

Abstract
The sphingomyelin-ceramide pathway is an evolutionarily conserved ubiquitous signal transduction system that regulates many cell functions including apoptosis. Sphingomyelin (SM) is hydrolyzed to ceramide by different sphingomyelinases. Ceramide serves as a second messenger in mediating cellular effects of cytokines and stress. In this study, we find that acid sphingomyelinase (SMase) activity was induced by UVA in normal JY lymphoblasts but was not detectable in MS1418 lymphoblasts from Niemann-Pick type D patients who have an inherited deficiency of acid SMase. We also provide evidence that UVA can induce apoptosis by activating acid SMase in normal JY cells. In contrast, UVA-induced apoptosis was inhibited in MS1418 cells. Exogenous SMase and its product, ceramide (10–40 μm), induced apoptosis in JY and MS1418 cells, but the substrate of SMase, SM (20–80 μm), induced apoptosis only in JY cells. These results suggest that UVA-induced apoptosis by SM is dependent on acid SMase activity. We also provide evidence that induction of apoptosis by UVA may occur through activation of JNKs via the acid SMase pathway.
Cell death is an irreversible process that culminates in cessation of biological activity (1-3) and can occur through apoptosis or necrosis (4-11). Apoptosis is an active and physiological mode of cell death and is well characterized by morphological changes including cell shrinkage, cytoplasmic blebbing, chromatin condensation, and DNA fragmentation (2). In recent years, substantial progress has been made in understanding the multistep regulatory mechanisms that are associated with the propensity of a cell to respond to various stimuli with apoptosis (1, 2, 7, 8). The regulatory system involves the presence of at least two distinct checkpoints, one controlled by the Bcl-2/Bax family of proteins (5-7) and the other by the cysteine and possibly serine proteases (2, 7, 8, 12, 20-22). In addition, mitochondria (9, 13) and the sphingomyelin (SM)1-ceramide pathway (11, 12, 14-17) play important roles in apoptotic signal transduction. These systems interact with the machinery regulating cell proliferation and DNA repair through several oncogenes and tumor suppressor genes such as p53 (2). Hence, antitumor strategies based on modulation of the propensity of the cell to undergo apoptosis attract great interest in oncology.
Sphingolipids such as SM had been previously regarded as metabolically inactive, functioning only as structural components of the membrane (17, 18). However, besides its structural role in biomembranes, SM plays a pivotal role in signal transduction and regulation of cellular functions including growth, differentiation, proliferation, and apoptosis (14-18). A number of studies have demonstrated that extracellular cytokines and stress stimuli, such as TNFα, interleukin-1β, FAS ligand, heat shock, γ-radiation (23), and UVC irradiation (12, 14), cause the activation of sphingomyelinase (SMase) and the release of ceramide. Ceramide, a product of SMase-catalyzed hydrolysis of SM, was shown to act as a lipid second messenger or biomodulator of diverse stress-related responses including cell cycle arrest, cell senescence, and apoptosis (14-22). This pathway is referred to as the SM cycle, SM-ceramide pathway (11, 17), or the SMase pathway. To date, at least seven classes of mammalian SMases have been described, differing in subcellular location, pH optimum, cation dependence, and roles in cell regulation (11, 15, 17). Two forms of SMases, distinguishable by their pH optima, are capable of initiating signal transduction (12). The acid SMase (pH optimum 4.5–5.0) is activated in cells exposed to ionizing radiation, FAS, CD28, interleukin-1, or TNFα (23, 24). The neutral SMase (pH optimum 7.4) has been implicated in mediating apoptosis in cells exposed to serum starvation, anti-FAS antibody, vitamin D, TNFα, or cytosine arabinoside (25, 26).
Solar ultraviolet (UV) radiation is known to be one of the most common environmental carcinogens leading to skin cancer (27-29). Also, UV exposure induces apoptosis in cultured cells and in vivo (29, 30). Most research has focused on the UVC (200–290 nm) and UVB (290–320 nm) induction of apoptosis (31, 32), and little is known about the effect of UVA (320–400 nm), which comprises over 90% of the solar UV. Here, we observe that acid SMase is activated by UVA, and we provide evidence that UVA-induced apoptosis is dependent on acid SMase activity. Exogenous sphingomyelinase and its product, ceramide, also induce apoptosis independent of activation of intracellular SMase, but induction of apoptosis by SM is dependent on the SMase activity. Our data further indicate that UVA-induced apoptosis may occur through activation of JNKs via the SMase pathway.
MATERIALS AND METHODS
Reagents
Dulbecco's modified Eagle's medium (DMEM), RPMI 1640, penicillin, streptomycin, l-glutamine, and fetal bovine serum (FBS) were purchased from Life Technologies, Inc. C2-ceramide (N-acetyl-d-erythro-sphingosine), a biologically active cell-permeant ceramide analog, C2-dihydroceramide (N-acetyl-d-erythro-sphinganine) that is inactive and may be used as a negative control for C2-ceramide, and SM from bovine brain used as a precursor to ceramide second messengers via the action of sphingomyelinase, were purchased from BioMol Inc. (Plymouth Meeting, PA). These three sphingolipids were dissolved in dimethyl sulfoxide (Me2SO) from Pierce. Acid SMase, pH 4.5, from human placenta, phosphocholine, aprotinin, leupeptin, 12-O-tetradecanoylphorbol-13-acetate (TPA), PD98059, SB2020190, and Hoechst 33258 (bisbenzimide, 2-(4-hydroxyphenyl)-5-(4-methyl-1-piperazinyl)-2,5-bi-1H-benzimidazole trihydrochloride pentahydrate) were from Sigma.
Cell Culture
Epstein-Barr virus-transformed normal human lymphoblast cell lines, JY, or Niemann-Pick lymphoblast cell lines, MS1418 (23), were a generous gift from Dr. Richard Kolesnick, Laboratory of Signal Transduction, Memorial Sloan-Kettering Cancer Center, New York. The two cell lines were maintained in a mixture of RPMI 1640 and DMEM (1:1, v/v) containing 15% FBS, 2 mm l-glutamine, 100 units/ml penicillin, and 100 μg/ml streptomycin, and cultured in a humidified atmosphere of 5% CO2, 95% air at 37 °C.
