

Abstract
A new approach wherein steric interactions between substituents of unsymmetrical bis(4-pyridyl)acetylene ligands dictate the self-selection of single isomers of [4+4] self-assembled squares is presented. Each [4+4] self-assembly is characterized by multinuclear 31P and 1H NMR spectroscopies and Electrospray Ionization mass spectrometry (ESI-MS). NMR spectroscopic studies are used to provide a means of evaluating the efficiency of bulky substituents at proximal or remote positions relative to the Pt-N bonding motif to direct self-selection. Molecular modeling using the MMFF force field is utilized to determine the relative energy of different isomers of each assembly, and modeling results reasonably explain the trend in self-selectivity with varying pyridyl substitution.
Keywords: Coordination Chemistry, Self-Assembly, Self-Selection
Introduction
The rational design and synthesis of discrete metallosupramolecular architectures is of intense interest due to the fascinating structural diversity and potential applications of these novel functional materials.1,2 By using the common, directional bonding geometries of transition metal centers - typically octahedral, square planar or tetrahedral - in combination with a variety of organic nucleophilic ligands of specific geometry and size, a wealth of nanometer-sized supramolecular polygons and polyhedra can be successfully obtained via the bottom-up synthetic approach.1,3 Multiple examples of coordination-driven self-assembly rely upon the use of symmetric dipyridyl ligands together with Pd(II) and Pt(II) building blocks to construct a range of supramolecular architectures.4,5 So far, few studies have investigated the use of unsymmetrical dipyridyl ligands6 to construct discrete supramolecules.
Statistically, unsymmetrical dipyridyl ligands would give rise to a mixture of isomers as a result of random combinations of different orientations of ligands provided that there is no driving bias, such as specific molecular information or interactions encoded in or between the building blocks that would lead to self-sorting.7 It is a formidable challenge, therefore, to control and isolate one single, pure isomer of a self-assembled metallacycle that contains an unsymmetrical ligand as one of its building units.6,8 Our group has previously reported the preferential self-assembly of single [2+2] rhomboidal isomers from ambidentate pyridyl/carboxylate donor ligands when combined with 90° or 0° ditopic organoplatinum acceptors.9 Exclusive self-selectivity resulted from a combination of geometric and electronic factors: in each case the rhomboid isomer that was the least geometrically strained and had the greatest separation of charge was the only isomer formed. This charge separation approach has been further extended to include the exclusive [3+3] self-assembly of one isomer of a heterobimetallic Pd3-Fe3 triangle via the coordination of a singular, ambidentate 4-isonicotinate ligand.10 This method of relying on charge-charge interactions and geometric strain, however, is likely to reach its limit upon the exploration into larger self-assemblies given that charge-charge interactions decrease with the square of their separation and larger systems are better able to distribute, and therefore tolerate, geometric strain. Herein, we describe a new strategy for the self-selection of a single isomer of a [4+4] supramolecular square composed of four 90° Pt(II) acceptors and four substituted, unsymmetrical bis(4-pyridyl)acetylene ligands. The self-selection process is governed by steric and electronic interactions.
Among the many 2D supramolecular polygon geometries, supramolecular squares have been extensively studied as they can be easily prepared using square-planar Pd(II) and Pt(II) metal centers as 90° corner units.1b,1f,11 Typically, such square-planar Pd(II) or Pt(II) metals are combined with symmetrical donor ligands, such as linear dipyridyl donors, to produce identical discrete supramolecular metallacycles. Discrete metallacycles may still be formed when unsymmetrical donor ligands are used; however they are considerably less likely to be identical. As a general model, one can envision an unsymmetrical, linear dipyridyl ligand as shown in Scheme 1. Both electron-rich pyridyl moieties are depicted as blue hexagons, however the two pyridyl moieties are colored different shades of blue to reflect their asymmetry. In the absence of any biasing interactions, there are four different isomers that may be formed upon the self-assembly of supramolecular [4+4] squares from an unsymmetrical ditopic donor and a 90° Pt(II) acceptor unit, depicted as a red sphere. The four different squares are shown as isomers A-D in Scheme 1. Isomer A, for example, has all four unsymmetrical donors oriented in an alternating fashion such that one of each type of pyridyl moiety - one light blue and one dark blue - is coordinated with each Pt(II) center. The resulting [4+4] metallacycle has C4 symmetry. In isomer B, one of the donor ligands is rotated 180° relative to isomer A, resulting in a supramolecular square with Cs symmetry. In this isomer, two of the Pt(II) acceptors will be coordinated by two different pyridyl moieties (one dark blue and one light blue, upper and lower left corners of B) while the remaining two Pt(II) acceptors are coordinated with two of the same pyridyl donors, either two light blue (upper right corner) or two dark blue (lower right corner). An analogous situation arises in isomer C, however C is more symmetric than isomer B and belongs to the C2 point group. This difference in symmetry results from the relative orientations of unsymmetrical donors in each [4+4] square, which gives isomer C a two-fold axis of rotational symmetry. The final supramolecular square structure that can be made is isomer D, which has D2 symmetry. In isomer D, every Pt(II) acceptor is coordinated by two of the same pyridyl moieties, either two dark blue or two light blue.
Scheme 1.

Schematic representation of the self-assembly of four Pt(II) acceptors with four 180° unsymmetrical dipyridyl donors to make four isomeric supramolecular squares. The asymmetry of the dipyridyl donor is reflected in the two shades of blue. Point group symmetries of each supramolecular square are given. Arrows are drawn around the perimeter of each metallacycle to further aid in visualizing the different orientations of the unsymmetrical donors in each isomer.
