

Abstract
Database search tools identify peptides by matching tandem mass spectra against a protein database. We study an alternative approach when all plausible de novo interpretations of a spectrum (spectral dictionary) are generated and then quickly matched against the database. We present a new MS-Dictionary algorithm for efficiently generating spectral dictionaries and demonstrate that MS-Dictionary can identify spectra that are missed in the database search. We argue that MS-Dictionary enables proteogenomics searches in six-frame translation of genomic sequences that may be prohibitively time-consuming for existing database search approaches. We show that such searches allow one to correct sequencing errors and find programmed frameshifts.
In 1994, Mann and Wilm (1) proposed the peptide sequence tag approach and outlined its applications for protein identification. However, it took 10 years for this approach to result in accurate tag-based tools like InsPecT (2) and Paragon (3), currently among the fastest MS/MS database search tools. The reason for this delay is that although generating some peptide sequence tags is easy, such tags are of little use unless they contain at least one correct tag with high probability. Generating small covering sets of tags (i.e. the sets of tags that almost surely contain a correct tag) turned out to be a more difficult problem that has recently been addressed (2–5).
Similar to generating the covering set of tags (that in most applications limited to tags of length 3), one can try to generate the covering sets of full-length peptide reconstructions that with high probability contain the correct peptide (spectral dictionary). Spectral dictionaries take the peptide sequence tag approach one step further by generating peptide reconstructions and ensuring that one of them is correct. They also have the potential to improve the filtration efficiency of tag-based tools (2, 3); for example, the filtration efficiency of 1000 de novo reconstructions of length 10 is orders of magnitude higher than even a single tag of length 3. However, although spectral dictionaries have important advantages over spectral tags, generating them remains an open problem.
The spectral dictionaries could be searched efficiently against a protein database resulting in a hybrid approach to peptide identification (Fig. 1). Although the idea of spectral dictionaries is almost as old as the idea of peptide sequence tags (6), the software tool RAId based on this approach was described only recently (7). However, although RAId generated promising initial results, it was based on a heuristic exhaustive search and turned out to be rather slow (2–4 min per spectrum) thus limiting its applicability. Also RAId was benchmarked on a small sample thus making it difficult to evaluate its performance on large MS/MS data sets. Here we describe a fast approach to generating spectral dictionaries that takes ≈0.1 s per spectrum and benchmark it on a data set of over 20,000 peptides.
Fig. 1.

Two approaches to peptide identification: traditional approach based on comparing spectra with the database (red) and the hybrid approach based on constructing spectral dictionaries and fast database lookup (blue). The red lines illustrate that in traditional searches every spectrum should be compared with every peptide in the database with a given parent mass (the running time scales linearly with the database size). The blue lines illustrate that every peptide in the spectral dictionary should be checked for presence in the database (the running time is negligible if the database is preprocessed as a hash table or a suffix tree). The running time of the de novo-based approaches is nearly independent of the database size (it is dominated by the time required to generate the spectral dictionaries). The fast database lookup can be implemented either as exact matching or as error-tolerant lookup (to search for mutations/polymorphisms).
Spectral dictionaries may have an edge over the traditional MS/MS approaches in searching very large databases, e.g. six-frame translations of entire genomes. Various proteogenomics studies (8–15) demonstrated that MS/MS search against a six-frame translation of the genome allows one to use MS/MS data for finding new genes, predicting programmed frameshifts, correcting DNA sequencing errors, etc. However, existing MS/MS database search tools are impractical for searches against the six-frame translation of large genomes like human (≈3 billion amino acids after removing repeats). Indeed most of the previous proteogenomics studies were limited to searches against the six-frame translations of bacterial genomes. The largest proteogenomics analysis conducted so far was the search against the six-frame translation of Arabidopsis thaliana that resulted in the discovery of nearly 800 new genes using InsPecT.1 However, even fast tag-based tools like InsPecT become impractical in searches of the 20 times larger six-frame translation of the human genome. Below we show that MS-Dictionary is able to search the six-frame translation of the human genome in roughly the same time as it takes to search the 100 times smaller database of all human proteins.
Spectral dictionaries make the size of the database almost irrelevant because the spectral dictionary can be matched against the six-frame translation as efficiently as against a much smaller database of known proteins. Because many genes remain unidentified even in the well studied organisms (see Siepel et al. (16) and Stark et al. (17) for the recent discovery of over 1000 new protein-coding genes in human and fruit fly genomes), the searches in six-frame translation represent a valuable tool for proteogenomics annotations.2
De novo peptide sequencing represents a fast alternative to MS/MS database search. Although the best de novo algorithms are orders of magnitude faster than the fastest database search tools (even on moderately sized databases), they are less accurate. However, the superior accuracy of the database search tools becomes less pronounced with the increase in the database size. Moreover we show that for very large databases our de novo peptide sequencing algorithm compares favorably to MS/MS database search tools. Thus, searches in very large databases represent an important niche where de novo-based approaches are accurate and orders of magnitude faster than the traditional database search approaches. A number of de novo methods have been developed, including Lutefisk (6, 19), Sherenga (20), PepNovo (21), PEAKS (22), EigenMS (23), NovoHMM (24), AUDENS (25), MSNovo (26), and PILOT (27) (see also Refs. 28–30). Most de novo tools use the spectrum graph approach where a spectrum is represented as a graph with peaks as vertices that are connected by edges if their mass difference corresponds to the mass of an amino acid.
De novo peptide sequencing can also be viewed as a database search in the database of all possible peptides. Even if this time-consuming search were feasible, it would remain unclear which peptide in the database of all peptides represents the real peptide that generated the spectrum. We estimate that in 50–95% of the cases (depending on the peptide length), the existing database search tools (2, 31–35) will fail to identify the correct peptide in such an ultimate test because its score will be lower than the score of an incorrect peptide. We therefore argue that any de novo peptide sequencing algorithm should output multiple peptide reconstructions rather than a single reconstruction. Matching these peptides against a database results in a hybrid spectral dictionary approach that bypasses the time-consuming matching of spectra against the database.
Spectral dictionaries allow one to turn every MS/MS database search tool into a de novo peptide sequencing software (by simply running this tool on all peptides from the spectral dictionary and selecting the top scoring peptide). After such “conversion,” one can estimate how well both database search tools and de novo tools would perform on very large databases. This experiment reveals a disappointing performance of both de novo and database search tools. Only 35–42% of peptides of length 10 (charge 2) are correctly reconstructed in such experiments (35, 38, and 42% for X!Tandem, PepNovo, and InsPecT, correspondingly). Our MS-Dictionary algorithm correctly reconstructs 50% of such peptides, a significant improvement over existing approaches.3 We further show that MS-Dictionary can search a six-frame translation of the entire human genome, the largest database ever searched for spectral interpretations.
The key problem in the spectral dictionary approach is deciding which and how many reconstructions must be generated. Generating too few peptides will lead to high false negative error rates, whereas generating too many peptides will lead to high false positive error rates. Some de novo algorithms output a single or a fixed number (decided before the search) of peptides. For example, RAId (7) generates 1000 de novo reconstructions and matches them against a database.4 We argue that for some spectra generating only one reconstruction is sufficient for finding the correct peptide, whereas in other cases (even with the same parent mass), a thousand reconstructions may be insufficient. We propose an approach for dynamically determining how many reconstructions must be generated for each spectrum and then actually generating them.5
Our MS-Dictionary software (available as open source) generates spectral dictionaries based on the recently introduced concept of the generating function of tandem mass spectra borrowed from statistical mechanics. The generating function approach efficiently analyzes the peptide reconstructions with the optimal and suboptimal scores and determines the statistical significance (spectral probability of those reconstructions (for more details, refer to Ref. 38).6
EXPERIMENTAL PROCEDURES
Peptide Sequencing Problem for Boolean Spectra
Dancik et al. (20) put de novo peptide sequencing in a probabilistic framework and described how to learn the parameters of the model and optimally solved it. Although the Dancik model was further extended in a number of studies (21, 24, 39, 40), it remains unclear how to design a rigorous probabilistic model for peak intensities. We start by introducing an abstract model that seemingly has nothing to do with de novo peptide sequencing but rather describes a very general probabilistic process that transforms one Boolean string into another. We will show later that this process generalizes the probabilistic model for de novo peptide sequencing from Dancik et al. (20) and also allows one to compute the spectral probability and the generating function of tandem mass spectra (38).