UVA, UVB, and UVC Irradiation of Cells
The UVA source used was a Philips TL100 watt/10R lamp obtained from Ultraviolet Resources International (Lakewood, OH). UVA irradiation, filtered through about 6 mm of plate glass to eliminate UVB and UVC generated by the UVA lamps, was administered to cultured cells in the UVA box with two ventilation fans installed to eliminate UVA thermal stimulation. The doses of UVA irradiation were 20, 40, 60, or 80 kJ/m2 as designated. The UVB irradiation was carried out in a UVB chamber that was fitted with a Kodak Kodacel K6808 filter that eliminates all wavelengths below 290 nm. The UVC radiation was from germicidal lamps. The cultured cells were or were not starved by replacing medium with 0.5% FBS/DMEM/RPMI mixed medium (for JY or MS1418 cells), and then cells were exposed to UVA, UVB, or UVC at various doses as described.
C2-ceramide, SM, and SMase Treatments
Normal human lymphoblast JY cells or acid SMase-deficient lymphoblast MS1418 cells (3 × 106 to 5 × 106) (23, 33) were seeded into 100-mm dishes or wells of 6-well plates and cultured for 24 h in the mixture of RPMI 1640 and DMEM (1:1, v/v) containing 15% FBS, 2 mm l-glutamine, 100 units/ml penicillin, and 100 μg of streptomycin/ml in a humidified atmosphere of 5% CO2, 95% air at 37 °C. After having been replaced with freshly mixed medium containing 0.5% FBS, these cells were cultured for an additional 30 min and were treated with C2-ceramide (10–40 μm), C2-dihydroceramide (10–40 μm), SM (20–80 μm), SMase (0.5–2.5 units/ml), or phosphocholine (20 or 200 μm). Me2SO was used as an internal negative control. These differentially treated cells were incubated and then harvested after various times as indicated.
Internucleosomal DNA Fragmentation Ladder Assay
DNA fragmentation visualized by agarose gel electrophoresis (21, 30, 33) is considered a biochemical hallmark for apoptosis. Briefly, JY or MS1418 cells (3 × 106 to 5 × 106) were cultured in 100-mm dishes and treated with UVA, UVB, or UVC irradiation or with C2-ceramide, C2-dihydroceramide, SM, SMase, or phosphocholine and then harvested by pipetting and centrifuging at 1,500 × g. The cells were lysed with buffer containing 5 mm Tris-HCl, pH 8.0, 20 mm EDTA, and 0.5% Triton X-100, and left on ice overnight at 4 °C. After centrifugation at 17,000 × g for 45 min at 4 °C, fragmented DNA in the supernatant fraction was extracted twice with phenol/chloroform/isopropyl alcohol (25:24:1, v/v) and once with chloroform and then precipitated overnight at −20 °C with 100% ethanol and 5 m NaCl. The DNA pellet was saved by centrifuging at 17,000 × g for 45 min at 4 °C and then washed once with 70% ethanol and resuspended in Tris-HCl, pH 8.0, with 100 μg/ml RNase at 37 °C for 2 h. The DNA fragments were separated by 1.8% agarose gel electrophoresis. DNA laddering in the gel was stained with ethidium bromide and photographed in UV light.
Detection of Nuclear Chromatin Condensation and Nuclear Fragmentation
Morphological changes characteristic of apoptosis were detected with Hoechst staining (34, 35). Briefly, exponentially growing JY or MS1418 cells (1 × 106) were irradiated with UVA, UVB, or UVC at the doses indicated. At 2–12 h after irradiation, cells were harvested by centrifuging at 1,500 × g for 5 min at 4 °C and washed twice with ice-cold PBS and then fixed in 1% formaldehyde in PBS, pH 7.4, for 15 min on ice and postfixed in 70% ethanol and stored at −20 °C up to 4 days. Again, the cells were washed twice in PBS, and pellets were resuspended in Hoechst staining buffer containing 20 μg/ml Hoechst 33258 and 2% Me2SO in PBS and incubated for 30 min at 37 °C in the dark. Nuclei were visualized using a fluorescence microscope. In each sample, a minimum of 400 cells was counted. The condensed, compacted, and fragmented nuclei were counted and expressed as a percentage of the total nuclei.
Assay for Acid SMase Activity in Preparations from Irradiated Cells
The enzymatic hydrolysis of SM to ceramide and phosphocholine by SMase was measured at pH 5.0 with the Amplex Red reaction kit (Molecular Probes Inc., Eugene, OR) (38). Briefly, after irradiation of JY or MS1418 cells (2.3 × 106) with UVA (80 kJ/m2), UVB (8 kJ/m2), or UVC (60 J/m2), cells were pelleted by centrifugation at 1,500 × g for 10 min at 4 °C and washed twice with ice-cold PBS. The cell pellet was resuspended and lysed in 0.6 ml of buffer containing 50 mm sodium acetate, pH 5.0, 1% Triton X-100, 1 μg/ml aprotinin, 1 mm EDTA, and 100 μg/ml phenylmethylsulfonyl fluoride for 60 min on ice. Then the supernatant fraction was saved by centrifugation at 17,000 × g for 10 min at 4 °C to remove nuclei. The protein concentration in the supernatant fraction was measured with the Bio-Rad Protein Assay. JY or MS1418 cell membrane-free supernatant fractions (adjusted to pH 5.0) were assayed for SMase activity in a two-step reaction system. First, to generate phosphocholine and ceramide, 0.5 mm SM was added to the supernatant fraction and incubated for 60 min at 37 °C. The reaction was then placed on ice and the fluorogenic probe Amplex Red reagent (10-acetyl-3,7-dihydroxyphenoxazine), which is sensitive to H2O2, was added and further incubated at 37 °C for 60 min to generate H2O2 (through alkaline phosphatase hydrolysis of phosphocholine and choline oxidation by choline oxidase to generate betaine and H2O2). H2O2 in the presence of horseradish peroxidase reacts with Amplex Red to generate the fluorescent resorufin. Each reaction mixture contained 5 μm Amplex Red reagent, 1 unit/ml horseradish peroxidase, 0.1 unit/ml choline oxidase, 4 units/ml alkaline phosphatase, all supplied with the kit. For a detailed description of the assay see Zhou et al. (38). The fluorescence intensity was measured immediately at 610 nm (excitation at 560 nm) with a SPEX Fluoromax instrument (Instruments S.A., Inc. Edina, NJ), temperature-controlled to 37 ± 0.1 °C (Neslab, RTE-111, Neslab Instruments, Portsmouth, NH).
