.6.1 had no effect on the vasculature of the murine adenocarcinoma MCaIV. The mean values of the vascular permeability (10−7 cm/s) of the antibody-treated tumors vs matched controls were 5.20 (N = 2) vs 6.74 (N = 2) and 3.65 (N = 3) vs 3.10 (N = 4) at 6 h and 48 h post-i.v. injections, respectively. The differences between control and antibody treatments were not statistically significant, indicating that the observed effect of the antibody on vascular permeability in human tumor xenografts was unlikely due to host immune response. The cytotoxicity of the antibody was assessed through the clonogenic assay (23). The number of colonies formed at three different plating densities of U87 cells was nearly identical between antibody-treated and control groups (data not shown). Thus, the cytotoxicity of the antibody at the dose used in our experiments was negligible.
Morphology of Tumor Vessels.
The vessels of LS174T tumors became smaller and less tortuous after anti-VEGF/VPF treatment. The histogram of the vessel diameter shifted to smaller sizes in response to a bolus i.v. injection of the anti-VEGF/VPF antibody (Fig. 1). The diameters of both the top and lower 10% of the vessels in the controls were significantly larger than those in the antibody-treated groups (P < 0.01). The medians (μm) of the histograms (control vs antibody treatment) were 14.5 vs 7.3, 12.6 vs 9.5, and 15.8 vs 9.5 on days 1, 3, and 5, respectively. In addition, the diameter of vessels longer than 200 μm decreased significantly (Fig. 2); few vessels were larger than 20 μm in diameter after the antibody treatment. Furthermore, the vascular or blood volume at the tumor surface was significantly smaller on days 1 and 3 compared with that of the matched controls (Table 4). However, at day 5, after a bolus i.v. injection of the anti-VEGF/VPF antibody, no significant difference was observed between controls and the treated groups. This observation was consistent with the permeability data (shown in Table 2), reflecting the shifted balance between antibody clearance and VEGF/VPF production in tumors. The reduction in vessel volume after the anti-VEGF/VPF treatment significantly decreased the blood supply in LS174T tumors, as shown in Fig. 3, but the tumors still grew in both groups. The vascular density, defined as the total vessel length per unit tissue surface area, was not changed significantly (Table 5). Taken together, these results suggested that the vascular volume was reduced after a bolus antibody treatment and that the reduction was largely due to the decrease in vessel diameter. However, the vascular density can be decreased after multiple injections of the antibody. For U87 tumors, more than 95% of the vessels on the tumor surface disappeared after four consecutive i.p. injections in 11 days. Only a few vessels could be observed at the edge of the tumors, and other regions on the entire tumor surface were avascular and decorated with aggregates of red blood cells from the regressed vessels (data not shown).
Figure 1.
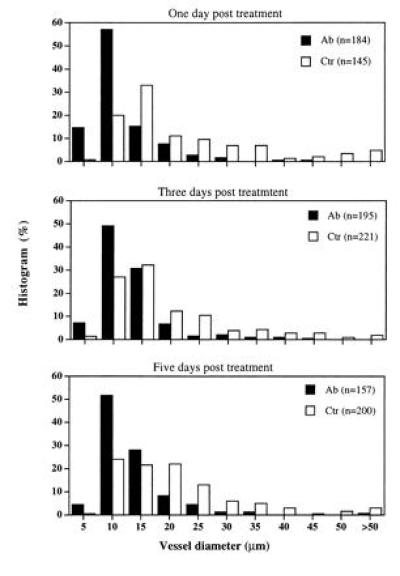
Changes in vessel diameter of LS174T tumors transplanted in dorsal skinfold chambers in SCID mice. Vessel diameter was measured at various time points post-i.v. bolus injection of the anti-VEGF/VPF antibody (N = 3) or PBS (N = 3). The values of n in the parentheses indicate the number of vessels used to generate the histogram. These results demonstrate that anti-VEGF/VPF treatment significantly reduces tumor vessel diameter.
Figure 2.