Let s = s1 … sn be a Boolean string called a spectrum and π = π1 … πn be a Boolean string called a peptide. The probability of peptide π generating spectrum s is defined as Prob(s|π) = Πi = 1n Prob(si|πi) where Prob(x|y) is a 2 × 2 matrix (see Fig. 2).
Fig. 2.

Left, probability Prob (x|y) of a peptide symbol y generating a spectrum symbol x. Right, the amino acid graph G for all peptides with parent mass 7 and only two possible “amino acids” A and B with masses 2 and 3, correspondingly. The highlighted path corresponds to the G-peptide 0101001 corresponding to AAB (masses of consecutive amino acid masses are 2, 2, and 3). Two other G-peptides with parent mass 7 are 0100101 (ABA) and 0010101 (BAA). The probability of a spectrum s = s1 … sn being generated by a peptide π = π1 … πn is defined as Prob(s|π) = Πi = 1n Prob(si|πi). This is illustrated above with π = 0101001 and s = 0001101 (Prob(s = 0001101, π = 0101001) = θ·(1 − θ)3 ρ2 (1 − ρ)).
Given a spectrum s and a set of strings Π, we are interested in solving the problem of finding maxπεΠ Prob(s|π). Below we focus on the sets Π that are relevant to tandem mass spectrometry. Let V = {0, 1, … , n} and G(V, E) be a topological ordering of a directed acyclic graph (DAG) such that i < j for every directed edge (i, j) in E. Every path from 0 to n in G corresponds to a G-peptide π = π1 … πn such that πi = 1 if vertex i belongs to the path (see Fig. 2). We are interested in the following Peptide Sequencing Problem (41): given a spectrum s and a DAG7 G, find a G-peptide π maximizing Prob(s|π) over all G-peptides.
In de novo peptide sequencing it is assumed that (i, j) ε E if (j − i) equals the integer mass of an amino acid. Such graphs are referred to as amino acid graphs (38) (compare with spectrum graphs (20, 42)). As a first approximation, an MS/MS spectrum with parent mass n can be represented as a string of ones (peak present) and zeros (peak missing) with a 0/1 for every 1-Da interval. Similarly sequences of amino acid masses (peptides) can also be represented as strings of zeros and ones. An amino acid with an integer mass α is represented as a string of α − 1 zeros followed by a single one. Then a peptide is simply a concatenation of Boolean strings corresponding to its amino acids. In this context, θ ≈ 0.05 (probability of observing a noise peak) and ρ ≈ 0.7 (probability of observing a b-ion) represent typical values of θ and ρ for ion trap MS/MS spectra (Fig. 2). This somewhat simplistic Boolean model can be modified for any mass resolution, peptide fragmentation rules, and peak intensities (4, 28, 29) (see below). Moreover the more realistic model can be analyzed with exactly the same algorithm as the Boolean model (20).
The model above does not capture the fact that MS/MS spectra represent both prefix ions (b-ions series) and suffix ions (y-ions series). To reflect this we represent peptides as strings in three-letter alphabet: 1 (theoretical b-cut), −1 (theoretical y-cut), and 0 (no cut). Given a peptide π = π1 … πn, we define its reverse as the peptide π* = −πn … −π1, i.e. πi* = −πn − i+ 1. We now redefine the probability of peptide π generating spectrum s as Prob(s|π) = Πi = 1n Prob(si|πi)·Prob(si|πi*), where Prob(x|y) is a 2 × 3 matrix.
From Boolean Spectra to MS/MS Spectra
Accounting for Peak Intensities—
Although the simple model described above led to an accurate peptide sequencing algorithm (20), it does not capture the intensities of fragment ion in MS/MS spectra. The experimental spectra represent real valued vectors s1 … sn rather than Boolean vectors (si is the peak intensity at mass i). One can argue that the same model based on probabilities p(x, y) where x is a (real valued) peak intensity and y ε {−1, 0, +1} would take into account the intensities of mass spectra. However, this model faces difficulties because (i) intensities vary between different spectra of the same peptide and (ii) the value of intensity seems to be less important than the distribution of intensities over different peaks (26). As a result, most peptide sequencing algorithms use heuristic approaches and do not try to come up with a rigorous model of spectra generation that accounts for intensities. We argue that peak ranks rather than peak intensities may lead to an adequate model of spectra generation. Peak ranks proved to be valuable in peptide identification; for example InsPecT (2) utilizes peak ranks in its scoring function. Below we show how to rigorously utilize peak ranks in de novo peptide sequencing and to solve the corresponding Peptide Sequencing Problem.
We now define a spectrum s = s1 … sn as a string in the alphabet I (ranks of peaks) and a peptide π = π1 … πn as a string in the alphabet F (types of neutral losses). The probability of peptide π generating spectrum s is defined as Prob(s|π) = Πi = 1n Prob(si|πi)·Πi = 1n Prob(si|πi*), where Prob(x|y) is an arbitrary |I| × |F| matrix representing the probability that a symbol y in the peptide generates a symbol x in the spectrum.
The spectrum strings s = s1 … sn are generated from tandem mass spectra as follows. For simplicity, we retain top k peaks from every MS/MS spectrum (up to k = 150 in our implementation). Spectra are filtered to remove noisy peaks as follows: given a peak at mass M, we retain the peak if it is among the top five peaks within a window of size 100 Da around M. Let us say this procedure gives t peaks, which are ranked from 1 to t. If t > k, we keep only the top k peaks; if t < k, we reinsert the top k − t peaks that were filtered out and assign them ranks t + 1 to k. We define si as the rank of the peak at mass i (if there is a peak at mass i) and define si = 0 if there is no peak at mass i.
The peptide strings π = π1 … πn are generated from amino acid sequences as follows. We define an alphabet of fragment ions as a set of integers corresponding to neutral losses, for example ion fragments b, b − H2O, and b − NH3 correspond to neutral losses {0, 18, 17}. Given a set of neutral losses {x1 … xt}, we represent every amino acid of mass α as a string s1 … sα of length α with α − t zeros and t non-zero symbols 1, 2, …, t located at positions α − x1, α − x2, … α − xt. The peptide string π = π1 … πk is simply a concatenation of strings corresponding to amino acids from the peptide. To make the model more accurate, we further added the doubly charged b- and y-ions as additional types of ions generated by the peptide strings.
MS-Dictionary Scoring Function—
When applying the above model for peptide identification, we are interested in the ratio of probabilities that a spectrum is generated by a given peptide π versus probability that a spectrum is generated by a string consisting of all zeros (noise). This can be represented as Prob(s|π)/Prob(s|0) = Πi=1n Prob(si|πi)/Πi=1n Prob(si|0). We further express it as the sum of log odds ratios as follows.
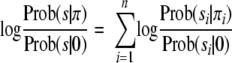 |
(Eq. 1) |
Using the training data set (described below), we learn the values of (Prob(si|πi))/(Prob(si|0)). The learning is done separately for the lower and the higher halves of the mass range (peaks corresponding to doubly charged ions only appear in the lower part of the spectrum). A smoothing function was applied on these values for lower intensity peaks (ranks 11–150); for each ion type, the value at any rank was set to the average value in a window of five ranks around the given rank. The distribution of these values for each peak rank is shown in supplemental Table S1 for three different spectrum lengths for the low and the high mass region. These statistics vary with the length; however, the differences between similar lengths (like 7 and 8) are typically small as compared with differences between very different lengths (like 7 and 20). Thus, specific length-dependent scoring can be applied using the approximate length inferred from the parent mass of the spectrum.
The MS-Dictionary scoring function described here was compared with the scoring functions of the popular database search tools SEQUEST (33), X!Tandem (31), and InsPecT (2). 50,000 spectra were chosen randomly from the Shewanella data set and searched with Sequest, X!Tandem, and InsPecT. The score of the best peptide for each spectrum from the database search was compared with the MS-Dictionary score for the same spectrum-peptide pair. We found good correlation between the MS-Dictionary scoring function and the scoring functions used in the database search tools; the correlation coefficients are 0.87 for SEQUEST, 0.90 for X!Tandem, and 0.96 for InsPecT (Fig. 3). These correlations are even better than the correlation between the database search tools themselves (for example, InsPecT and X!Tandem raw scores have a correlation coefficient of only 0.75).
Fig. 3.

Correlation between InsPecT and MS-Dictionary scores computed on randomly selected 50,000 spectra (correlation coefficient is 0.96).