Measurement of Cellular Ceramide Levels
JY or MS1418 cells were starved for 12 h in medium containing 0.5% FBS. The cells were then harvested at 5, 15, or 30 min following irradiation with UVA (80 kJ/m2), UVB (8 kJ/m2), or UVC (60 J/m2). Lipids were extracted from the cell pellets with chloroform:methanol (2:1, v/v) (39). The lipids dissolved in chloroform were applied to solid phase extraction cartridges (50 mg Extract-Clean Silica, Alltech, Deerfield, IL), and ceramides were eluted with 2.5 ml of chloroform:methanol (98:2, v/v) after the neutral lipids had been eluted with 2 ml of chloroform. After acid hydrolysis of the ceramide fraction (5% v/v HCl:methanol, 70 °C, 3 h), free sphingosine was extracted into chloroform in the presence of the internal standard, 0.1 μg of C16 dihydrosphingosine (Matreya, Pleasant Gap, PA). The extracts were dried and derivatized with tertiary butyldimethylchlorosilane/imidazole reagent (Alltech). Tertiary butyldimethylchlorosilane/imidazole derivatives were analyzed by gas chromatography/mass spectrometry (Hewlett-Packard 5892 GC, 5972 Mass Selective Detector, and 7673 Autosampler, HP5 MS capillary column) in the selected ion-monitoring mode. The base peaks were monitored as follows: m/z 472 for C16 dihydrosphingosine (M-57; loss of tert-butyl moiety) and m/z 353 for sphingosine (cleavage between C2 and C3). The amount of ceramides was calculated from the ratio of peak areas and normalized to cell number.2
Phosphorylation of ERKs, JNKs, and p38
Mitogen-activated protein kinases (MAPKs), including ERKs, JNKs and p38 kinases, are known to be activated via phosphorylation (40). Therefore, the phosphorylated levels of MAPKs reflect MAPKs activity. Here, immunoblot analysis of phosphorylated proteins for ERKs, JNKs, and p38 kinase was carried out using the specific antibodies against phosphorylated sites of ERKs (Tyr-204 of p44 and p42), JNKs (Thr-183/Tyr-185), and p38 kinase (Thr-180/Tyr-182) (New England Biolabs, Inc., Beverly, MA). Briefly, JY or MS1418 cells (3 × 106) growing exponentially were seeded in a 100-mm dish and starved for 24 h in a mixed medium of DMEM and RPMI 1640 (1:1, v/v) containing 0.5% FBS. After irradiation with UVA (80 kJ/m2), UVB (8 kJ/m2), or UVC (60 J/m2), the cell pellets were harvested by centrifugation at 1,500 × g and then washed once with ice-cold PBS and lysed in 300 μl of SDS sample lysis buffer containing 62.5 mM Tris-HCl, pH 6.8, 2% (w/v) SDS, 10% (v/v) glycerol, 50 mM dithiothreitol, and 0.1% bromphenol blue. The lysed samples were sonicated for 5–10 s. Samples containing equal amounts of protein (Bio-Rad protein assay) were loaded in each lane of an 8% SDS-polyacrylamide gel for electrophoresis and subsequently transferred onto Immobilon-P transfer membrane. The phosphorylated MAPK proteins were detected by Western immunoblotting using a chemiluminescent detection system and visualized using the Storm840 PhosphorImaging system. Intensity of bands in Western blots was calculated and analyzed using the Image-QuaNT™ software (Molecular Dynamics, Sunnyvale, CA).
Preparation and Analysis of Normal, jnk1−/−, and jnk2−/− Primary Embryo Fibroblasts
Embryonic fibroblasts from normal, jnk1−/−, and jnk2−/− knockout mice were isolated and prepared according to the procedure of Loo and Cotman (41). Cells were established in culture in DMEM supplemented with 10% FBS, 2 mM l-glutamine, 100 units/ml penicillin, and 100 μg/ml streptomycin in a humidified atmosphere of 5% CO2 at 37 °C. For analysis of apoptosis, the cells were irradiated with UVA in serum-free DMEM, and the cells were lysed, and the DNA laddering assay was performed as described above.
RESULTS
Activation of Acid SMase by UVA, UVB, and UVC Irradiation
Acid SMase plays an important role in cellular response to extracellular stimuli by transmitting the signal into cells through the acid SMase pathway (11, 12, 14-18, 23, 24). The Amplex™ SMase assay kit (38) was used for measuring acid SMase activity in vitro in UVA-, UVB-, and UVC-irradiated cell preparations. As shown in Fig. 1, in normal JY cells, acid SMase was activated almost immediately after UVA, UVB, or UVC irradiation, but in acid SMase-deficient MS1418 cells the induction was comparatively weaker. In normal JY cells, the peak of acid SMase activity appeared at 15 min following UVA irradiation, and the activity was maintained at least 30 min. In JY cells treated with UVB or UVC, the activation peak occurred almost immediately at 5 min following irradiation and was maintained at that level or slightly higher for at least 30 min. In MS1418 cells, the peaks induced by UVA or UVB occur only at 15 min and by UVC only at 5 min and then the activity goes back to unstimulated levels. The peak level of acid SMase activity in normal JY cells was at least 3–4-fold greater than that observed in SMase-deficient MS1418 cells (Fig. 1).
Fig. 1. Induction of acid SMase activity in UV-irradiated cells.

JY and MS1418 cells (2.3 × 106) were grown in a mixture of RPMI 1640 and DMEM (1:1, v/v) containing 15% FBS. The cells were starved for 12 h in medium containing 0.5% FBS and then irradiated with UVA (80 kJ/m2), UVB (8 kJ/m2), or UVC (60 J/m2). After lysis of cells, protein concentration and acid SMase activity in supernatant fractions were measured as described under “Materials and Methods.” The figure shows the relative fluorescence units in JY and MS1418 cells normalized to mg of cellular protein. The value for control cells was subtracted from each sample. Each bar indicates the mean and S.D. from four assay samples.
Increased Cellular Ceramide Level following UVA, UVB, or UVC Irradiation
Ceramide is generated by the hydrolysis of SM by SMase (14-22). Ceramide levels were measured by gaschromatography/mass spectrometry2 In JY cells, ceramide levels increased almost immediately after UVA, UVB, or UVC irradiation, but no increase was observed in the SMase-deficient MS1418 cells (Fig. 2). These data and those in Fig. 1 indicate that the increase in ceramide in JY cells following exposure to UV irradiation is caused by acid SMase activation.
Fig. 2. Increase in ceramide level in UV-irradiated JY cells.