Along these lines, we have adopted the organoplatinum ligand cis-(Me3P)2Pt(OTf)2 (1) as a 90° acceptor for constructing 2D square architectures because of its ditopic bonding motif and relatively simple proton NMR, the latter ensuring that the NMR spectra of resulting single or multiple isomers are discernible. We have also prepared three new unsymmetrical bis(4-pyridyl)acetylene donors: 2,6-dimethyl-4-(pyridin-4-ylethynyl)pyridine (2), 2-chloro-4-(pyridin-4-ylethynyl)pyridine (3), and 3,5-dichloro-4-(pyridin-4-ylethynyl)pyridine (4); which were synthesized via Sonogashira coupling reactions of their respective pyridyl halides and 4-ethynylpyridine (Scheme 2). Mixing acceptor 1 with donors 2-4 in a 1:1 molar ratio results in the spontaneous self-assembly of supramolecular square compounds 5-7, respectively, as shown in Scheme 2. Ditopic donors 2-4 were chosen because of the variation in the size and regiochemistry of substituents on their pyridyl rings. As outlined above, each unsymmetrical donor ligand is capable, in theory, of forming four isomeric [4+4] assemblies when combined with ditopic acceptor 1. In the case of mono-chloro donor 3, however, an even more complex set of isomeric metallacycles is possible depending upon the relative orientations of the α-Cl atoms within each square isomer (A-D). For example, isomer A of metallacycle 6 may have all α-Cl atoms aligned on the same face of the metallacycle, two on each side, or three on one side and one on the other (see Supporting Information for further details). Once assembled, the restricted rotation around the Pt-N bond12 does not allow for α-Cl pyridyl moieties to rotate from one face of the square to the other. The varying size and location of substituents of 2-4 should play a key role in affecting product formation and, potentially, self-selection during self-assembly.13 It is important to note that the self-selectivity study described herein is different from previous reports that relied primarily upon geometric and electronic effects and utilized non-linear ambidentate donor ligands. In the present study, all donors are linear and are the same length, precluding any geometric bias, and both coordination sites of each ligand are pyridyl nitrogen atoms, precluding a charge bias that arises when ambidentate ligands are used. In this article we show that the series of unsymmetrical donors undergo various degrees of self-sorting due to the influence of steric and electronic effects during the self-assembly of [4+4] metallacyclic squares.
Scheme 2.

Synthesis of unsymmetrical dipyridyl donors 2-4 and their combination with 90° Pt(II) acceptor 1 in CD3NO2 leading to the self-assembly of supramolecular squares 5-7, respectively.
Results and Discussion
Supramolecular squares 5-7 were prepared by mixing 90° Pt(II) acceptor 1 with unsymmetrical donors 2-4, respectively, in a 1:1 molar ratio in CD3NO2 at 298 K for 24 hr. As previously discussed and shown in Scheme 1, four isomeric supramolecular squares of different symmetry may result from each self-assembly. The symmetry of each isomer must be carefully considered and correlated with NMR spectra in order to acquire structural details from spectroscopic studies. For each metal center coordinated by two different pyridyl moieties, the 31P{1H} NMR of the PMe3 ligands will appear as two coupled doublets given that the trans effect results in two inequivalent, and consequently coupled, phosphorous nuclei. Therefore, two doublets for A the isomer and two singlets for D the isomer will be observed in their corresponding 31P{1H} NMR spectra because of the higher C4 and D2 symmetry of A and D, respectively. The 31P{1H} NMR spectra of isomer C can be discerned, based on its C2 symmetry, as two coupled doublets along with two singlets. The low Cs symmetry of isomer B causes every phosphorous nucleus to be in a dissimilar chemical environment and can be identified as such. Accordingly, whether each self-assembly results in a single supramolecular square isomer or a randomly distributed mixture of possible isomers, NMR spectroscopic data can provide sufficient evidence for affirming the occurrence and efficiency of self-selection.
The reaction of 1 and 2 in CD3NO2 yields a homogeneous pale yellow solution of 5. The 31P{1H} NMR spectra displays a pair of coupled doublets (CD3NO2, δ = -18.79 ppm, 1JPt-P = 3266.6 Hz, 2JP-P = 23.8 Hz; δ = -25.94 ppm, 1JPt-P = 3578.4 Hz, 2JP-P = 23.8 Hz) of approximately equal intensity with concomitant 195Pt satellites (Figure 1a). Compared with the phosphorous NMR of the starting material 1 (δ = -20.09 ppm, 1JPt-P = 3915 Hz), the doublet of 5 at -18.79 ppm is shift downfield by roughly 1.3 ppm, which can be assigned to the phosphorous nuclei trans to the dimethylpyridyl moiety, and forms a sharp contrast with the other doublet at δ = -25.94 ppm, which is up-shifted about 5.8 ppm. This can be rationalized by the fact that the stronger electron-donating dimethylpyridyl moiety will weaken back-donation from the Pt(II) center to the opposite PMe3 due to the trans effect. This deduction is further substantiated by the 1H NMR spectra, in which the NMR signals attributed to the pyridyl α-Me groups of the PyMe2 moiety, the β-H of PyMe2, and the β-H of the unsubstituted pyridine ring experienced downfield shifts of 0.32 ppm, 0.25 ppm and 0.32 ppm, respectively, much larger than the upfield shift of the α-H signals of the unsubstituted pyridine ring (Δδ = 0.02 ppm), indicating the loss of electron density upon coordination of the unsubstituted pyridyl moiety to the Pt(II) center. Furthermore, two characteristic coupled doublets corresponding to different PMe3 groups are also observed in the 1H NMR spectrum at δ = 1.97 and 1.73 ppm (Figure 1b), which exhibit a downshift of 0.11 ppm and an upshift by 0.13 ppm, respectively, relative to the PMe3 proton signal of 1. Consequently, the relative simplicity and symmetry of the coupled doublets in the 31P{1H} NMR spectrum of 5 are consistent with the formation of isomer A, showing that self-selection of one single isomer occurs in the self-assembly of 1 and unsymmetrical ligand 2. Self-selection in the self-assembly of 5 is likely the result of interligand α-Me-α-Me steric interactions that prohibit the coordination of two of the dimethyl pyridyl moieties to the same Pt(II) center as well as electronic effects resulting from the difference in electron-donating abilities, as evidenced by NMR chemical shift changes, between the dimethyl pyridyl and pyridyl moieties of ligand 2.