Suboptimal Peptide Reconstructions—
We use the dynamic programming algorithm for computing the spectral probability and the generating function from Kim et al. (38). The number of peptide reconstructions is computed for each mass value, and the optimal score is determined for a mass within specified error tolerance from the parent mass. We then generate top reconstructions such that their SpectralProbability (see Ref. 38 for details) adds up to a fixed threshold (we typically use 10−9). Starting from the topmost score, reconstructions at each score are selected until their cumulative probability exceeds the threshold (all reconstructions at the borderline score are selected; hence the total probability may marginally exceed the threshold). We limit the number of reconstructions generated for any spectrum to at most 100,000.
The dynamic programming table is constructed for all mass values between 0 and parent mass + 0.5 with a resolution of 0.1 Da. The number of reconstructions is computed by summing up the results for all mass values in a window of 1 Da around the exact parent mass to account for the low accuracy of ion trap mass spectrometers. In case of precision mass spectrometry (e.g. FTMS), accurate solutions (with low parent mass error) can be obtained by increasing the resolution and reducing the size of the window around the parent mass. For efficient computation, Ile and Leu are treated as the same amino acid, resulting in a 19-letter amino acid alphabet at the time of generating reconstructions. In the low accuracy setting, Gln and Lys are also treated as the same amino acid.
Symmetric Versus Antisymmetric de Novo Reconstructions—
Some de novo reconstructions may be symmetric, i.e. the same peak in the spectrum may contribute to the score up to four times as a singly charged or doubly charged b-ion or y-ion. The algorithm to alleviate this problem was proposed by Chen et al. (28) and further improved by others (22, 29). Later Lu and Chen (43) designed an algorithm for generating all antisymmetric peptide reconstructions. We have chosen not to use the antisymmetric path approach in MS-Dictionary because (i) it leads to a significant time overhead when many reconstructions are generated and (ii) it does not take into account doubly charged ion fragments that often have high intensities and thus contribute significantly to MS-Dictionary scores. To accurately score the symmetric reconstruction, MS-Dictionary rescores the obtained peptide reconstructions to exclude multiple contributions from the same peak. Starting with the highest scoring reconstructions, we check the peptide sequence to determine whether there are any peaks that have multiple contributions to the score. These peptides are rescored by using only the largest contributions from such peaks.
Template-free Spectral Recalibration—
Recalibration of tandem mass spectra is important for correcting systematic mass errors. All existing spectral recalibration tools use templates (interpreted spectra with known b/y-peaks) to perform linear recalibration using either least squares fit (19, 22, 44) or least median of squares fit (23). In the de novo peptide sequencing framework the reliable templates are hard to obtain thus reducing the utility of spectral recalibration to Q-TOF and LTQ-FT data. In the low mass accuracy setting, the applications of template-based spectral recalibration are mainly limited to validating candidate peptide identifications. As a result, de novo peptide sequencing programs commonly default to a rather high fragment mass tolerance (e.g. 0.5 Da for ion trap data) and thus result in many erroneous spectral interpretations. We describe a template-free spectral recalibration procedure for ion trap mass spectra and demonstrate that it reduces the required mass tolerance from 0.5 to 0.2 Da. We further show that this recalibration leads to significant improvement in MS-Dictionary accuracy.
The fractional masses of amino acids may be as large as 0.1 for arginine (mass, 156.1 Da). The first step of our MS-Recalibration tool is rescaling all peaks in the spectrum by multiplying all masses by 0.9995 to minimize the theoretical fractional masses of amino acids. After rescaling the fractional mass of arginine is 0.02 (156.02 Da), and the fractional masses of all other amino acids are below 0.04 (the average fractional mass is reduced 3-fold from 0.06 to 0.02).
MS-Calibration further filters the rescaled spectra to retain the high intensity peaks using a sliding window as described above. Using Int(m) and Frac(m) to denote the integer and fractional part of mass m (respectively), our goal is to find α and β minimizing the sum Σ(Frac(α·m + β))2 over all masses m in the rescaled filtered spectrum (Fig. 4a). The coefficients α and β are computed with the least squares fit algorithm and are used to recalibrate all peaks in the rescaled spectrum. Although MS-Recalibration has no information about the peptide that produced the spectrum, Fig. 4b illustrates that it achieves almost the same accuracy as the template-based approaches that recalibrate the spectra based on the information about the correct positions of b/y-ions. After applying MS-Recalibration, one can safely set the mass tolerance to 0.2 Da (and retain 96% of b/y-peaks) as compared with the 0.5 Da in existing approaches. Another advantage of our method is that it makes the mass error distributions centered around zero regardless of their positions in the spectrum. This feature is important for designing a new scoring function that carefully account for errors in peak positions (see below).
Fig. 4.

a, comparison of template-free (solid line) and template-based (dashed line) recalibrations for a single spectrum. Each black dot represents a two-dimensional point (m, Frac(m)) for a mass m (for every peak in the rescaled and filtered spectrum). Each white dot represents a two-dimensional point (m, Error(m)) for a b- or y-peak with mass m and the difference between the theoretical and experimental mass of the peak equal to Error(m) (for every b- and y-peak in the original spectrum). b, MS-Recalibration performance on 1745 identified spectra of length 10 in the Shewanella data set. The template-based recalibration uses the positions of theoretical b- and y-ions in the spectrum to fit the positions of b- and y-ions in the experimental spectrum using the least squares fit algorithm. The template-free MS-Recalibration does not require knowledge of the theoretical b- and y-ions. The error distribution for non-calibrated spectra is shown for comparison. The average error is 0.13 before recalibration, 0.07 after MS-Recalibration, and 0.06 after the template-based recalibration. Before recalibration, only 79% of b/y-ions are within a mass error of 0.2 Da as compared with 96% after MS-Recalibration (similar to 98% for the template-based recalibration).
Incorporating Mass Errors into the Scoring Function—
Most de novo peptide sequencing tools (4, 6, 19, 20, 22–24, 26, 27, 29, 45–47) set up a fixed mass error threshold (e.g. 0.5 Da for ion traps) and compute the scoring functions for all peaks within this error threshold. Bafna and Edwards (48) and Mo et al. (26) noticed that assigning the same scores to all peaks within the error threshold may not be the optimal way to score spectra in both database search and de novo peptide sequencing applications. For example, a high intensity peak with mass error 0.5 Da is typically less “reliable” than a medium intensity peak with mass error 0.1. Recent incorporation of mass errors into the scoring function (as a quantitative component rather than a cutoff) led to a significant improvement in MSNovo accuracy (26). MS-Dictionary also incorporates mass errors in the scoring functions and further improves MSNovo model as described below.
MSNovo uses a unified peak error model (Gaussian distribution) and peak rank model (exponential distribution) independent of the ion type, rank, and position of each peak. However, Fig. 5a illustrates that different fragment ions have different error models. Fig. 5b reveals that peak ranks and mass errors (that are assumed to be independent in MSNovo) are strongly correlated. Also Fig. 5b reveals subtle irregularity in noise peaks indicating that the noise model in Mo et al. (26) needs to be adjusted. MS-Dictionary takes these observations into account and incorporates the mass errors into its scoring function using a more adequate error model than Mo et al. (26). Below we briefly describe the error-dependent scoring for Boolean spectra (this model can be extended to MS/MS spectra as described above).
Fig. 5.

a, different fragment ions have different rank distributions (statistics are given for all spectra of length 10 from the Shewanella data set). b, distributions of mass errors of y-peaks depend on their intensity (statistics are given for all spectra of length 10 from the Shewanella data set). The high intensity peaks (solid curve) tend to have more accurate mass measurements than the lower intensity peaks (dashed curve). The fractional parts of very low intensity peaks (peaks of rank higher than 150) are centered around zero after rescaling (dashed-dot curve).
The Boolean spectra model assumes that a peptide symbol πi generates the spectrum symbol si at exactly the same position. We now extend this model by assuming that the peptide symbol πi can generate spectrum symbol si + ɛ where ɛ represents a mass measurement error. We assume that errors are “small,” i.e. they do not exceed a threshold ɛmax (ɛmax is typically 0.5 for ion trap spectra). Incorporating errors into the spectrum generation model requires introducing the three-dimensional matrix Prob(x, ɛ|y) where −ɛmax ≤ ɛ ≤ +ɛmax and x and y are Boolean as before. The probability of peptide π generating a spectrum s with error ɛ = ɛ1, … , ɛn can now be defined as Prob(s, ɛ|π). The Peptide Sequencing Problem can now be reformulated as the Peptide Sequencing Problem with Errors: given a spectrum s and a DAG G, find a G-peptide π and mass errors ɛ maximizing Prob(s, ɛ|π) = Πi=1n Prob(si+ɛi, ɛi|πi) over all G-peptides and over all mass errors ɛ.