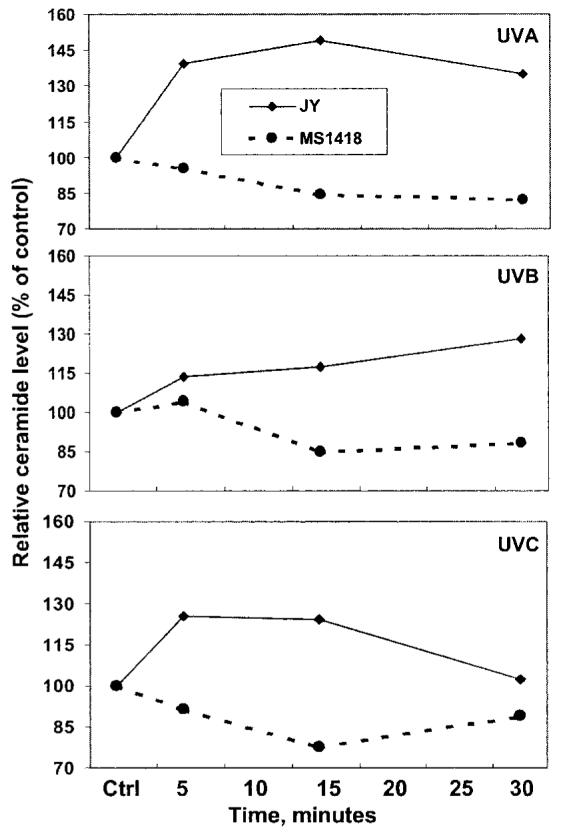
JY or MS1418 cells (3 × 106) were starved for 12 h in medium containing 0.5% FBS and then irradiated with UVA, UVB, or UVC as described in Fig. 1. Extraction of lipids and analysis of ceramides were performed as described under “Materials and Methods.” The ceramide level was normalized to cell number and is shown as % of control (value of 100). The figure shows that ceramide levels increase in UVA-, UVB-, or UVC-irradiated JY cells but not in MS1418 cells following irradiation. Each point represents the mean from triplicate incubations.
UVA-induced Apoptosis Occurs in Normal SMase Cells but Is Inhibited in SMase-deficient Cells
Apoptosis was analyzed according to morphological changes observed in apoptotic nuclei and DNA fragmentation laddering. As shown in Fig. 3A, UVA induced typical apoptosis in normal SMase JY cells, but significantly less DNA laddering induced by UVA was observed in SMase-deficient MS1418 cells. UVA-induced apoptosis was dose- (Fig. 3, A and C) and time-dependent (Fig. 3B, left panel) in SMase-normal JY cells. At 10 h following irradiation of cells with 40–80 kJ/m2 of UVA, DNA fragmentation laddering was evident in normal JY cells but almost not present in MS1418 cells (Fig. 3A). The number of apoptotic nuclei determined by Hoechst staining was directly related to doses of UVA irradiation (Fig. 3C). Typical apoptosis occurred in JY cells at 6 h after UVA irradiation (80 kJ/m2) and then increased until 12 h (Fig. 3B, left panel). In contrast, the time-dependent apoptotic response was significantly diminished in MS1418 cells (Fig. 3B, right panel). On the other hand, UVB- or UVC-induced apoptosis was not significantly different in either normal SMase or SMase-deficient cells (Fig. 3, A–C). These data suggest that UVA- but not UVB- or UVC-induced apoptosis may depend on activation of the acid SMase pathway.
Fig. 3. UVA induces apoptosis in normal SMase JY cells.

JY and MS1418 cells (3 × 106) were grown for 24 h in a mixture of RPMI 1640 and DMEM (1:1, v/v) containing 15% FBS and then irradiated with UVA, UVB, or UVC at the doses indicated. Following irradiation, the cells were harvested at the time points indicated. Fragmented DNA in supernatant fractions was extracted and separated after lysis and centrifugation, and quantitative analysis of apoptotic cells was determined by DNA laddering analysis (A and B) and Hoechst staining (C) as described under “Materials and Methods.” The upper (A) and lower (C) panels show that the dose-dependent apoptosis induced by UVA occurs in JY cells but not in MS1418 cells. On the other hand, a similar level of apoptosis induced by UVB and UVC occurs in both cell types. B, the middle left panel shows that apoptosis induced by UVA, UVB, or UVC is time-dependent in JY cells. The lower right panel shows that only apoptosis induced by UVB or UVC is time-dependent, and UVA-induced apoptosis is less significant in MS1418 cells.
Exogenous Acid SMase and Ceramide Can Induce Apoptosis in Both Normal and SMase-deficient Cells
Intracellular levels of ceramide have been shown to increase in response to various extracellular stimuli including UV exposure (12, 20, 42), and ceramide has been reported to induce apoptosis in target cells (15, 42). Here, our data show that apoptosis was induced after treatment of both normal JY and SMase-deficient MS1418 cells with exogenous SMase (Fig. 4B) or cell-permeant C2-ceramide (Fig. 4A). However, apoptosis did not occur in either cell line after treatment with C2-dihydroceramide, an internal negative control (Fig. 4A). Additionally, phosphocholine, another hydrolysis product of SMase, did not induce apoptosis in either cell line (data not shown). These data suggest that SMase and ceramide, but not phosphocholine, are implicated in mediating apoptosis.
Fig. 4. Exogenous ceramide and acid SMase can induce apoptosis in both normal JY and SMase-deficient MS1418 cells, but SM only induces apoptosis in JY cells.

JY and MS1418 cells (3 × 106) were cultured in a mixture of RPMI 1640 and DMEM (1:1, v/v) containing 15% FBS and then were treated for 14 h with C2-ceramide, C2-dihydroceramide, SM, or acid SMase at the doses indicated. Fragmented DNA was extracted and separated as described under “Materials and Methods.” A, the upper two panels show that C2-ceramide but not C2-dihydroceramide can induce apoptosis in both JY and MS1418 cells. B, the lower two panels show that SM only induces apoptosis in normal JY cells (lower left panel) and not in SMase-deficient MS1418 cells (lower right panel). On the other hand, addition of exogenous SMase induces apoptosis in both cell types (lower two panels).
Exogenous SM Can Induce Apoptosis in Normal SMase Cells but Not in SMase-deficient Cells
SM is hydrolyzed by SMase to generate ceramide and phosphocholine. As shown in Fig. 4, A and B, after treatment of cells with SM, apoptosis was induced in normal SMase cells (upper and lower left panels) but not in SMase-deficient cells (upper and lower right panels). These data indicate that acid SMase is required for SM-induced apoptosis.