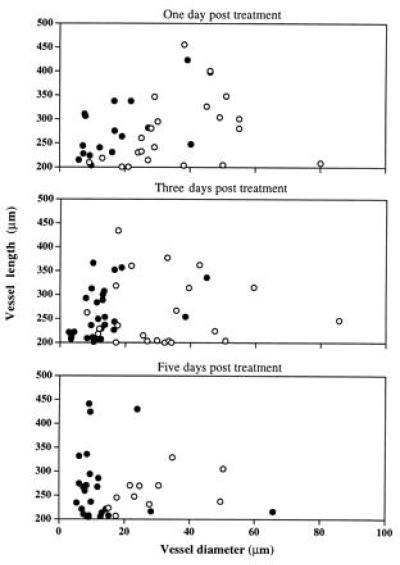
Diameter of long vessels (>200 μm) decreased significantly in the antibody-treated (solid symbols) LS174T tumors transplanted in dorsal skinfold chambers in SCID mice, compared with the controls (open symbols) (P < 0.0001). Vessel diameter and length were measured at various time points postbolus i.v. injection of the anti-VEGF antibody (N = 3) or PBS (N = 3).
Table 4.
Time-dependent changes in vascular volume in LS174T tumors in dorsal skinfold chambers: Total vascular volume/tumor surface area (10−4 cm3/cm2)*
| Control | Antibody-treated | Time, day |
|---|---|---|
| 6.87 (6.45–6.89) | 1.43 (1.00–2.37)† | 1 |
| 2.82 (2.57–11.23) | 1.82 (1.61–2.01)† | 3 |
| 3.72 (2.24–10.14) | 1.09 (0.70–2.85) | 5 |
The data are shown as median (range). The anti-VEGF antibody (492 μg/ml) (N = 3) or PBS (N = 3) was administered as a bolus i.v. approximately 3 weeks after tumor transplantation, depending on growth rate and vascularization of tumors; and the measurement was performed at various time points postinjections. The dose of each injection was 0.2 ml.
The volume per tumor surface area was calculated using the data of vessel diameter and length.
P < 0.05 vs the matched control.
Figure 3.
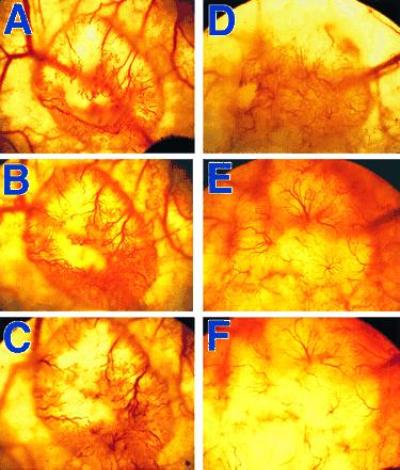
Photographs showing the reduction of vessel volume in LS174T tumors transplanted in dorsal skinfold chambers after the anti-VEGF/VPF treatment. The photos were taken at various time points both before (day 0; A and D) and after bolus i.p. injections of PBS (B and C) or the anti-VEGF/VPF antibody (E and F) on day 3 (B and E) or 7 (C and F). The injections were given at days 0 and 4, respectively. The size of tumors before treatments was ≈4 mm in diameter. There was a significant decrease in vessel volume of the antibody-treated tumor. Small vessels in this tumor were not visible on days 3 (E) or 7 (F) because of the lower magnification of these images.
Table 5.
Time-dependent changes in vascular density in LS174T tumors in dorsal skinfold chambers: Vessel density (cm/cm2)
| Control | Antibody-treated | Time, day |
|---|---|---|
| 101 (71–112) | 104 (100–124) | 1 |
| 117 (104–133) | 115 (93–155) | 3 |
| 111 (101–120) | 102 (94–117) | 5 |
See Table 4 legend for details.
DISCUSSION
Our study shows that tumor vascular permeability can be reduced by systemic anti-VEGF/VPF treatment. The effect of the antibody is time-dependent and related to the mode of injection. The vascular permeability may increase again if the anti-VEGF/VPF treatment is discontinued. Furthermore, neutralization of the endogenous VEGF/VPF causes a decrease in vessel diameter and tortuosity and eventually leads to vessel regression in solid tumors.