The matrix Prob(x, ɛ|y) was learned from the training sample, and the learned parameters were further used in the dynamic programming algorithm as described before. Table I compares the performance of MS-Dictionary with PepNovo version 1.03 and illustrates that MS-Dictionary outperforms PepNovo for all peptide lengths.
Table I.
Comparison of MS-Dictionary and PepNovo reveals that MS-Dictionary outperforms PepNovo for all peptide length (Shewanella data set)
| Length | Correct amino acids
|
Correct peptides
|
||
|---|---|---|---|---|
| PepNovo | MS-Dictionary | PepNovo | MS-Dictionary | |
| % | % | |||
| 8 | 88.7 | 92.2 | 51.1 | 58.1 |
| 10 | 85.8 | 91.2 | 38.2 | 49.6 |
| 12 | 79.7 | 87.2 | 23.1 | 34.5 |
| 14 | 71.1 | 81.7 | 11.8 | 17.8 |
| 16 | 61.1 | 79.0 | 3.8 | 12.9 |
| 18 | 56.8 | 74.2 | 1.5 | 7.6 |
| 20 | 49.8 | 65.6 | 0.3 | 3.3 |
RESULTS
Data Sets—
We used the previously published Shewanella oneidensis MR-1 spectral data set containing 14.5 million spectra. The experimental procedures8 for acquiring the spectra and identifications from this data set are described in Gupta et al. (14). 28,377 peptides were reliably identified with false discovery rate 5% using InsPecT (spectrum-level false discovery rate (FDR) is 1%). The InsPecT search was run using default parameter settings (fragment ion tolerance of 0.5 Da and parent mass tolerance of 2.5 Da). For this study, we selected 21,087 tryptic peptides with charge 2, obtained one representative spectra for each of these peptides (most peptides were identified from multiple spectra), and grouped these by the length of their peptide identifications to form a test data set for each length. We will refer to the length of the InsPecT identification of a spectrum as the spectrum length. For the sake of convenience, all lengths 7 through 10 and even lengths between 10 and 20 were considered. The trends across these lengths show smooth progression, and there is no reason to believe that the odd lengths between 10 and 20 would show any deviant behavior. To avoid computational artifacts introduced by errors in the parent mass, we have chosen to correct the parent masses according to the InsPecT identifications.
Generating Multiple de Novo Reconstructions—
A spectrum may have many reconstructions with the optimal score, and in these cases, reporting only one reconstruction is clearly deficient. For example, Fig. 6 shows a spectrum for which two distinct peptides, LHEALPDPEK and HLEALGAFYK, receive the optimal de novo score of 90.
Fig. 6.

Two optimal de novo interpretations, LHEALPDPEK (a) and HLEALGAFYK (b), for a particular spectrum.
We further argue that even generating all optimal reconstructions may not be sufficient for finding the correct peptide. For many spectra, the correct peptide has a lower score than an incorrect peptide. Fig. 7 shows a spectrum for which the correct peptide FINVIMQDGK (as identified reliably by InsPecT) has a score of 111, a high score that exceeds the average score of correct identifications. However, another reconstruction, YPNVMLQDGK (not present in the database), has an even higher score of 123. We note that for ≈60% of length 10 spectra, the correct peptide has a suboptimal PepNovo score (≈50% for MS-Dictionary score), and this fraction quickly increases with the peptide length (Fig. 8). Because the existing de novo approaches fail to identify the correct peptide as the optimal reconstruction in a large fraction of the spectra, a de novo method should consider multiple reconstructions with suboptimal scores.
Fig. 7.

Shown are the correct peptide FINVIMQDGK as identified by InsPecT database search (a) and YPNVMLQDGK, a de novo reconstruction, for a particular spectrum (b). The former gets a score of 111 compared with a higher score of 123 for the latter.
Fig. 8.

Fraction of the spectra for which the correct peptide (as identified by the database search) has a suboptimal de novo score (depending on the length of the spectra). The distribution is shown for MS-Dictionary and PepNovo scoring functions.
How Existing Database Search Approaches Fare While Searching Very Large Databases—
All database search tools we tested would fail to identify the correct peptide for more than half of the length 10 spectra if they were searching through the database of all possible peptides. This is an indication of limitations of the scoring functions of existing database search tools. Because actually searching a database of all peptides is impractical, we conservatively estimate the error rates of MS/MS database search tools by constructing a custom database for each spectrum containing all de novo reconstructions with MS-Dictionary scores better or equal to the correct peptide. Even if we used the theoretical database of all possible peptides, it is likely that the identified peptides would be one of those top reconstructions that we included in our custom database. The rate of finding the correct peptide would only drop if more peptides were added. InsPecT was able to identify the correct peptide (peptide identified in the Shewanella database in Gupta et al. (14)) in such a custom database in only 42% of the cases, and X!Tandem was able to identify the correct peptide in 35% of cases for length 10 peptides. Both InsPecT (version 2006.09.07) and X!Tandem (version 2007.01.01.2) were run with parent mass tolerance of 2.5 Da, fragment mass tolerance of 0.5 Da, fixed modification of Cys + 57, and no optional modifications and without any enzyme preference. The best match for each spectrum is reported. The parent masses of spectra were corrected according to the mass of the correct peptide. Table II illustrates that the accuracy of various tools decreases sharply with the increase in the spectrum length. PepNovo (a de novo search method) has similar or better accuracy than InsPecT in finding the correct peptide reconstruction. PepNovo version 1.03 was used with fixed Cys + 57 modification.
Table II.
Accuracy of InsPecT and X!Tandem against a database of all peptides estimated as the percentage of spectra for which the correct peptide will be identified with maximal score in the database search
PepNovo and MS-Dictionary accuracy (percentage of spectra for which the correct peptide is a top scoring peptide) is added for comparison. Peptides that differ by amino acid substitutions Ile/Leu and Lys/Gln with similar masses are considered valid reconstructions.
| Length | InsPecT | X!Tandem | PepNovo | MS-Dictionary |
|---|---|---|---|---|
| 7 | 63 | 51 | 54 | 57 |
| 8 | 59 | 47 | 51 | 58 |
| 9 | 48 | 41 | 45 | 51 |
| 10 | 42 | 35 | 38 | 50 |
| 12 | 18 | 22 | 23 | 35 |
| 14 | 16 | 11 | 12 | 18 |
We remark that in some applications (e.g. the search in large EST databases or using MS/MS for proteogenomics annotations (13, 14)), the databases are very large. It implies that the search in such databases (at least for shorter peptides) is not unlike the search in the database of all peptides. Table II leads to a surprising conclusion that for short peptides simply generating de novo reconstructions and matching them against the database may be a more accurate (and much faster) approach than X!Tandem/InsPecT in the case of very large databases. Below we show that MS-Dictionary leads to a better performance than InsPecT/X!Tandem/PepNovo in such applications (Fig. 9).
Fig. 9.

MS-Dictionary accuracy as a function of the spectrum length. The percentage of spectra that were correctly reconstructed by MS-Dictionary (i.e. the correct peptide was present in the spectral dictionary) is shown on the y axis. Accuracies are computed for three different values of SpectralProbability, viz. 10−10, 10−9, and 10−8. Comparison with PepNovo (counting the percentage of spectra for which PepNovo reconstructs the correct peptide) is shown. Because the number of reconstructions for length 14 aa and above is often larger than our allowed limit of 100,000 reconstructions per spectrum, the same set of reconstructions is generated for different SpectralProbability values.
Performance of MS-Dictionary—
The test data sets (all peptide identifications in Shewanella) were analyzed with MS-Dictionary for each peptide length. The size of the spectral dictionary depends on the SpectralProbability parameter of the generating function (38) that influences the error rate of peptide identifications if the spectrum was submitted to a database search. Because we deal with tryptic peptides, we only consider the reconstructions that end in Lys or Arg (although MS-Dictionary is not limited to tryptic peptides).9
As the spectrum length increases, the size of the peptide search space increases dramatically, making it harder to generate the spectral dictionary. Thus all de novo search methods yield lower accuracy for longer peptides. The generating function approach allows one to dynamically determine the number of peptide reconstructions and increase the chance of finding the correct peptide in the set of de novo reconstructions (see Fig. 9).