UVA-induced Phosphorylation of JNKs Is SMase-dependent
Activation of JNKs by UVB and UVC has been shown to be SMase-dependent (43). Here, we found that UVA-induced phosphorylation of JNKs was also SMase-dependent (Fig. 5, A and B) and the phosphorylation coincided with activation of acid SMase by UVA (Fig. 1). In addition, UVB- or UVC-induced phosphorylation of JNKs occurred in JY cells (Fig. 5, A and B), but in SMase-deficient MS1418 cells, JNKs phosphorylation was not significantly induced by UVA, UVB, or UVC (Fig. 5, A and B). These data suggest that SMase is required for activation of JNKs by UVA, UVB, or UVC, further indicating that JNKs are downstream kinases of the SMase signaling pathway. Additionally, TPA-induced phosphorylation of JNKs was observed in both cell lines (Fig. 5A), suggesting that TPA-induced activation of JNKs may be independent of SMase.
Fig. 5. Phosphorylation of JNKs is induced in UV-irradiated JY cells but not in MS1418 cells.

JY or MS1418 cells (3 × 106) were starved for 24 h in a mixture of RPMI 1640 and DMEM (1:1, v/v) containing 0.5% FBS. Cells were irradiated with UVA (80 kJ/m2), UVB (8 kJ/m2), or UVC (60 J/m2) and then harvested by centrifugation at time points as indicated. After lysis and sonication, samples containing equal amounts of protein were separated by 8% SDS-PAGE and analyzed by Western immunoblotting using anti-phospho-JNKs antibody as described under “Materials and Methods.” Nonirradiated cells were used as a negative control. A, The figure shows that phosphorylation of JNKs was induced in UVA-, UVB-, or UVC-irradiated JY cells but almost not in MS1418 cells. B, intensity of bands was quantitated with Image-QuanNT™ software. Each value indicates the mean and S.D. from three independent experiments and is expressed as percentage of increase in phosphorylation compared with control.
UVA-, UVB-, UVC-induced Phosphorylation of ERKs Is Inhibited in Normal SMase Cells Compared with SMase-deficient MS1418 Cells
Although ERKs were reported not to be activated by UVA irradiation (44), our data showed that UVA, like UVB and UVC, induced phosphorylation of ERKs in SMase-deficient MS1418 cells, and the phosphorylation was blocked in SMase-normal JY cells compared with control values (Fig. 6, A and B). The data suggest that ERKs phosphorylation may not be related to UV-induced activation of SMase signaling in JY cells.
Fig. 6. UV-induced phosphorylation of ERKs is inhibited in JY cells.

JY or MS1418 cells (3 × 106) were starved for 24 h in a mixture of RPMI 1640 and DMEM (1:1, v/v) containing 0.5% FBS. Cells were irradiated with UVA (80 kJ/m2), UVB (8 kJ/m2), or UVC (60 J/m2) and then harvested by centrifugation at time points as indicated. After lysis and sonication, samples containing equal amounts of protein were separated by 8% SDS-PAGE and analyzed by Western immunoblotting using anti-phospho-ERKs antibody as described under “Materials and Methods.” Nonirradiated cells were used as a negative control. A shows that UVA–, UVB-, or UVC-induced phosphorylation of ERKs was induced in MS1418 cells but was markedly less in JY cells. B, intensity of bands was quantitated with Image-QuanNT™ software. Each value indicates the mean and S.D. from three independent experiments and is expressed as percentage of increase in phosphorylation compared with control.
UVA-, but Not UVB- or UVC-induced Phosphorylation of p38 Kinase, Is Also Inhibited in Normal SMase Cells
Activation of p38 kinase was shown to be induced by UVA (44). Here, our data showed that UVA-induced phosphorylation of p38 kinase occurred in both JY and MS1418 cells, but the phosphorylation appeared to be less in JY cells (Fig. 7, A and B). On the other hand, UVB- or UVC-induced phosphorylation of p38 kinase was not significantly different overall in these two cell lines (Fig. 7, A and B). These data also suggest that UVA-induced p38 kinase activation is not related to UV-induced activation of the SMase signaling pathway.
Fig. 7. UVA- but not UVB- or UVC-induced phosphorylation of p38 kinase is inhibited in JY cells.

JY or MS1418 cells (3 × 106) were starved for 24 h in a mixture of RPMI 1640 and DMEM (1:1, v/v) containing 0.5% FBS. Cells were irradiated with UVA (80 kJ/m2), UVB (8 kJ/m2), or UVC (60 J/m2) and then harvested by centrifugation at time points as indicated. After lysis and sonication, samples containing equal amounts of protein were separated by 8% SDS-PAGE and analyzed by Western immunoblotting using anti-phospho-p38 kinase antibody as described under “Materials and Methods.” Nonirradiated cells were used as a negative control. A shows that phosphorylation of p38 kinase was induced in UVA, UVB, or UVC-irradiated JY and MS1418 cells, and the phosphorylation by UVA but not UVB or UVC was less in JY cells. B, intensity of bands was quantitated with Image-QuanNT™ software. Each value indicates the mean and S.D. from three independent experiments and is expressed as percentage of increase in phosphorylation compared with control.
PD98059 and SB202190 Do Not Inhibit UVA-induced Apoptosis in JY Cells
Whether activation of ERKs or p38 kinase is reported to be involved in apoptosis or anti-apoptosis depends on cell type and the kind of stimulation (45-47). Here, to study further the role of ERKs and p38 kinase in UVA-induced apoptosis, we used PD98059, a selective inhibitor of MEK1 resulting in inhibition of ERK activation (48), and SB202190, a selective inhibitor of p38 kinase activation (49). Our data showed that PD98059 and SB202190 did not inhibit UVA-induced apoptosis in JY cells (Fig. 8, left panel). On the other hand, UVA-induced apoptosis was observed in SMase-deficient MS1418 cells after pretreatment with PD98059 or SB202190 (Fig. 8, right panel). These data suggest that ERKs and p38 kinase are not required for UVA-induced apoptosis in normal JY cells.
Fig. 8. UVA-induced apoptosis is not inhibited by PD98059 or SB202190.

JY or MS1418 cells (3 × 106) were pretreated for 1.5 h with Me2SO as control, or with PD98059 or SB202190 and then irradiated with UVA (80 kJ/m2). Twelve hours after irradiation, cells were harvested by centrifugation. Fragmented DNA was extracted and separated as described under “Materials and Methods.” Nonirradiated cells were used as a negative control, and cells treated only with UVA irradiation were the positive control. The figure shows that PD98059 and SB202190 do not inhibit UVA-induced apoptosis in JY cells (left panel), and UVA-induced apoptosis occurs after pretreatment of MS1418 with PD98059 or SB202190 (right panel).