The reduction of tumor vascular permeability is observed within 6 h post-i.v. injection of the antibody. In addition, the antibody has no effect on the murine mammary adenocarcinoma MCaIV because the antibody does not recognize the murine form of VEGF/VPF. Thus, the vascular response in the human tumor xenografts observed in this study is unlikely due to the host immune response. The inhibitory effect on tumor vascular permeability via the neutralization of the endogenous VEGF/VPF is time-dependent and cumulative. It reaches a maximum at 3 days post-i.v. treatment in LS174T tumors, suggesting that endothelial cell remodeling is involved. However, the permeability is never reduced to the level of normal tissues (e.g., skin) (data not shown). It is possible that cytokines other than VEGF/VPF are involved in the regulation of vascular permeability, but we cannot exclude the possibility that the mouse VEGF/VPF released by host cells (e.g., macrophage and fibroblast) infiltrated into the human tumor xenografts is responsible for the remaining level of the vascular permeability (6, 7). The antibody used in our study was developed against human VEGF/VPF and has no effect on its mouse form, as shown above for the MCaIV tumors. Thus, the anti-VEGF/VPF treatment may not be able to neutralize all of the VEGF/VPF molecules in experimental tumors. Finally, neutralization of the growth factor may elicit endothelial cell remodeling to seal fenestrae induced by VEGF/VPF (13) and thus to lower the extravasation of macromolecules. However, the antibody treatment may not lead to the expression of adhesion molecules (e.g., vascular endothelial cadherin) (25) involved in forming normal junctions between endothelial cells. Defects in the junctional structure may lead to an increase in vascular permeability (2).
The time-dependent response of tumor vessels to the anti-VEGF/VPF treatment differs when the antibody is administered i.v. vs i.p. This discrepancy could be related to the pharmacokinetics of the antibody. Intravenous injection results in a rapid increase in the plasma concentration and a higher peak value of the antibody in comparison with the i.p. injection. Therefore, a similar pharmacokinetic profile may occur in tumors, which results in a higher rate of VEGF/VPF neutralization in tumors in the case of i.v. injection. The extent of permeability reduction is similar in both cases, suggesting that the dose of the antibody injection in our study is adequate to saturate the endogenous human VEGF/VPF in tumors.
Our previous studies (ref. 4 and unpublished data) indicate that tumor vascular permeability depends on the tissue microenvironment. The vascular barrier in a human glioblastoma (HGL21) transplanted in the cranial windows in SCID mice is comparable to the blood-brain barrier (4) and can be interrupted by topical application of a human recombinant VEGF/VPF (unpublished observation). However, when the tumor is transplanted s.c., the vessels become leaky (unpublished observation). On the other hand, if vessels are leaky at both places, the ones in the cranial windows exhibit higher permeability than the vessels in the dorsal skinfold chambers. This phenomenon has been observed for LS174T tumors (Tables 2 and 3) as well as for the vessels induced by different growth factors sequestered in collagen gels (26). Anti-VEGF/VPF treatment eliminates such a difference between sites of tumor transplantation (Tables 2 and 3), suggesting that the apparent difference in the vascular permeability of LS174T tumors transplanted at two locations could be attributed to the extent of VEGF/VPF expression.
Previous studies have shown that anti-VEGF/VPF treatment inhibits angiogenesis and thus retards tumor growth (6, 7). However, we have found that this antibody also induces regression of preformed vessels in solid tumors, implying that the anti-VEGF/VPF treatment may cause tumor shrinkage in addition to growth inhibition. The mechanism(s) of vessel regression is still unknown. Two possible scenarios are suggested here. First, endothelial cells may undergo apoptosis after removal of the VEGF/VPF (27) or after anti-angiogenic therapy (28). Thus, VEGF/VPF is also referred to as a survival factor for vascular endothelial cells (27). Second, endothelial cells may become smaller in size after neutralization of endogenous VEGF/VPF, which will result in a decrease in vessel diameter as observed in the present study (Figs. 1 and 2). The second scenario is proposed based on the study of Augustin et al. (29) who have demonstrated that the normal bovine corpus luteum regression is characterized by the rounding, condensation, and detachment of endothelial cells and does not apparently involve endothelial cell apoptosis. Once the vessel diameter is smaller than the minimum diameter of erythrocytes (≈2.8 μm), the blood flow will be stopped in that vessel and the in the down stream vascular network. In some cases, vessels larger than 2.8 μm could also be blocked by tumor cells or leukocytes because they are much more rigid than erythrocytes (30) and interact with vascular endothelial cells via adhesion molecules (31–33). The progression of these processes may stop blood flow in the entire tumor vascular network. Therefore, we propose that both pathways of the vessel regression are present in solid tumors after the anti-VEGF/VPF treatment and that tumor vessels require constant stimulation with VEGF/VPF to maintain their morphology and endothelial cell proliferation.