The number of reconstructions obtained for these same length spectra varies over orders of magnitude. Although the peak of the distribution of the number of reconstruction is at log2(size of spectral dictionary) ≈ 10 (comparable to the number of reconstructions generated in Alves and Yu (7)), some of these spectra have fewer than 100 or more than 10,000 reconstructions. This remarkable variance in the size of spectral dictionaries illustrates the point that different spectra have a different number of plausible reconstructions and raises a concern about de novo methods that return a fixed number of peptides.
Recently Frank et al. (46) described de novo peptide sequencing for data acquired from FT-ICR instruments when both the parent mass and the peak positions are accurate. However, acquiring such spectra remains time-consuming, and an intermediate approach that is gaining prominence is to acquire mass spectra with high precision at MS1 stage and lower precision at MS/MS stage, giving accurate parent mass but inaccurate peak positions. However, the existing de novo search methods are aimed toward ion traps or other low accuracy mass spectrometers, which may have parent mass errors on the order of 1 dalton. Because vertices in the spectrum graph are constructed based on low accuracy peaks, it is not clear how to exploit the accurate parent mass information that is available from new high accuracy instruments. Availability of accurate parent mass values can be effectively utilized in MS-Dictionary to filter the reconstructions. The number of reconstructions for 5-ppm accuracy is typically 4–16 times smaller than the corresponding numbers for 0.5-dalton accuracy (data are not shown).
Using MS-Dictionary for Database Search—
In any database search, a large number of spectra remain unidentified. This may happen due to several reasons: these spectra may have many missing or noisy peaks making them difficult to interpret, the corresponding peptide may not be present in the database, or the peptide may have a post-translational modification not captured by the search algorithm. In the case of S. oneidensis MR-1, only ≈10% of the 14.5 million spectra were reliably identified (14). We show that MS-Dictionary is able to find identifications for some previously unidentified spectra.
We selected all (≈600 thousands) spectra of charge 2 from the Shewanella data set within the parent mass range from 1100 to 1200 Da (the typical mass range for length 10 peptides). All these spectra were searched against the Shewanella proteome with MS-Dictionary (generated with spectral probability 1e−9), InsPecT, and X!Tandem. The same analysis was repeated with a decoy database of the same size. A spectrum is considered identified if any of the reconstructions are present in the Shewanella database (target database). Fig. 10 demonstrates that InsPecT and MS-Dictionary significantly improve on X!Tandem (at 5% FDR, X!Tandem, InsPecT, and MS-Dictionary identified 3272, 4184, and 4137 peptides, respectively). We further rescored InsPecT identifications using MS-GF spectral probabilities achieving an even better performance for the hybrid InsPecT ⊕ MS-GF hybrid tool (4299 peptide identifications at 5% FDR). Fig. 11 shows the Venn diagrams of peptides identified by X!Tandem, InsPecT, MS-Dictionary, and InsPecT ⊕ MS-GF. To further illustrate the applicability of MS-Dictionary in proteogenomics applications we extended the analysis of Shewanella proteome described above (Fig. 10) to the 7 times larger six-frame translation of Shewanella. We selected all spectra from the Shewanella data set that were not identified in the InsPecT database search with the ParentMass range from 1100 to 1200 Da and with MS-GF scores above 50 (24,814 spectra).10 MS-Dictionary generated spectral dictionaries for these spectra at three different values of SpectralProbability. The same analysis was repeated with a decoy database of the same size. A spectrum is considered identified if any of the reconstructions are present in the six-frame translation of the Shewanella genome (target database). Table III shows the number of new peptides identified by MS-Dictionary in each database that were not found in the earlier database search. For SpectralProbability = 10−10, 1007 new peptides are identified from 6211 spectra in the target database, whereas only six peptides (from six spectra) are identified in the decoy database, corresponding to a peptide-level false discovery rate of 0.6%. As the SpectralProbability is lowered, the false discovery rate turns into zero at 2 × 10−11) with 794 peptide identifications. 280 of them were identified previously by InsPecT (from other higher quality spectra), but 514 represent new peptide identifications. Interestingly 512 (99.6%) of them map to the known protein sequences (including contaminants), providing further confirmation that these identifications are correct. Indeed because the size of the Shewanella protein database is only ≈15% of the size of six-frame Shewanella translation, one expects that only 15% of these proteins would hit the Shewanella database by chance. Moreover of 512 peptides, 508 are matched to expressed proteins (confirmed by at least two InsPecT identifications in Gupta et al. (14)), and two are matched to proteins with a single identified peptide, confirming the expression of these proteins. Supplemental Table S2 lists each of the new peptide identifications.
Fig. 10.

Comparison of the number of peptide identifications by various approaches, viz. InsPecT, X!Tandem, MS-Dictionary, and InsPecT ⊕ MS-GF. The searches were performed with spectra of charge 2 from the Shewanella data set within the parent mass range from 1100 to 1200 Da. The curves display the number of peptide identifications for different score thresholds (corresponding to different false discovery rates).
Fig. 11.

Venn diagram showing the overlap between peptides identified by different approaches at 5% false discovery rate. a, overlap between InsPecT, X!Tandem, and MS-Dictionary. b, overlap between InsPecT, X!Tandem, and InsPecT ⊕ MS-GF.
Table III.
MS-Dictionary identification of Shewanella spectra that were not identified in the InsPecT search in Gupta et al. (14)
For different values of SpectralProbability (first column), the number of peptide identifications (IDs) on the target database (second column) and the decoy database (third column) are reported. The numbers in parentheses represent the corresponding number of spectral identifications (many spectra correspond to the same peptide identification). The target database here is the six-frame translation of the whole Shewanella genome containing ≈10 million aa, and a decoy database of the same size is used. The fourth column provides the FDR at the peptide level (ratio of decoy and target database peptide identifications), and the fifth column specifies the number of new peptides identified in the target database that were not observed in the InsPecT search. The number in parentheses in the last column shows the number of new peptides mapped to the protein-coding regions and illustrates that although the protein database is only 15% of the size of the six-frame translation 97.1–99.6% of these peptides are mapped to the protein database. These peptides are missed by InsPecT either because of borderline p values (as shown in Kim et al. (38), the generating function of MS-Dictionary results in better separation between correct and erroneous hits than the scoring functions of InsPecT and X!Tandem) or because of the absence of good peptide sequence tags.
| SpectralProbability | IDs
|
FDR | New peptides | |
|---|---|---|---|---|
| Target | Decoy | |||
| 1e−9 | 1169 (8771) | 29 (64) | 0.025 | 768 (746) |
| 1e−10 | 995 (6171) | 6 (6) | 0.006 | 652 (646) |
| 5e−11 | 914 (5327) | 2 (2) | 0.002 | 595 (591) |
| 2e−11 | 794 (4269) | 0 (0) | 0 | 514 (512) |
A closer look at the two peptides that fall outside the annotated proteins reveals two frameshifts. The first peptide, IAVGLSSANFGR, maps downstream of the gene SO_2754, which is annotated as “hypothetical sodium-type flagellar protein MotY” and has length 122 aa. Basic local alignment search tool (BLAST) (49) query of the peptide against other Shewanella strains shows that the peptide is conserved in four other strains and contained in longer proteins of length 289. By aligning the nucleotide sequence of S. oneidensis MR-1 against these other strains, we find a sequencing error (insertion of an extra A at nucleotide position 362) that results in a stop codon and early truncation of the gene with only 122 amino acids. The second peptide, SDIGWGSQIR, falls in the region of the gene SO_0991 (peptide chain release factor 2), which is now annotated in The Institute for Genomic Research (TIGR) as a programmed frameshift (but has the correct protein sequence missing from fasta files because of the frameshift). These examples show that new peptide identifications from MS-Dictionary not only increase coverage for annotated genes but also provide clues for correcting gene annotations.
We note that peptide identifications reported here are based on the spectra in the 1100–1200-Da parent mass range only, and their number is expected to be much larger if spectra of other masses are also included. Supplemental Table S3 shows that spectra in lower or higher mass ranges also show similar trends as spectra in the 1100–1200-Da range. MS-Dictionary thus has the potential to provide a significant number of new peptide identifications from spectra that were missed in the traditional database searches.