UVA-induced Apoptosis Is Blocked in jnk1−/− and jnk2−/− Cells
Activation of JNKs is known to result in phosphorylation of c-Jun and to be involved in the induction of apoptosis (12, 45, 46). JNKs were activated by UVA via the acid SMase pathway as described above (Fig. 5). Here our data showed further that UVA-induced apoptosis was markedly blocked in jnk1−/− and jnk2−/− cells compared with wild-type jnk+/+ cells (Fig. 9B), although induction of acid SMAse activity occurred immediately following UVA irradiation of the three cell lines (Fig. 9A). Taken together, our data indicate that UVA-induced apoptosis occurs through the SMase signaling activation of JNK1 and JNK2 and not through activation of ERKs or p38 kinases.
Fig. 9. UVA-induced apoptosis is abolished in jnk1−/− and jnk2−/− cells albeit UVA induces acid smase activity.

Primary embryonic fibroblasts were prepared from wild-type and jnk1−/− and jnk2−/− knockout mice. The cells were starved for 12 h in serum-free DMEM and then irradiated with UVA (80 kJ/m2). A, the cells were harvested at indicated times following irradiation. Then acid SMase activities in supernatant fractions were determined as described in Fig. 1, except that the fluorescence intensity was measured with a Lab-systems Fluoroskan (Franklin, MA) with filter combination 527 nm (excitation) and 620 nm (emission). The UVA-induced SMase activity was normalized to nonirradiated control (value of 100) and is shown as percent. The data are representative of three independent experiments, and each bar represents the mean and S.D. from triplicate samples. B, 14 h following irradiation, detached and attached cells were harvested by scraping and centrifuging. Fragmented DNA in supernatant fractions was extracted and separated as described under “Materials and Methods.” Nonirradiated cells were used as negative control. The figures show that induction of acid SMase activity by UVA occurs (A) but UVA-induced apoptosis is completely blocked (B) in jnk1−/− and jnk2−/− cells.
DISCUSSION
Exposure to UV radiation can cause cell cycle arrest (50, 51), alterations in mitochondrial membrane permeability (30), and cell death by necrosis or apoptosis (42, 51). In the present study, we observed that UVA, like UVB and UVC, induced apoptosis in normal lymphoblast cells (JY) but not in MS1418 lymphoblasts from Niemann-Pick type D patients who have an inherited deficiency of acid SMase. UVA radiation is known to induce an array of stress proteins quite distinct from those induced by UVB or UVC (51). UVB and UVC were clearly shown to mimic growth factor responses and stimulate signal transduction including the SMase pathway (12, 20, 32, 43). Noncytotoxic exposure to UVA can also up-regulate several signal molecules (32, 42), but the role of the SMase pathway in UVA-induced apoptosis is not well understood.
In this study, we provide evidence that UVA, UVB, and UVC can all activate acid SMase (Fig. 1) and lead to an increase in ceramide levels (Fig. 2) in normal JY cells. Analysis of DNA fragmentation laddering and morphological changes in apoptotic cells showed that sublethal doses of UVA (20–80 kJ/m2), similar to lethal doses of UVB and UVC, can also induce apoptosis in normal JY cells, but UVA-induced apoptosis is prevented in acid SMase-deficient MS1418 cells. On the other hand, the difference in UVB- or UVC-induced apoptosis was less apparent in normal and SMase-deficient cells. These results suggest that UVA-induced apoptosis occurs through activation of acid SMase, whereas UVB- or UVC-induced apoptosis occurs through SMase-independent pathways. However, additional pathways involved in mediating UVA-induced apoptosis cannot be disregarded, including neutral SMase (25, 26), stress-activated protein kinase/JNKs (12), caspases (30), and mitochondria (30), because UVA-induced apoptosis is not completely blocked in acid SMase-deficient MS1418 cells.
Klotz et al. (44) reported that UVA did not activate ERKs, and we found that UVA, like UVB and UVC, did not induce phosphorylation (Fig. 6) and activation (data not shown) of ERKs in SMase-normal JY cells. However, UVA strongly induced ERKs phosphorylation in SMase-deficient MS1418 cells suggesting that in the absence of SMase, UVA may stimulate alternate pathways (e.g. ERKs) that may protect against UVA-induced apoptosis in these cells. We also found that UVA, as well as UVB and UVC, induced phosphorylation and activation of p38 kinase (Fig. 7). However, UVA-induced phosphorylation of p38 kinase appeared to be less in normal SMase JY cells compared with SMase-deficient MS1418 cells, whereas UVB- or UVC-induced phosphorylation was similar between the two cell lines (Fig. 7). Neither inhibition of UVA-induced ERKs activation by PD98059 (48) nor inhibition of p38 kinase activation by SB202190 (49) blocked UVA-induced apoptosis in normal JY cells (Fig. 8, left panel). However, MS1418 cells pre-treated with PD98059 or SB202190 now showed typical apoptosis following UVA irradiation (Fig. 8, right panel). These results further confirm that activation and phosphorylation of ERKs and p38 kinase do not appear to be involved in UVA-induced apoptosis in normal JY cells, but in the absence of SMase, UVA irradiation may induce ERKs and p38 kinase leading to inhibition of apoptosis.
Acid SMase was previously shown to be involved in UVB- and UVC-induced activation of JNKs (43). In the present study, we observed that UVA, like UVB and UVC, induced phosphorylation of JNKs in acid SMase normal JY cells and that the phosphorylation of JNKs was markedly inhibited in SMase-deficient cells (Fig. 5). However, TPA-induced JNKs phosphorylation was not different between the two cell lines. These results suggest that UVA-, UVB-, or UVC-, but not TPA-induced phosphorylation and activation of JNKs, is acid SMase-dependent. In addition, we observed that UVA-induced apoptosis was completely blocked in jnk1−/− and jnk2−/− cells (Fig. 9B) further indicating that JNKs are a downstream kinase family of the acid SMase pathway (45, 46). Overall, these results strongly suggest that UVA-induced apoptosis in normal SMase JY cells occurs primarily through activation of JNKs via the acid SMase pathway.
Acknowledgments
We thank Drs. Xun Li and Zbigniew Darzynkiewicz at the Cancer Research Institute and Department of Pathology, New York Medical College, Valhalla, NY, for providing some details on a direct DNA strand break labeling (36, 37). We also thank Drs. Nanyue Chen and Qing-Bai She for their help on the assays for the DNA fragmentation ladder assay; Randy Krebsbach for assistance with the ceramide assays, and Andria Hansen for secretarial assistance.