The expression of VEGF/VPF and its receptor (kinase insert domain receptor) is directly correlated with the incidence of metastasis (34); and anti-VEGF/VPF treatments inhibit the tumor cell dissemination (19, 20). The mechanism has been suggested to be due to the decrease in tumor vessel density and, thus, to the decrease in the probability of tumor cell intravasation. The results of the present study propose a new possible mechanism for interpreting the data of tumor metastasis: Anti-VEGF/VPF treatment reduces the diameter of tumor vessels, which leads to a significant increase in resistance to blood flow and cancer cell movement within tumor vessels and eventually can block tumor blood flow as discussed above and thus can inhibit blood-borne metastasis.
Acknowledgments
We thank Drs. Leo Gerweck and Brian Seed for helping with the clonogenic assay and Northern blot analysis, respectively, and Ms. Julia Kahn for preparation of dorsal skinfold chambers. The study was supported by National Institutes of Health Grant R35-CA-56591.
Footnotes
Abbreviations: VEGF/VPF, vascular endothelial growth factor/vascular permeability factor; SCID, severe combined immunodeficient.
Preliminary results of this work were presented at the Annual Meeting of the Microcirculatory Society, April 13–14, 1996, Bethesda, Md, and at the Annual Meeting of Experimental Biology, April 14–17, 1996, Washington, DC.
References
- 1.Gerlowski L E, Jain R K. Microvasc Res. 1986;31:288–305. doi: 10.1016/0026-2862(86)90018-x. [DOI] [PubMed] [Google Scholar]
- 2.Jain R K. Cancer Metastasis Rev. 1987;6:559–593. doi: 10.1007/BF00047468. [DOI] [PubMed] [Google Scholar]
- 3.Yuan F, Leunig M, Berk D A, Jain R K. Microvasc Res. 1993;45:269–289. doi: 10.1006/mvre.1993.1024. [DOI] [PubMed] [Google Scholar]
- 4.Yuan F, Salehi H A, Boucher Y, Vasthare U S, Tuma R F, Jain R K. Cancer Res. 1994;54:4564–4568. [PubMed] [Google Scholar]
- 5.Wu N Z, Klitzman B, Rosner G, Needham D, Dewhirst M W. Microvasc Res. 1993;46:231–253. doi: 10.1006/mvre.1993.1049. [DOI] [PubMed] [Google Scholar]
- 6.Dvorak H F, Brown L F, Detmar M, Dvorak A M. Am J Pathol. 1995;146:1029–1039. [PMC free article] [PubMed] [Google Scholar]
- 7.Ferrara N, Heinsohn H, Walder C E, Bunting S, Thomas G R. Ann NY Acad Sci. 1995;752:246–256. doi: 10.1111/j.1749-6632.1995.tb17435.x. [DOI] [PubMed] [Google Scholar]
- 8.Roberts W G, Hasan T. Cancer Res. 1993;53:153–157. [PubMed] [Google Scholar]
- 9.Rak J, Mitsuhashi Y, Bayko L, Filmus J, Shirasawa S, Sasazuki T, Kerbel R S. Cancer Res. 1995;55:4575–4580. [PubMed] [Google Scholar]
- 10.Pötgens A J G, van Altena M C, Lubsen N H, Ruiter D J, de Waal R M W. Am J Pathol. 1996;148:1203–1217. [PMC free article] [PubMed] [Google Scholar]
- 11.Kim K J, Li B, Houck K, Winer J, Ferrara N. Growth Factors. 1992;7:53–64. doi: 10.3109/08977199209023937. [DOI] [PubMed] [Google Scholar]
- 12.Senger D R, Perruzzi C A, Feder J, Dvorak H F. Cancer Res. 1986;46:5629–5632. [PubMed] [Google Scholar]
- 13.Roberts W G, Palade G E. J Cell Sci. 1995;108:2369–2379. doi: 10.1242/jcs.108.6.2369. [DOI] [PubMed] [Google Scholar]