Searching the Six-frame Translation of the Human Genome with MS-Dictionary—
Although mass spectrometry has been successfully used for bacterial gene predictions (8–11, 14, 15, 50), the proteogenomics studies of large eukaryotic genomes are still in infancy. Even the fastest MS/MS database search tools become impractical in such studies because they require searches in huge databases resulting from the six-frame translations of eukaryotic genomes (≈2.5 billion amino acids for repeat-masked human genome). Tanner et al. (13) and Edwards (51) made a step toward proteogenomics searches of the human genome by combining the EST and MS/MS analysis. Although this approach is very valuable it can only be successful if the same exons are supported by both EST and MS/MS data. The largest proteogenomics analysis conducted so far is the search of the six-frame translation of A. thaliana that resulted in the discovery of nearly 800 new genes using InsPecT.2 Although InsPecT is 10 times faster than X!Tandem and 60 times faster than SEQUEST (see Payne et al. (52)), it becomes too slow in searches of the translated mammalian genomes. Because neither InsPecT nor X!Tandem can search the translated human genome,11 we ran InsPecT on a 124 times smaller database and assumed that its running time is proportional to the database size. The running time of InsPecT is estimated at 42 s per spectrum,12 whereas MS-Dictionary takes less than 1 s per spectrum on average on a desktop machine with a 2.16-GHz Intel processor. Below we demonstrate that MS-Dictionary can search the translated human genome and identify over 10,000 human peptides with low FPR. Recently Tanner et al. (13) demonstrated that such peptides can significantly improve the accuracy of traditional de novo gene prediction tools and boosted the accuracy of GeneID predictions by 0.65 correct exons per gene on average.
MS-Dictionary generates the spectral dictionary for each spectrum and uses fast pattern matching to match the spectral dictionary against the indexed database.13 We used a simple partitioning/indexing that divides the translated human genome into 124 equally sized subgenomes. Generating a spectral dictionary with 10,000 reconstructions takes 0.1 s per spectrum, and pattern matching of a spectral dictionary against all 124 databases (including file input and output overhead) takes 0.8 s per spectral dictionary on average. This results in less than 1-s running time, a 40-fold speedup over InsPecT.14
To benchmark MS-Dictionary we used the human HEK293 MS/MS data set generated in Steve Briggs’ laboratory. We focus on 48,926 doubly charged peptides with tryptic C terminus identified by InsPecT15 (InsPecT version 20070613, human IPI database version 3.18) with 2.5% false discovery rate (for a detailed description see Refs. 13 and 53). We removed 17,821 peptides that span the exon boundaries (these peptides cannot be identified by searching the translated human genome) resulting in 31,105 peptides. Because most peptides in HEK293 are represented by multiple spectra, we randomly selected one spectrum of all spectra of the same peptide. We further searched 31,105 spectra against the translated human genome (version 48 from Ensembl) with masked repeats and with corrected parent mass as described before. For each spectrum, we generated a spectral dictionary with SpectralProbability = 10−11 and limited the maximum size of spectral dictionaries to 10,000. Each peptide in the spectral dictionary was matched (without errors) against the translated human genome.
The searches in the translated human genome are not expected to identify all spectra reliably identified in the human protein database. Indeed Castellana et al.2“lost” ≈30% of all identifications of peptides falling within exons after switching from the protein database to the translated genome database of A. thaliana. Such losses are unavoidable because many reliable identifications in the protein database turn into statistically insignificant identifications in the much larger translated genome. For example, although SpectralProbability = 10−10 makes sense for searching the human protein database, it results in very high error rates (FPR = 25%) in a ≈100 times larger translated human genome. Therefore, all peptide identifications with SpectralProbability ≥ 10−10 will be lost after switching from the protein database to the translated human genome.16 We have therefore chosen SpectralProbability = 10−11 as a threshold resulting in estimated FPR = DatabaseSize·SpectralProbability = 2.5·109·10−11 = 0.025. Because 9470 of 31,105 peptides (30%) have SpectralProbability exceeding 10−11, they cannot be identified in any sensible database search against the translated human genome. It leaves us with 21,635 peptides that can be potentially identified in the translated human genome.
MS-Dictionary identified 10,266 of 21,635 spectra in the translated human genome. 98.9% of the identified peptides fall into the human proteins, and only 1.1% fall into non-coding regions.17 To further estimate FPR of our experiment, we selected a single run (25,746 spectra), picked out unidentified doubly charged spectra in this run (16,205 spectra), and used MS-Dictionary to generate spectral dictionaries and match them against the translated human genome. MS-Dictionary identified only 71 spectra in this experiment, corresponding to an FPR of 0.44%.
Therefore, MS-Dictionary reliably identifies ≈10,000 peptides from human proteins without knowing the human proteome. However, it also “loses” ≈11,000 peptides that can be potentially identified in searches of the translated human genome. Fig. 12 illustrates that although MS-Dictionary identifies a large fraction of peptides of length 10–13 the performance deteriorates for shorter and longer peptides. Because the SpectralProbability threshold has to be low in proteogenomics applications, only very high quality spectra of shorter peptides represent reliable identifications (only 23% of spectra of length 9). This does not indicate the poor performance of MS-Dictionary but rather reflects the stringent threshold. For the spectra of length more than 14 aa, the performance of MS-Dictionary deteriorates because of the limited size of spectral dictionaries. Further algorithmic developments (e.g. generating dictionaries of long tags) are needed to address this shortcoming of MS-Dictionary.
Fig. 12.

The percentage of peptides identified by MS-Dictionary in the translated human genome as compared with all peptides identifies in searches of human protein database. Spectral dictionaries were generated for the 21,635 selected spectra from HEK293 data set and searched against the translated human genome. For each spectrum, if the correct peptide is contained in the dictionary of the spectrum, we regarded the spectrum as identified.
DISCUSSION
Here we demonstrate the importance of obtaining multiple de novo peptide reconstructions and describe the MS-Dictionary tool for generating these reconstructions. We emphasize that the number of generated reconstructions must not be fixed a priori, as done by existing de novo tools, but decided dynamically for the given spectrum because the number of plausible reconstructions varies from spectrum to spectrum. We use the generating function approach (38) that allows one to determine the set of reconstructions that must be reported. The ability to generate spectral dictionaries makes this method useful for hybrid de novo-based database search by increasing the likelihood of finding the correct peptide while keeping the number of false identifications low. MS-Dictionary identifies new peptides from spectra that were not identified with a regular database search. MS-Dictionary can be modified to search for mutations and polymorphisms by simply substituting the exact pattern matching by error-tolerant pattern matching of spectral dictionaries against databases.
Future work will focus on developing this hybrid approach into a viable tool for peptide identification by extending it to highly charged spectra and improving the efficiency of this approach in the case of longer peptides. Deteriorated performance for highly charged and long peptides is an important limitation of all de novo approaches to spectral interpretations. The existing de novo peptide sequencing tools are aimed at charge 2 peptides with the single exception of the greedy best strong tag algorithm (30) that is best suited for tag generation rather than full-length de novo peptide sequencing, which is the focus of this study. All tools we tested also deteriorated while searching longer peptides in very large databases. For example, InsPecT and X!Tandem would correctly identify only 16 and 11% of all length 14 peptides in the de novo peptide sequencing framework (Table II). Although MS-Dictionary improves on these tools, its accuracy is also rather low (18%). This observation reveals the shortcomings of existing de novo and database search tools that often score the incorrect peptides higher than the correct peptides. Frank et al. (46) recently discussed the “homeometric peptides” that represent the key obstacle for developing better de novo algorithms (they become more pronounced with the increase in the peptide length). This problem is partially alleviated by generating all reconstructions with a given SpectralProbability and further matching them against a database (Fig. 9).
Supplementary Material
Acknowledgments
We thank Vineet Bafna, Ari Frank, Sam Payne, and Stephen Tanner for useful discussions. We thank Steve Briggs and Dick Smith for sharing MS/MS data generated in their laboratories.
Footnotes
Published, MCP Papers in Press, August 14, 2008, DOI 10.1074/mcp.M800103-MCP200
N. Castellana, S. Payne, Z. Shen, M. Stanke, S. Briggs, and V. Bafna, submitted manuscript.
Spectral dictionaries are also helpful in searches for fusion peptides that are common in tumor proteomes but not explicitly present in protein databases (18).
Although MS-Dictionary compares well with X!Tandem and InsPecT for charge 2 spectra, the performance of all existing de novo tools (including MS-Dictionary and PepNovo) deteriorates for highly charged peptides (3+). The problem of de novo analysis of highly charged spectra has been addressed recently by Cao and Nesvizhskii (36).
Although it may appear that matching 1000 peptides against the database is rather time-consuming, the combinatorial pattern-matching algorithms (37) are able to do it in negligible time.
The problem of generating varying numbers of reconstructions for each spectrum becomes particularly important for long peptides. For instance, PepNovo (4) accurately reconstructs 54% of peptides of length 7 and only 0.4% of peptides of length 20.