Footnotes
This work was supported by The Hormel Foundation, National Institute of Health Grants CA77646, CA81064, GM45741, and GM45928, and the Academy of Finland. The costs of publication of this article were defrayed in part by the payment of page charges. This article must therefore be hereby marked “advertisement” in accordance with 18 U.S.C. Section 1734 solely to indicate this fact.
The abbreviations used are: SM, sphingomyelin; SMase, sphingomyelinase; UVA, ultraviolet light A; DMEM, Dulbecco's modified Eagle's medium; FBS, fetal bovine serum; Me2SO, dimethyl sulfoxide; PBS, phosphate-buffered saline; MAPKs, mitogen-activated protein kinases; ERKs, extracellular signal-regulated kinases; JNKs, c-Jun N-terminal kinases; p38, p38 MAPK or p38 kinases; TPA, 12-O-tetradecanoylphorbol-13-acetate; TNFα, transforming growth factor-α.
REFERENCES
- 1.Saikumar P, Dong Z, Mikhailov V, Denton M, Weinberg JM, Venkatachalam MA. Am. J. Med. 1999;107:489–506. doi: 10.1016/s0002-9343(99)00259-4. [DOI] [PubMed] [Google Scholar]
- 2.Darzynkiewicz Z, Juan G, Li X, Gorczyca W, Murakami T, Traganos F. Cytometry. 1997;27:1–20. [PubMed] [Google Scholar]
- 3.Allen RT, Hunter WJI, II, Agrawal DK. J. Pharmacol. Toxicol. Methods. 1997;37:215–228. doi: 10.1016/s1056-8719(97)00033-6. [DOI] [PubMed] [Google Scholar]
- 4.Kitanaka C, Kuchino Y. Cell Death Differ. 1999;6:508–515. doi: 10.1038/sj.cdd.4400526. [DOI] [PubMed] [Google Scholar]
- 5.Tsujimoto Y, Shimizu S. FEBS Lett. 2000;466:6–10. doi: 10.1016/s0014-5793(99)01761-5. [DOI] [PubMed] [Google Scholar]
- 6.Fadeel B, Zhivotovsky B, Orrenius S. FASEB J. 1999;13:1647–1657. doi: 10.1096/fasebj.13.13.1647. [DOI] [PubMed] [Google Scholar]
- 7.Hofmann K. Cell. Mol. Life Sci. 1999;55:1113–1128. doi: 10.1007/s000180050361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Earnshaw WC, Martins LM, Kaufmann SH. Annu. Rev. Biochem. 1999;68:383–424. doi: 10.1146/annurev.biochem.68.1.383. [DOI] [PubMed] [Google Scholar]
- 9.Green DR, Reed JC. Science. 1998;281:1309–1312. doi: 10.1126/science.281.5381.1309. [DOI] [PubMed] [Google Scholar]
- 10.Godar DE. J. Investig. Dermatol. Symp. Proc. 1999;4:17–23. doi: 10.1038/sj.jidsp.5640175. [DOI] [PubMed] [Google Scholar]
- 11.Okazaki T, Kondo T, Kitano T, Tashima M. Cell. Signal. 1998;10:685–692. doi: 10.1016/s0898-6568(98)00035-7. [DOI] [PubMed] [Google Scholar]
- 12.Verheij M, Bose R, Lin XH, Yao B, Jarvis WD, Grant S, Birrer MJ, Szabo E, Zon LI, Kyriakis JM, Haimovitz-Friedman A, Fuks Z, Kolesnick RN. Nature. 1996;380:75–79. doi: 10.1038/380075a0. [DOI] [PubMed] [Google Scholar]
- 13.Susin SA, Lorenzo HK, Zamzami N, Marzo I, Snow BE, Brothers GM, Mangion J, Jacotot E, Costantini P, Loeffler M, Larochette N, Goodlett DR, Aebersold R, Siderovski DP, Penninger JM, Kroemer G. Nature. 1999;397:441–446. doi: 10.1038/17135. [DOI] [PubMed] [Google Scholar]
- 14.Hannun YA. Science. 1996;274:1855–1859. doi: 10.1126/science.274.5294.1855. [DOI] [PubMed] [Google Scholar]
- 15.Levade T, Jaffrézou JP. Biochim. Biophys. Acta. 1999;1438:1–17. doi: 10.1016/s1388-1981(99)00038-4. [DOI] [PubMed] [Google Scholar]
- 16.Mathias S, Peña LA, Kolesnick RN. Biochem. J. 1998;335:465–480. doi: 10.1042/bj3350465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Igarashi Y. J. Biochem. (Tokyo) 1997;122:1080–1087. doi: 10.1093/oxfordjournals.jbchem.a021865. [DOI] [PubMed] [Google Scholar]
- 18.Merrill AH, Jr., Schmelz EM, Dillehay DL, Spiegel S, Shayman JA, Schroeder JJ, Riley RT, Voss KA, Wang E. Toxicol. Appl. Pharmacol. 1997;142:208–225. doi: 10.1006/taap.1996.8029. [DOI] [PubMed] [Google Scholar]
- 19.Okazaki T, Bell RM, Hannun YA. J. Biol. Chem. 1989;264:19076–19080. [PubMed] [Google Scholar]
- 20.Jarvis WD, Fornari FA, Jr., Auer KL, Freemerman AJ, Szabo E, Birrer MJ, Johnson CR, Barbour SE, Dent P, Grant S. Mol. Pharmacol. 1997;52:935–947. doi: 10.1124/mol.52.6.935. [DOI] [PubMed] [Google Scholar]
- 21.Cuvillier O, Pirianov G, Kleuser B, Vanek PG, Coso OA, Gutkind JS, Spiegel S. Nature. 1996;381:800–803. doi: 10.1038/381800a0. [DOI] [PubMed] [Google Scholar]
- 22.Kawano T, Cui J, Koezuka Y, Toura I, Kaneko Y, Motoki K, Ueno H, Nakagawa R, Sato H, Kondo E, Koseki H, Taniguchi M. Science. 1997;278:1626–1629. doi: 10.1126/science.278.5343.1626. [DOI] [PubMed] [Google Scholar]
- 23.Santana P, Peña LA, Haimovitz-Friedman A, Martin S, Green D, McLoughlin M, Cordon-Cardo C, Schuchman EH, Fuks Z, Kolesnick R. Cell. 1996;86:189–199. doi: 10.1016/s0092-8674(00)80091-4. [DOI] [PubMed] [Google Scholar]