Figure 1.
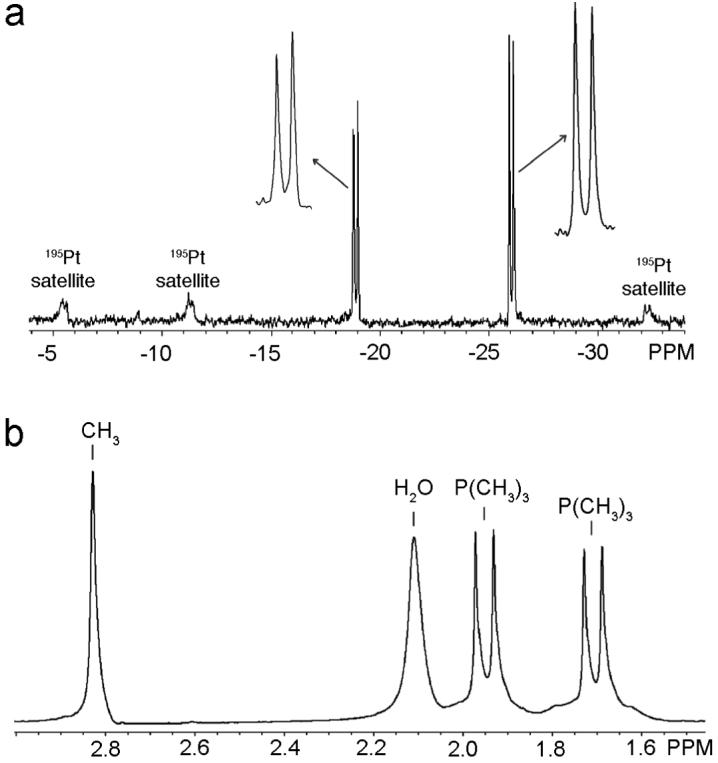
(a) Partial 31P{1H} NMR spectrum of 5 showing only two sets of doublets. (b) Partial 1H NMR spectrum of 5 showing a set of coupled doublets arising from the two inequivalent PMe3 substituents. The splitting pattern and high symmetry of both spectra strongly support the formation of only one isomer, that of isomer A, in the self-assembly of supramolecular square 5.
Mixing monochloro-substituted ligand 3 in a 1:1 stoichiometric ratio with acceptor 1 generates [4+4] self-assembly 6. Sharp 31P{1H} and 1H NMR spectroscopic peaks indicate that only discrete structures are obtained, however, the complicated splitting of 31P{1H} NMR spectra suggest that multiple supramolecular isomers are formed. As shown in Figure 2, three pairs of doublets (blue: -26.59 ppm, -26.79 ppm, 2JP-P = 24.4 Hz; red: -26.67 ppm, -26.87 ppm, 2JP-P = 24.4 Hz; green: -26.92 ppm, -27.13 ppm, 2JP-P = 25.2 Hz) are observed in the downfield region of the 31P{1H} NMR spectrum of 6, indicating the existence of inequivalent coordination modes for the Pt(II) centers. The small difference of approximately 1.0 ppm between the downfield and upfield shifts of signals in the 31P{1H} NMR of 6 denotes similar electron density between the two pyridyl moieties, in contrast to the shifts observed for 5 (∼7.0 ppm) where the difference in electron density is greater. A statistical analysis involving random variations of the orientation of donor ligands 3 within the supramolecular square 6 would suggest that the four possible isomers A:B:C:D would be formed in a 1:4:2:1 ratio in the absence of any inter-ligand effects. However, the 31P{1H} NMR spectrum demonstrates that the single isomer corresponding to the doublet highlighted in blue is the dominant species in the mixture and the other two doublets (red and green) are of lower but roughly equal intensity. The PMe3 signals in the 1H NMR spectrum lend further support to the formation of one predominant isomer, one in which the two coupled doublets highlighted in blue at δ = 1.79 ppm (2JP-H = 5.7 Hz) and 1.75 ppm (2JP-H = 5.7 Hz) have the same shape as those of assembly 5 and their symmetry implies that the resulting dominant species is also highly symmetric. The main species formed in the self-assembly of 6 can therefore be identified as isomer A. However, the monosubstitution of the less sterically bulky Cl atom and more similar electron-donating character of the monochloro and unsubstituted pyridyl moieties of 3 result in a decrease in the efficiency of self-selection in 6 relative to the high degree of self-selection observed in the self-assembly of 5.
Figure 2.