Although the accuracy of MS-Dictionary in the standard de novo peptide sequencing improves on the state-of-the-art tool PepNovo (21), optimizing de novo peptide sequencing is an important but not the crucial goal for our main application. As Alves and Yu (7) pointed out, de novo peptide sequencing and spectral dictionary approaches have similar but distinct goals: an outstanding de novo algorithm is not a prerequisite for the spectral dictionary approach to perform well.
The abbreviations used are: DAG, directed acyclic graph; FDR, false discovery rate; EST, expressed sequence tag; aa, amino acids; FPR, false positive rate; Prob, probability.
The spectra were acquired on an ion trap MS (LCQ, ThermoFinnigan, San Jose, CA) using ESI. The program extract_msn (ThermoFinnigan) was used to generate the dta files with standard options.
Although this analysis loses peptides at the C terminus of proteins, it will have a minor effect on the reported statistics.
Although most spectra with MS-GF scores above 50 correspond to high quality peptide identifications by both InsPecT and X!Tandem, a significant portion of them may have borderline InsPecT/X!Tandem scores. As discussed previously (38), such low scores may reflect deficiencies of the underlined scoring approaches.
Both tools report unexpected errors on the translated human genome.
This is a lower bound that does not account for overhead caused by indexing/partitioning of large databases.
Indexing the entire six-frame translation of the human genome takes less than an hour.
We estimate that optimized indexing/partitioning or running MS-Dictionary on a large shared memory machine would further reduce the running time.
Although MS-Dictionary generates both tryptic and non-tryptic peptides, we selected doubly charged peptides with tryptic C terminus to simplify benchmarking.
In particular, all peptides of length 8 and shorter are likely to be lost because SpectralProbability even of a single peptide of length 8 is rather high (≈0.4·10−10).
Although most spectral dictionaries have zero or one hit in the human genome, 1.8% of them have multiple hits (typically two hits).
This work was supported, in whole or in part, by National Institutes of Health Grant 5R01RR016522-05. This work was also supported a by Howard Hughes Medical Institute professor award. The costs of publication of this article were defrayed in part by the payment of page charges. This article must therefore be hereby marked “advertisement” in accordance with 18 U.S.C. Section 1734 solely to indicate this fact.
The on-line version of this article (available at http://www.mcponline.org) contains supplemental material.
REFERENCES
- 1.Mann, M., and Wilm, M. ( 1994) Error-tolerant identification of peptides in sequence databases by peptide sequence tags. Anal. Chem. 66, 4390–4399 [DOI] [PubMed] [Google Scholar]
- 2.Tanner, S., Shu, H., Frank, A., Wang, L., Zandi, E., Mumby, M., Pevzner, P., and Bafna, V. ( 2005) InsPecT: identification of posttranslationally modified peptides from tandem mass spectra. Anal. Chem. 77, 4626–4639 [DOI] [PubMed] [Google Scholar]
- 3.Shilov, I., Seymour, S., Patel, A., Loboda, A., Tang, W., Keating, S., Hunter, C., Nuwaysir, L., and Schaeffer, D. ( 2007) The Paragon Algorithm: a next generation search engine that uses sequence temperature values and feature probabilities to identify peptides from tandem mass spectra. Mol. Cell. Proteomics 6, 1638–1655 [DOI] [PubMed] [Google Scholar]
- 4.Frank, A., Tanner, S., Bafna, V., and Pevzner, P. ( 2005) Peptide sequence tags for fast database search in mass-spectrometry. J. Proteome Res. 4, 1287–1295 [DOI] [PubMed] [Google Scholar]
- 5.Liu, C., Yan, B., Song, Y., Xu, Y., and Cai, L. ( 2006) Peptide sequence tag-based blind identification of post-translational modifications with point process model. Bioinformatics 22, e307–e313 [DOI] [PubMed] [Google Scholar]
- 6.Taylor, J., and Johnson, R. ( 1997) Sequence database searches via de novo peptide sequencing by tandem mass spectrometry. Rapid Commun. Mass Spectrom. 11, 1067–1075 [DOI] [PubMed] [Google Scholar]
- 7.Alves, G., and Yu, Y. ( 2005) Robust accurate identification of peptides (RAId): deciphering MS2 data using a structured library search with de novo based statistics. Bioinformatics 21, 3726–3732 [DOI] [PubMed] [Google Scholar]
- 8.Jaffe, J., Berg, H., and Church, G. ( 2004) Proteogenomic mapping as a complementary method to perform genome annotation. Proteomics 4, 59–77 [DOI] [PubMed] [Google Scholar]
- 9.Kalume, D., Peri, S., Reddy, R., Zhong, J., Okulate, M., Kumar, N., and Pandey, A. ( 2005) Genome annotation of Anopheles gambiae using mass spectrometry-derived data. BMC Genomics 6, 128–138 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Wang, R., Prince, J., and Marcotte, E. ( 2005) Mass spectrometry of the M. smegmatis proteome: protein expression levels correlate with function, operons, and codon bias. Genome Res. 15, 1118–1126 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Fermin, D., Allen, B., Blackwell, T., Menon, R., Adamski, M., Xu, Y., Ulintz, P., Omenn, G., and States, D. ( 2006) Novel gene and gene model detection using a whole genome open reading frame analysis in proteomics. Genome Biol. 7, R35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Savidor, A., Donahoo, R., Hurtado-Gonzales, O., VerBerkmoes, N., Shah, M., Lamour, K., and McDonald, W. ( 2006) Expressed peptide tags: an additional layer of data for genome annotation. J. Proteome Res. 5, 3048–3058 [DOI] [PubMed] [Google Scholar]
- 13.Tanner, S., Shen, Z., Ng, J., Florea, L., Guigo, R., Briggs, S., and Bafna, V. ( 2007) Improving gene annotation using peptide mass spectrometry. Genome Res. 17, 231–239 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Gupta, N., Tanner, S., Jaitly, N., Adkins, J., Lipton, M., Edwards, R., Romine, M., Osterman, A., Bafna, V., Smith, R., and Pevzner, P. ( 2007) Whole proteome analysis of post-translational modifications: applications of mass-spectrometry for proteogenomic annotation, Genome Res. 17, 1362–1377 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Gupta, N., Benhamida, J., Bhargava, D., Goodman, E., Kain, I., Nguyen, N., Ollikainen, N., Rodriguez, J., Wang, J., Lipton, M., Romine, M., Bafna, V., Smith, R., and Pevzner, P. ( 2008) Comparative proteogenomics: combining mass spectrometry and comparative genomics to analyze multiple genomes. Genome Res. 18, 1133–1142 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Siepel, A., Diekhans, M., Brejová, B., Langton, L., Stevens, M., Comstock, C., Davis, C., Ewing, B., Oommen, S., Lau, C., Yu, H., Li, J., Roe, B., Green, P., Gerhard, D., Temple, G., Haussler, D., and Brent, M. ( 2007) Targeted discovery of novel human exons by comparative genomics. Genome Res. 17, 1763–1773 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Stark, A., Lin, M., Kheradpour, P., Pedersen, J., Parts, L., Carlson, J., Crosby, M., Rasmussen, M., Roy, S., Deoras, A., Ruby, J., Brennecke, J., Hodges, E., Hinrichs, A., Caspi, A., Paten, B., Park, S., Han, M., Maeder, M., Polansky, B., Robson, B., Aerts, S., van Helden, J., Hassan, B., Gilbert, D., Eastman, D., Rice, M., Weir, M., Hahn, M., Park, Y., Dewey, C., Pachter, L., Kent, W., Haussler, D., Lai, E., Bartel, D., Hannon, G., Kaufman, T., Eisen, M., Clark, A., Smith, D., Celniker, S., Gelbart, W., and Kellis, M. ( 2007) Discovery of functional elements in 12 Drosophila genomes using evolutionary signatures. Nature 450, 219–232 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Ng, J., and Pevzner, P. ( 2008) Algorithm for identification of fusion proteins via mass spectrometry. J. Proteome Res. 7, 89–95 [DOI] [PubMed] [Google Scholar]