- 24.Grassmé H, Gulbins E, Brenner B, Ferlinz K, Sandhoff K, Harzer K, Lang F, Meyer TF. Cell. 1997;91:605–615. doi: 10.1016/s0092-8674(00)80448-1. [DOI] [PubMed] [Google Scholar]
- 25.Adam D, Wiegmann K, Adam-Klages S, Ruff A, Krönke M. J. Biol. Chem. 1996;271:14617–14622. doi: 10.1074/jbc.271.24.14617. [DOI] [PubMed] [Google Scholar]
- 26.Adam-Klages S, Adam D, Wiegmann K, Struve S, Kolanus W, Schneider-Mergener J, Krönke M. Cell. 1996;86:937–947. doi: 10.1016/s0092-8674(00)80169-5. [DOI] [PubMed] [Google Scholar]
- 27.De Laat JM, De Gruijl FR. Cancer Surv. 1996;26:173–191. [PubMed] [Google Scholar]
- 28.Scharffetter-Kochanek K, Wlaschek M, Brenneisen P, Schauen M, Blaudschun R, Wenk J. Biol. Chem. 1997;378:1247–1257. [PubMed] [Google Scholar]
- 29.Slominski A, Pawelek J. Clin. Dermatol. 1998;16:503–515. doi: 10.1016/s0738-081x(98)00023-6. [DOI] [PubMed] [Google Scholar]
- 30.Tada-Oikawa S, Oikawa S, Kawanishi S. Biochem. Biophys. Res. Commun. 1998;247:693–696. doi: 10.1006/bbrc.1998.8869. [DOI] [PubMed] [Google Scholar]
- 31.Beissert S, Granstein RD. Crit. Rev. Biochem. Mol. Biol. 1996;31:381–404. doi: 10.3109/10409239609108723. [DOI] [PubMed] [Google Scholar]
- 32.Bender K, Blattner C, Knebel A, Iordanov M, Herrlich P, Rahmsdorf HJ. J. Photochem. Photobiol. B Biol. 1997;37:1–17. doi: 10.1016/s1011-1344(96)07459-3. [DOI] [PubMed] [Google Scholar]
- 33.DeMaria R, Lenti L, Malisan F, d'Agostino F, Tomassini B, Zeuner A, Rippo MR, Testi R. Science. 1997;277:1652–1655. doi: 10.1126/science.277.5332.1652. [DOI] [PubMed] [Google Scholar]
- 34.Li H, Zhu H, Xu CJ, Yuan J. Cell. 1998;94:491–501. doi: 10.1016/s0092-8674(00)81590-1. [DOI] [PubMed] [Google Scholar]
- 35.Suschek CV, Krischel V, Bruch-Gerharz D, Berendji D, Krutmann J, Kröncke KD, Kolb-Bachofen V. J. Biol. Chem. 1999;274:6130–6137. doi: 10.1074/jbc.274.10.6130. [DOI] [PubMed] [Google Scholar]
- 36.Li X, Traganos F, Melamed MR, Darzynkiewicz Z. Cytometry. 1995;20:172–180. doi: 10.1002/cyto.990200210. [DOI] [PubMed] [Google Scholar]
- 37.Li X, Melamed MR, Darzynkiewicz Z. Exp. Cell Res. 1996;222:28–37. doi: 10.1006/excr.1996.0004. [DOI] [PubMed] [Google Scholar]
- 38.Zhou M, Diwu Z, Panchuk-Voloshina N, Haugland RP. Anal. Biochem. 1997;253:162–168. doi: 10.1006/abio.1997.2391. [DOI] [PubMed] [Google Scholar]
- 39.Folch J, Lees M, Sloane Stanley GH. J. Biol. Chem. 1957;226:497–507. [PubMed] [Google Scholar]
- 40.Cano E, Mahadevan LC. Trends Biochem. Sci. 1995;20:117–122. doi: 10.1016/s0968-0004(00)88978-1. [DOI] [PubMed] [Google Scholar]
- 41.Loo DT, Cotman CW. In: Cell Biology: A Laboratory Handbook. 2nd Ed. Celis JE, editor. Academic Press, Inc.; San Diego: 1998. pp. 65–72. [Google Scholar]
- 42.Obeid LM, Hannun YA. J. Cell. Biochem. 1995;58:191–198. doi: 10.1002/jcb.240580208. [DOI] [PubMed] [Google Scholar]
- 43.Huang C, Ma W-Y, Ding M, Bowden GT, Dong Z. J. Biol. Chem. 1997;272:27753–27757. doi: 10.1074/jbc.272.44.27753. [DOI] [PubMed] [Google Scholar]
- 44.Klotz L-O, Pelliuex C, Briviba K, Pierlot C, Aubry J-M, Sies H. Eur. J. Biochem. 1999;260:917–922. doi: 10.1046/j.1432-1327.1999.00255.x. [DOI] [PubMed] [Google Scholar]
- 45.Ichijo H, Nishida E, Irie K, ten Dijke P, Saitoh M, Moriguchi T, Takagi M, Matsumoto K, Miyazono K, Gotoh Y. Science. 1997;275:90–94. doi: 10.1126/science.275.5296.90. [DOI] [PubMed] [Google Scholar]
- 46.Xia Z, Dickens M, Raingeaud J, Davis RJ, Greenberg ME. Science. 1995;270:1326–1331. doi: 10.1126/science.270.5240.1326. [DOI] [PubMed] [Google Scholar]
- 47.Assefa Z, Vantieghem A, Declercq W, Vandenabeele P, Vandenheede JR, Merlevede W, de Witte P, Agostinis P. J. Biol. Chem. 1999;274:8788–8796. doi: 10.1074/jbc.274.13.8788. [DOI] [PubMed] [Google Scholar]
- 48.Deak M, Clifton AD, Lucocq JM, Alessi DR. EMBO J. 1998;17:4426–4441. doi: 10.1093/emboj/17.15.4426. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Blair AS, Hajduch E, Litherland GJ, Hundal HS. J. Biol. Chem. 1999;274:36293–36299. doi: 10.1074/jbc.274.51.36293. [DOI] [PubMed] [Google Scholar]
- 50.Herrlich P, Blattner C, Knebel A, Bender K, Rahmsdorf HJ. Biol. Chem. 1997;378:1217–1229. doi: 10.1515/bchm.1997.378.11.1217. [DOI] [PubMed] [Google Scholar]
- 51.Tyrrell RM. In: (UV activation of mammalian stress proteins) in Stress-inducible cellular Responses. Feige U, Morimoto RI, Yahara I, Polla B, editors. Birkäuser Verlag; Basel: 1996. pp. 255–271. [Google Scholar]