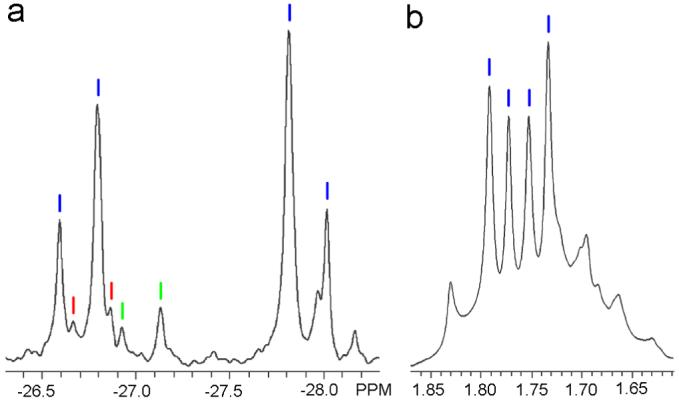
Partial 31P{1H} NMR (a) and 1H NMR (b) spectra of the self-assembly of 6. In both spectra, pairs of coupled doublets are indicated by blue, red, and green lines. Based upon the splitting patterns, symmetry, and the intensities of spectral signals, it can be concluded that one predominant species (isomer A) is formed during self-assembly though small amounts of other isomers are present as well.
To investigate the steric interactions between substituents at the β position of pyridyl moieties and their effect on self-selection, we mixed dichloro donor ligand 4 with acceptor 1 in a 1:1 stoichiometric ratio, thus yielding a colorless solution of 7. The resulting 31P NMR spectrum is significantly more complex than those of 5 and 6, with most peaks having near equal intensity. One pair of singlets (blue) and one pair of coupled doublets (red) can be distinguished as shown in Figure 3. The coupled doublets (red) are located at δ = -26.58 ppm (2JP-P = 23.8 Hz) and -27.67 (2JP-P = 23.8 Hz). These 31P{1H} NMR signals relate to the pair of coupled signals highlighted in red that are observed in the 1H NMR spectrum at δ = 1.77 ppm (2JP-H = 4.2 Hz) and 1.75 (2JP-P = 4.5 Hz). By analogy to the NMR spectrum and analysis of 6, it is clear that such coupled doublets (red) in the 31P{1H} and 1H NMR spectra of 7 can be attributed to isomer A. Unlike self-assemblies 5 and 6, however, the intensities of peaks associated with the A isomer of 7 are almost equivalent to adjacent peaks. Due to significant overlapping of signals in both the 31P and 1H NMR spectra, it is difficult to determine which other isomers (B-D) are present in the mixture and in what relative amounts. These observations indicate that the β-dichloro substituted ligand (4) has considerably less influence on the self-selection of a single isomer because the two chlorine atoms are too far removed from the Pt-N coordination site to have significant steric or electronic influence.
Figure 3.
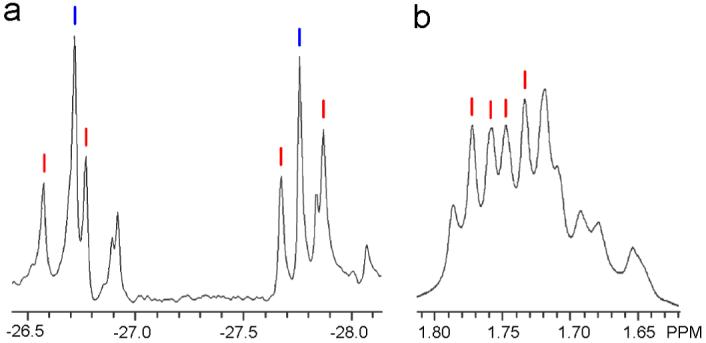
(a) Partial 31P{1H} NMR spectrum of self-assembly 7. Pairs of coupled doublets are indicated by red and blue lines. (b) Partial 1H NMR spectrum of self-assembly 7. The set of two coupled doublets highlighted by red lines corresponds to the same species that gives rise to the coupled doublets (red) in the 31P{1H} NMR spectrum and can be ascribed to isomer A.
The formation of discrete supramolecular squares during the self-assembly of 5-7 is further confirmed by electrospray ionization mass spectrometry (ESI-MS). As shown in Figure 4, the peaks resulting from each self-assembly after the loss of three triflate anions are found at: [5 - 3CF3SO3- + CD3NO2]3+ (m/z 1009.7), [6 - 3CF3SO3-]3+ (m/z 997.0) and [7 - 3CF3SO3-]3+ (m/z 1043.0). It is noteworthy that one nitromethane molecule is consistently found in the ionization species of self-assembly 5 though not in the other two 6 and 7. This may likely result from C-H· · · O hydrogen-bonding interaction between the pyridyl methyl moieties and CD3NO2, an interaction not present in assemblies 6 and 7. All observed peaks are isotopically resolved and in good agreement with their corresponding theoretical distributions (red). ESI-MS results unambiguously identify 5-7 as [4+4] supramolecular squares, however they cannot distinguish the relative abundances of different isomers of each square.
Figure 4.

Theoretical (top, red) and experimental (bottom, blue) ESI-MS of self-assembled squares (a) 5 [M-3OTf + CD3NO2]3+, (b) 6 [M-3OTf]3+, (c) 7 [M-3OTf]3+.