- 19.Taylor, J., and Johnson, R. ( 2001) Implementation and uses of automated de novo peptide sequencing by tandem mass spectrometry. Anal. Chem. 73, 2594–2604 [DOI] [PubMed] [Google Scholar]
- 20.Dancık, V., Addona, T., Clauser, K., Vath, J., and Pevzner, P. ( 1999) De novo protein sequencing via tandem mass-spectrometry. J. Comp. Biol. 6, 327–341 [DOI] [PubMed] [Google Scholar]
- 21.Frank, A., and Pevzner, P. ( 2005) PepNovo: de novo peptide sequencing via probabilistic network modeling. Anal. Chem. 77, 964–973 [DOI] [PubMed] [Google Scholar]
- 22.Ma, B., Zhang, K., Hendrie, C., Liang, C., Li, M., Doherty-Kirby, A., and Lajoie, G. ( 2003) PEAKS: powerful software for peptide de novo sequencing by tandem mass spectrometry. Rapid Commun. Mass Spectrom. 17, 2337–2342 [DOI] [PubMed] [Google Scholar]
- 23.Bern, M., and Goldberg, D. ( 2006) De novo analysis of peptide tandem mass spectra by spectral graph partitioning. J. Comput. Biol. 13, 364–378 [DOI] [PubMed] [Google Scholar]
- 24.Fischer, B., Roth, V., Roos, F., Grossmann, J., Baginsky, S., Widmayer, P., Gruissem, W., and Buhmann, J. ( 2005) NovoHMM: a hidden Markov model for de novo peptide sequencing. Anal. Chem. 77, 7265–7273 [DOI] [PubMed] [Google Scholar]
- 25.Grossmann, J., Roos, F., Cieliebak, M., Liptak, Z., Mathis, L., Muller, M., Gruissem, W., and Baginsky, S. ( 2005) AUDENS: a tool for automated peptide de novo sequencing. J. Proteome Res. 4, 1768–1774 [DOI] [PubMed] [Google Scholar]
- 26.Mo, L., Dutta, D., Wan, Y., and Chen, T. ( 2007) MSNovo: a dynamic programming algorithm for de novo peptide sequencing via tandem mass spectrometry. Anal. Chem. 79, 4870–4878 [DOI] [PubMed] [Google Scholar]
- 27.Dimaggio, P., Jr., and Floudas, C. ( 2007) De novo peptide identification via tandem mass spectrometry and integer linear optimization. Anal. Chem. 79, 1433–1446 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Chen, T., Kao, M., Tepel, M., Rush, J., and Church, G. ( 2001) A dynamic programming approach to de novo peptide sequencing via tandem mass spectrometry. J. Comput. Biol. 8, 325–337 [DOI] [PubMed] [Google Scholar]
- 29.Bafna, V., and Edwards, N. ( 2003) On de novo interpretation of tandem mass spectra for peptide identification, in Proceedings of the Seventh Annual International Conference on Computational Molecular Biology, Berlin, April 10–13, 2003, pp. 9–18, Association for Computing Machinery, New York
- 30.Chong, K., Ning, K., Leong, H., and Pevzner, P. ( 2006) Modeling and characterization of multi-charge mass spectra for peptide sequencing. J. Bioinform. Comput. Biol. 4, 1329–1352 [DOI] [PubMed] [Google Scholar]
- 31.Craig, R., and Beavis, R. ( 2004) TANDEM: matching proteins with tandem mass-spectra. Bioinformatics 20, 1466–1467 [DOI] [PubMed] [Google Scholar]
- 32.Creasy, D., and Cottrell, J. ( 2002) Error tolerant searching of uninterpreted tandem mass spectrometry data. Proteomics 2, 1426–1434 [DOI] [PubMed] [Google Scholar]
- 33.Eng, J., McCormack, A., and Yates, J. ( 1994) An approach to correlate tandem mass-spectral data of peptides with amino acid sequences in a protein database. J. Am. Soc. Mass Spectrom. 5, 976–989 [DOI] [PubMed] [Google Scholar]
- 34.Clauser, K., Baker, P., and Burlingame, A. ( 1999) Role of accurate mass measurement (±10 ppm) in protein identification strategies employing MS or MS/MS and database searching. Anal. Chem. 71, 2871–2882 [DOI] [PubMed] [Google Scholar]
- 35.Geer, L., Markey, S., Kowalak, J., Wagner, L., Xu, M., Maynard, D., Yang, X., Shi, W., and Bryant, S. ( 2004) Open mass spectrometry search algorithm. J. Proteome Res. 3, 958–964 [DOI] [PubMed] [Google Scholar]
- 36.Cao, X., and Nesvizhskii, A. ( 2008) Improved sequence tag identification method for peptide identification in tandem mass spectrometry. J. Proteome Res. 7, 4422–4434 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Gusfield, D. ( 1997) Algorithms on Strings, Trees and Sequences: Computer Science and Computational Biology, pp. 87–123, Cambridge University Press, Cambridge, UK
- 38.Kim, S., Gupta, N., and Pevzner, P. ( 2008) Spectral probabilities and generating functions of tandem mass spectra: a strike against decoy databases. J. Proteome Res. 7, 3354–3363 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Havilio, M., Haddad, Y., and Smilansky, Z. ( 2003) Intensity-based statistical scorer for tandem mass spectrometry. Anal. Chem. 75, 435–444 [DOI] [PubMed] [Google Scholar]
- 40.Colinge, J., Masselot, A., Giron, M., Dessingy, T., and Magnin, J. ( 2003) OLAV: towards high-throughput tandem mass spectrometry data identification. Proteomics 3, 1454–1463 [DOI] [PubMed] [Google Scholar]
- 41.Bandeira, N., Olsen, J., Mann, M., and Pevzner, P. ( 2008) Multi-spectra peptide sequencing and its applications to multistage mass spectrometry. Bioinformatics 24, i416–i423 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Bartels, C. ( 1990) Fast algorithm for peptide sequencing by mass spectroscopy. J. Proteome Res. 19, 363–368 [DOI] [PubMed] [Google Scholar]
- 43.Lu, B., and Chen, T. ( 2003) A suffix tree approach to the interpretation of tandem mass spectra: applications to peptides of non-specific digestion and post-translational modifications. Bioinformatics 19, Suppl. 2, ii113–ii121 [DOI] [PubMed] [Google Scholar]
- 44.Matthiesen, R., Trelle, M., Højrup, P., Bunkenborg, J., and Jensen, O. ( 2005) Vems 3.0: algorithms and computational tools for tandem mass spectrometry based identification of post-translational modifications in proteins. J. Proteome Res. 4, 2338–2347 [DOI] [PubMed] [Google Scholar]
- 45.Ma, B., Zhang, K., and Liang, C. ( 2005) An effective algorithm for peptide de novo sequencing from MS/MS spectra. J. Comput. Syst. Sci. 70, 418–430 [Google Scholar]
- 46.Frank, A., Savitski, M., Nielsen, M., Zubarev, R., and Pevzner, P. ( 2007) De novo peptide sequencing and identification with precision mass spectrometry. J. Proteome Res. 6, 114–123 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Zhang, Z. ( 2004) De novo peptide sequencing based on a divide-and-conquer algorithm and peptide tandem spectrum simulation. Anal. Chem. 76, 6374–6383 [DOI] [PubMed] [Google Scholar]
- 48.Bafna, V., and Edwards, N. ( 2001) Scope: a probabilistic model for scoring tandem mass spectra against a peptide database. Bioinformatics 17, Suppl. 1, 13–21 [DOI] [PubMed] [Google Scholar]
- 49.Altschul, S., Gish, W., Miller, W., Myers, E., and Lipman, D. ( 1990) Basic local alignment search tool. J. Mol. Biol. 215, 403–410 [DOI] [PubMed] [Google Scholar]
- 50.Jaffe, J., Stange-Thomann, N., Smith, C., DeCaprio, D., Fisher, S., Butler, J., Calvo, S., Elkins, T., FitzGerald, M., Hafez, N., Kodira, C., Major, J., Wang, S., Wilkinson, J., Nicol, R., Nusbaum, C., Birren, B., Berg, H., and Church, G. ( 2004) The complete genome and proteome of Mycoplasma mobile. Genome Res. 14, 1447–1461 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Edwards, N. ( 2007) Novel peptide identification from tandem mass spectra using ESTs and sequence database compression. Mol. Syst. Biol. 3, 102–108 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Payne, S., Yau, M., Smolka, M., Tanner, S., Zhou, H., and Bafna, V. ( 2008) Phosphorylation specific ms/ms scoring for rapid and accurate phospho-proteome analysis. J. Proteome Res. 7, 3373–3381 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Frank, A., Bandeira, N., Shen, Z., Tanner, S., Briggs, S., Smith, R., and Pevzner, P. ( 2008) Clustering millions of tandem mass spectra. J. Proteome Res. 7, 113–122 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.