- 14.Senger D R. Am J Pathol. 1996;149:1–8. [PMC free article] [PubMed] [Google Scholar]
- 15.Carmellet P, Ferreira V, Breier G, Pollefeyt S, Kieckens L, Gertsenstein M, Fahrig M, Vandenhoeck A, Harpal K, Eberhardt C, Declercq C, Pawling J, Moons L, Collen D, Risau W, Nagy A. Nature (London) 1996;380:435–439. doi: 10.1038/380435a0. [DOI] [PubMed] [Google Scholar]
- 16.Ferrara N, Carver-Moore K, Chen H, Dowd M, Lu L, O’Shea K S, Powell-Braxton L, Hillan K J, Moore M W. Nature (London) 1996;380:439–442. doi: 10.1038/380439a0. [DOI] [PubMed] [Google Scholar]
- 17.Asano M, Yukita A, Matsumoto T, Kondo S, Suzuki H. Cancer Res. 1995;55:5296–5301. [PubMed] [Google Scholar]
- 18.Kim K J, Li B, Winer J, Armanini M, Gillett N, Phillips H S, Ferrara N. Nature (London) 1993;362:841–844. doi: 10.1038/362841a0. [DOI] [PubMed] [Google Scholar]
- 19.Warren R S, Yuan H, Matli M R, Gillett N A, Ferrara N. J Clin Invest. 1995;95:1789–1797. doi: 10.1172/JCI117857. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Melnyk O, Shuman M A, Kim K J. Cancer Res. 1996;56:921–924. [PubMed] [Google Scholar]
- 21.Leunig M, Yuan F, Menger M D, Boucher Y, Goetz A E, Messmer K, Jain R K. Cancer Res. 1992;52:6553–6560. [PubMed] [Google Scholar]
- 22.Endrich B, Reinhold H S, Gross J F, Intaglietta M. J Natl Cancer Inst. 1979;62:387–395. [PubMed] [Google Scholar]
- 23.Gerweck L E, Kornblith P L, Burlett P, Wang J, Sweigert S. Radiology. 1977;125:231–234. doi: 10.1148/125.1.231. [DOI] [PubMed] [Google Scholar]
- 24.Gramza A W, Lucas J M, Mountain R E, Schuller D E, Lang J C. BioTechniques. 1995;18:228–229. [PubMed] [Google Scholar]
- 25.Lampugnani M G, Resnati M, Raiteri M, Pigott R, Pisacane A, Houen G, Ruco L P, Dejana E. J Cell Biol. 1992;118:1511–1522. doi: 10.1083/jcb.118.6.1511. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Dellian M, Witwer B P, Salehi H A, Yuan F, Jain R K. Am J Pathol. 1996;149:59–72. [PMC free article] [PubMed] [Google Scholar]
- 27.Alon T, Hemo I, Itin A, Pe’er J, Stone J, Keshet E. Nat Med. 1995;1:1024–1028. doi: 10.1038/nm1095-1024. [DOI] [PubMed] [Google Scholar]
- 28.Holmgren L, O’Reilly M S, Folkman J. Nat Med. 1995;1:149–153. doi: 10.1038/nm0295-149. [DOI] [PubMed] [Google Scholar]
- 29.Augustin H G, Braun K, Telemenakis I, Modlich U, Kuhn W. Am J Pathol. 1995;147:339–351. [PMC free article] [PubMed] [Google Scholar]
- 30.Jain R K. Cancer Res. 1988;48:2641–2658. [PubMed] [Google Scholar]
- 31.Melder R J, Koenig G C, Witwer B P, Safabakhsh N, Munn L L, Jain R K. Nat Med. 1996;2:992–997. doi: 10.1038/nm0996-992. [DOI] [PubMed] [Google Scholar]
- 32.Jain R K, Koenig G C, Dellian M, Fukumura D, Munn L L, Melder R J. Cancer Metastasis Rev. 1996;15:195–204. doi: 10.1007/BF00437472. [DOI] [PubMed] [Google Scholar]
- 33.Springer T A. Annu Rev Physiol. 1995;57:827–872. doi: 10.1146/annurev.ph.57.030195.004143. [DOI] [PubMed] [Google Scholar]
- 34.Takahashi Y, Kitadai Y, Bucana C D, Cleary K R, Ellis L M. Cancer Res. 1995;55:3964–3968. [PubMed] [Google Scholar]