Molecular force field modeling was used to investigate the differences between the A-D isomers of supramolecular squares 5-7 and to gain insight into the preference, if any, for forming one isomer at the expense of others. Each individual isomer of squares 5-7 were built within the input mode of the program Maestro v9.51.09 and subjected to a 1 ns molecular dynamics simulation (MMFF force field, gas phase, 300K) in order to equilibrate the structures. The output of each simulation was then minimized to full convergence. In every case it was found that isomer A of supramolecular squares 5-7 was the lowest energy, and therefore most favored isomer. A comparison of the energy differences between isomers B-D relative to isomer A (taken as the zero point of relative energy for each assembly) is shown in Figure 5 for each respective supramolecular square 5-7. These energy differences of isomers A-D provide strong support for the experimental NMR spectroscopic results. In the case of self-assembly 5, for example, only one isomer is observed spectroscopically. Molecular modeling suggests a large difference in energy between isomer A of self-assembly 5 and the other isomers (B, C: 20.3 kcal/mol; D: 40.3 kcal/mol) on account of unfavorable interligand Me-Me steric interactions between ligands oriented such that their pyridyl α-methyl groups are coordinated to the same Pt(II) center. These steric effects occur at one Pt(II) center in isomers B and C and at two Pt(II) centers for isomer D, which accounts for the trend in the energy differences. As a result of interligand Me-Me interactions, isomer D must distort to adopt a more rhomboidal shape as shown in Figure 6a.
Figure 5.

Graph of the energy differences between isomers A-D of supramolecular squares 5-7 obtained from molecular modeling. Energies are expressed in Kcal/mol. For all squares (5, 6, and 7) isomer A was found to be the most stable and is therefore listed as 0.0 Kcal/mol. The energies of isomers B-D of square 5 are given relative to isomer A of square 5 and so on for each supramolecular square. For the α-Cl square 6, isomers indicated with as prime (’) are secondary structural isomers with α-Cl atoms on the same face of 6 as discussed in the text.
Figure 6.

Representative examples of isomers of supramolecular squares 5 (a), 6 (b), and 7 (c) obtained from molecular force field modeling. Methyl and chloro substituents on the “front” face of each square are labeled as “Me” or “Cl.” The difference in steric interactions between isomers A and D of bis α-Me square 5 can be seen in (a). In (b) is an example of secondary isomers that are possible in α-Cl square 6: isomer C (top) has all four Cl atoms on alternating faces of the square while isomer C’ (bottom) has two Cl atoms coordinated to the same Pt(II) center on the same face of the square, inducing steric and electrostatic repulsion. The structural differences between isomers A and D of bis β-Cl square 7 in (c) are very minor, giving rise to a difference of only 2.7 Kcal/mol between the two isomers.
As mentioned previously, a number of secondary structural isomers are possible for self-assembled supramolecular square 6 due to the reduced symmetry of linear donor 3 compared to donors 2 and 4. The four pyridyl α-Cl atoms may align all on one side of square 6, two on each side, or three on one side and one on the other. Those isomers having α-Cl atoms coordinated to the same Pt(II) center and on the same face of 6 experience repulsive interligand (α-Cl)-(α-Cl) steric and electronic interactions and have higher relative energies than those having the α-Cl atoms on opposite faces of the supramolecular square. An example comparing the C and C’ isomers of 6 is shown in Figure 6b. Molecular modeling indicates that B and C isomers having α-Cl atoms of pyridyl moieties attached to the same Pt(II) center but oriented on opposite faces of the square have mean relative energies of about 0.9 Kcal/mol compared to isomer A. In the case of isomer D, the lowest energy conformation (that with α-Cl atoms on alternating faces of square 6) is 6.8 Kcal/mol above isomer A. This decrease in relative energy differences between isomers A, B, C, and D is in contrast to those observed for supramolecular square 5 and is in agreement with the observation that a limited self-selection process occurs in the self-assembly of 6. Isomers with adjacent α-Cl atoms on the same face of square 6 (i.e. B’, C’, D’, and D”, see Supporting Information) have higher relative energies ranging from 11.4 to 28.5 Kcal/mol above the lowest energy isomer A. Lastly, when the substituents are moved far away from Pt-N bonding center as they are in square 7 (Figure 6c), the relative energy gaps between A and B-D are all within 2.7 Kcal/mol, too small to efficiently control the self-selection process, thus generating a mixture of many isomers.
In summary, we have utilized steric effects between substituents of unsymmetrical bis(4-pyridyl)acetylene ligands (2-4) to control the degree of self-selection in the self-assembly of [4+4] supramolecular squares. The collective 31P and 1H NMR spectra indicate that the efficiency of self-selection of isomers is greater when substituents of greater steric bulk are located at positions proximal to the Pt-N bonding motif and efficiency decreases as the size of substituents is decreased or the substituents are moved further from the Pt-N coordination site. Molecular modeling using the MMFF force field allows for the relative energy of each isomer of the supramolecular squares to be compared and the results support the experimental trend in self-selectivity from complete self-selectivity, as in supramolecular square 5, to the absence of self-selectivity, as in supramolecular square 7.
Experimental Section
Methods and Materials
The organoplatinum acceptor ligand cis-(PMe3)2Pt(OTf)2 (1) was synthesized according to literature methods.14 2-Chloro-4-iodopyridine and 3,5-dichloro-4-iodopyridine were purchased from Aldrich and used without further purification. 4-Bromo-2,6-lutidine was prepared according to published procedure.15 4-Ethynylpyridine was fleshly prepared prior to a reaction by neutralization of commercially available its HCl salt.
General method for the synthesis of unsymmetrical bipyridyl ligands 2-4
A 10 ml Schlenk flask was charged with 4-ethynylpyridine (0.20 mmol), aryl halide (0.20 mmol), bis(triphenylphosphine) palladium(II) dichloride (0.005 mmol) and copper(I) bromide (0.009 mmol) under the stream of nitrogen. Fleshly distilled triethylamine (3 ml) was added to the flask via syringe, and the reaction mixture was stirred for 24 hrs at 80∼90°C. The solvent was then evaporated in vacuo, and the resulting residue was dissolved in CH2Cl2 (50 ml). The organic phase was washed with saturated aqueous NaHCO3 (15ml) and dried over anhydrous Na2SO4. Purification by column chromatography with silica gel yields the corresponding unsymmetrical bipyridyl ligands.
2,6-Dimethyl-4-(pyridin-4-ylethynyl)pyridine (2)
dark brown solid, 32% yield, mp 157∼160□; 1HNMR (300MHz, CDCl3) δ 2.51 (s, 6H), 7.29 (s, 2H), 7.34 (dd, J = 4.5, 1.5Hz, 2H), 8.61 (dd, J = 4.5, 1.5Hz, 2H); 13C-NMR (75MHz, CDCl3) δ 18.4, 90.7, 91.0, 118.0, 125.7, 126.6, 130.8, 149.3, 149.9; MS(EI): 208(M+, 100), 192(15), 180(10), 166(7), 77(10), 51(8). Anal. Calcd for C14H12N2: C, 80.74; H, 5.81; N, 13.45. Found: C, 80.31; H, 5.50; N, 13.22.
2-Chloro-4-(pyridin-4-ylethynyl)pyridine (3)
pale yellow solid, 69% yield, mp 110∼112□; 1HNMR (300MHz, CDCl3) δ 7.31 (dd, J = 5.1, 1.2Hz, 1H), 7.38 (dd, J = 4.5, 1.5Hz, 2H), 7.45 (s, 1H), 8.40 (d, J = 5.1Hz, 1H), 8.64 (dd, J = 4.5, 1.5Hz, 2H); 13C-NMR (75MHz, CDCl3) δ 89.4, 92.1, 124.5, 125.8, 126.4, 129.9, 133.2, 150.0, 150.2, 152.1; MS(EI): 216(41), 214(M+, 100), 179(65), 152(16), 126(10), 99(11), 74(11), 51(5). Anal. Calcd for C12H7ClN2: C, 67.15; H, 3.29; N, 13.05. Found: C, 67.05; H, 3.23; N, 12.71.
3,5-Dichloro-4-(pyridin-4-ylethynyl)pyridine (4)
white solid, 31% yield, mp 169∼171□; 1HNMR (300MHz, CDCl3) δ 7.47 (dd, J = 4.5, 1.5Hz, 2H), 8.55 (s, 2H), 8.68 (dd, J = 4.5, 1.5Hz, 2H); 13C-NMR (75MHz, CDCl3) δ 85.0, 100.9, 125.9, 129.7, 129.8, 133.7, 147.3, 150.3; MS(EI) 252(12), 250(64), 248(M+,100), 213(20), 186(12), 151(14), 124(12), 98(15), 74(7). Anal. Calcd for C12H6Cl2N2: C, 57.86; H, 2.43; N, 11.25. Found: C, 57.60; H, 2.38; N, 10.87.
Self-assembly of Square 5
A CD3NO2 solution of 2,6-dimethyl-4-(pyridin-4-ylethynyl)pyridine 2 (1.85 mg, 8.88 μmmol) was transferred into a vial charged with 0.2 mL CD3NO2 solution of cis-(Me3P)2Pt(OTf)2 1 (5.73 mg, 8.88 μmmol) and washed with excess CD3NO2 (3 × 0.15 mL) to ensure quantitative transfer. The mixture was stirred for 24 hours to generate a clear pale yellow solution, which was then placed into a NMR tube for analysis. Evaporating the solvent under the vacuum pump resulted in the solid product. Yield: 95%. 1H NMR (CD3NO2, 300 MHz): δ 8.65 (m, 8H, Hα-Py), 7.68 (d, J = 5.7 Hz, 8H, Hβ-Py), 7.54 (s, 8H, Hβ’-Py), 2.83 (s, 24H, CH3-Py), 1.97 (d, 2JP-H = 12 Hz, 36H, PCH3), 1.73 (d, 2JP-H = 12 Hz, 36H, PCH3). 31P {1H} NMR (CD3NO2, 121.4 MHz): δ-18.79 (d, 1JPt-P = 3266.6 Hz, 2JP-P’ = 23.8 Hz), -25.94 (d, 1JPt-P = 3578.4 Hz, 2JP-P’ = 23.8 Hz).
Self-assembly of Square 6
The dipyridyl donor ligand 2-chloro-4-(pyridin-4-ylethynyl)pyridine 3 (2.17 mg, 10.10 μmmol) and the organoplatinum 90° acceptor cis-(Me3P)2Pt(OTf)2 1 (6.52 mg, 10.10 μmmol) were weighed and added to respective glass vials. To the vial containing donor ligand 3 was charged 0.2 mL of CD3NO2 solvent, which was subsequently transferred to the vial filled with a 0.2 mL CD3NO2 solution of 1. This process was repeated for three times (3 × 0.15 mL) to ensure quantitative transfer of the donor to the acceptor. The reaction solution was then stirred at room temperature for 24 h to yield a homogeneous colorless solution. The solution was transferred into the NMR tube to collect 1H and 31P NMR spectra. Solid product was obtained by removing the solvent under vacuum pump. Yield: 96%. 1H NMR (CD3NO2, 300 MHz): δ 8.96-9.02 (m, 12H, Hα- and Hα’-Py), 7.76-7.98 (m, 16H, Hβ- and Hβ’-Py), 1.66-1.83 (m, 72H, PCH3). 31P {1H} NMR (CD3NO2, 121.4 MHz): δ -26.59 - -27.13 (three doublets, 1JPt-P = 3302.8 Hz; -26.59, -26.79, 2JP-P’ = 24.4 Hz; -26.67, -26.87, 2JP-P’ = 24.4 Hz; -26.92, -27.13, 2JP-P’ = 25.2 Hz), -27.82 - -28.17 (two doublets, 1JPt-P = 3142.8 Hz; -27.82, -28.02, 2JP-P’ = 24.4 Hz; -27.97, -28.17, 2JP-P’ = 24.4 Hz).
Self-assembly of Square 7
To a 0.2 mL CD3NO2 solution of cis-(Me3P)2Pt(OTf)2 1 (6.01 mg, 9.31 μmmol) in a vial was added a 0.2 mL CD3NO2 solution of 3,5-dichloro-4-(pyridin-4-ylethynyl)pyridine 4 (2.32 mg, 9.31 μmmol). This process was repeated for three times (3 × 0.15 mL) to complete transfer of the donor to the acceptor. The reaction mixture was stirred at ambient temperature for 24 h to produce a clear colorless solution. The solution was transferred into the NMR tube for 1H and 31P NMR spectra collection. The solid product was obtained by removing CD3NO2 under vacuum pump. Yield: 93%. 1H NMR (CD3NO2, 300 MHz): δ 8.98-9.28 (m, 16H, Hα- and Hα’-Py), 7.82-7.89 (m, 8H, Hβ-Py), 1.65-1.79 (m, 72H, PCH3). 31P {1H} NMR (CD3NO2, 121.4 MHz): δ -26.58 - -26.92 (m, 1JPt-P = 3284.1 Hz), -27.67 - -28.07(m, 1JPt-P = 3156.1 Hz).
Supplementary Material
Acknowledgement
P.J.S. thanks the NIH (Grant GM-057052) and the NSF (Grant CHE-0306720) for financial support. B.H.N. thanks the NIH (Grant GM-080820) for financial support. This work was also supported by the 2008 Research Fund of the University of Ulsan.
References
- (1).(a) Stang PJ, Olenyuk B. Acc. Chem. Res. 1997;30:502–518. [Google Scholar]; (b) Leininger S, Olenyuk B, Stang PJ. Chem. Rev. 2000;100:853–908. doi: 10.1021/cr9601324. [DOI] [PubMed] [Google Scholar]; (c) Seidel SR, Stang PJ. Acc. Chem. Res. 2002;35:972–983. doi: 10.1021/ar010142d. [DOI] [PubMed] [Google Scholar]; (d) Fujita M. Chem. Soc. Rev. 1998;27:417–425. [Google Scholar]; (e) Swiegers GF, Malefetse TJ. Chem. Rev. 2000;100:3483–3537. doi: 10.1021/cr000023w. [DOI] [PubMed] [Google Scholar]; (f) Holliday BJ, Mirkin CA. Angew. Chem., Int. Ed. 2001;40:2022–2043. [PubMed] [Google Scholar]; (g) Cotton FA, Lin C, Murillo CA. Acc. Chem. Res. 2001;34:759–771. doi: 10.1021/ar010062+. [DOI] [PubMed] [Google Scholar]; (h) Fujita M, Tominaga M, Hori A, Therrien B. Acc. Chem. Res. 2005;38:369–378. doi: 10.1021/ar040153h. [DOI] [PubMed] [Google Scholar]; (i) Fiedler D, Leung DH, Bergman RG, Raymond KN. Acc. Chem. Res. 2005;38:349–358. doi: 10.1021/ar040152p. [DOI] [PubMed] [Google Scholar]; (j) Severin K. Chem. Commun. 2006:3859–3867. doi: 10.1039/b606632c. [DOI] [PubMed] [Google Scholar]; (k) Pitt MA, Johnson DW. Chem. Soc. Rev. 2007;36:1441–1453. doi: 10.1039/b610405n. [DOI] [PubMed] [Google Scholar]
- (2).(a) Cotton FA, Lin C, Murillo CA. J. Am. Chem. Soc. 2001;123:2670–2671. doi: 10.1021/ja004149m. [DOI] [PubMed] [Google Scholar]; (b) Yoshizawa M, Takeyama Y, Kusukawa T, Fujita M. Angew. Chem., Int. Ed. 2002;41:1347–1349. doi: 10.1002/1521-3773(20020415)41:8<1347::aid-anie1347>3.0.co;2-x. [DOI] [PubMed] [Google Scholar]; (c) Murase T, Sato S, Fujita M. Angew. Chem., Int. Ed. 2007;46:1083–1085. doi: 10.1002/anie.200603561. [DOI] [PubMed] [Google Scholar]; (d) Balzani V, Bergamini G, Campagna S, Puntoriero F. Top. Curr. Chem. 2007;280:1–36. [Google Scholar]; (e) Yang H-B, Ghosh K, Zhao Y, Northrop BH, Lyndon MM, Muddiman DC, White HS, Stang PJ. J. Am. Chem. Soc. 2008;130:839–841. doi: 10.1021/ja710349j. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (3).(a) Caulder DL, Raymond KN. Angew. Chem., Int. Ed. Engl. 1997;36:1440–1442. [Google Scholar]; (b) Stang PJ. Chem. Eur. J. 1998;4:19–27. [Google Scholar]; (c) Gianneschi NC, Tiekink ERT, Rendina LM. J. Am. Chem. Soc. 2000;122:8474–8479. [Google Scholar]; (d) Tabellion FM, Seidel SR, Arif AM, Stang PJ. J. Am. Chem. Soc. 2001;123:7740–7741. doi: 10.1021/ja015784a. [DOI] [PubMed] [Google Scholar]; (e) Iengo E, Zangrando E, Minatel R, Alessio E. J. Am. Chem. Soc. 2002;124:1003–1013. doi: 10.1021/ja016162s. [DOI] [PubMed] [Google Scholar]; (f) Campbell K, Kuehl CJ, Ferguson MJ, Stang PJ, Tykwinski RR. J. Am. Chem. Soc. 2002;124:7266–7267. doi: 10.1021/ja025773x. [DOI] [PubMed] [Google Scholar]; (g) Iengo E, Zangrando E, Geremia S, Graff R, Kieffer B, Alessio E. Chem. Eur. J. 2002;8:4670–4674. doi: 10.1002/1521-3765(20021018)8:20<4670::AID-CHEM4670>3.0.CO;2-X. [DOI] [PubMed] [Google Scholar]; (h) Su C-Y, Cai Y-P, Chen C-L, Smith MD, Kaim W, Zur Loye H-C. J. Am. Chem. Soc. 2003;125:8595–8613. doi: 10.1021/ja034267k. [DOI] [PubMed] [Google Scholar]; (i) Addicott C, Das N, Stang PJ. Inorg. Chem. 2004;43:5335–5338. doi: 10.1021/ic049326p. [DOI] [PubMed] [Google Scholar]; (j) Natarajan R, Savitha G, Moorthy JN. Cryst. Growth Des. 2005;5:69–72. [Google Scholar]; (k) Steel PJ. Acc. Chem. Res. 2005;38:243–250. doi: 10.1021/ar040166v. [DOI] [PubMed] [Google Scholar]; (l) Yang H-B, Ghosh K, Northrop BH, Stang PJ. Org. Lett. 2007;9:1561–1564. doi: 10.1021/ol070371l. [DOI] [PubMed] [Google Scholar]; (m) Yamaguchi T, Tashiro S, Tominaga M, Kawano M, Ozeki T, Fujita M. Chem. Asian J. 2007;2:468–476. doi: 10.1002/asia.200600429. [DOI] [PubMed] [Google Scholar]; (n) Wang Y, Cheng P, Song Y, Liao D-Z, Yan S-P. Chem. Eur. J. 2007;13:8131–8138. doi: 10.1002/chem.200700431. [DOI] [PubMed] [Google Scholar]
- (4).(a) Yang H-B, Das N, Huang F, Hawkridge AM, Muddiman DC, Stang PJ. J. Am. Chem. Soc. 2006;128:10014–10015. doi: 10.1021/ja063377z. [DOI] [PubMed] [Google Scholar]; (b) Yang H-B, Hawkridge AM, Huang SD, Das N, Bunge SD, Muddiman DC, Stang PJ. J. Am. Chem. Soc. 2007;129:2120–2129. doi: 10.1021/ja066804h. [DOI] [PubMed] [Google Scholar]; (c) Yang H-B, Ghosh K, Northrop BH, Zheng Y-R, Lyndon MM, Muddiman DC, Stang PJ. J. Am. Chem. Soc. 2007;129:14187–14189. doi: 10.1021/ja073744m. [DOI] [PMC free article] [PubMed] [Google Scholar]; (d) Northrop BH, Glöckner A, Stang PJ. J. Org. Chem. 2008;73:1787–1794. doi: 10.1021/jo702380b. [DOI] [PubMed] [Google Scholar]
- (5).(a) Suzuki K, Kawano M, Sato S, Fujita M. J. Am. Chem. Soc. 2007;129:10652–10653. doi: 10.1021/ja073629b. [DOI] [PubMed] [Google Scholar]; (b) Murase T, Sato S, Fujita M. Angew. Chem. Int. Ed. 2007;46:5133–5136. doi: 10.1002/anie.200700793. [DOI] [PubMed] [Google Scholar]
- (6).Rang A, Engeser M, Maier NM, Nieger M, Lindner W, Schalley CA. Chem. Eur. J. 2008;14:3855–3859. doi: 10.1002/chem.200800113. [DOI] [PubMed] [Google Scholar]
- (7).(a) Wu A, Isaacs L. J. Am. Chem. Soc. 2004;126:10035–10043. doi: 10.1021/ja0486972. [DOI] [PubMed] [Google Scholar]; (b) Liu S-M, Ruspic C, Mukhopadhyay P, Chakrabarti S, Zavalij PY, Isaacs L. J. Am. Chem. Soc. 2005;127:15959–15967. doi: 10.1021/ja055013x. [DOI] [PubMed] [Google Scholar]; (c) Mukhopadhyay P, Zavalij PY, Isaacs L. J. Am. Chem. Soc. 2006;128:14093–14102. doi: 10.1021/ja063390j. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (8).(a) Barboiu M, Petit E, van der Lee A, Vaughan G. Inorg. Chem. 2006;45:484–486. doi: 10.1021/ic0517220. [DOI] [PubMed] [Google Scholar]; (b) Nitschke JR. Acc. Chem. Res. 2007;40:103–112. doi: 10.1021/ar068185n. [DOI] [PubMed] [Google Scholar]; (c) Legrand Y-M, van der Lee A, Barboiu M. Inorg. Chem. 2007;46:9540–9547. doi: 10.1021/ic701122a. [DOI] [PubMed] [Google Scholar]; (d) Hutin M, Cramer CJ, Gagliardi L, Shahi ARM, Bernardinelli G, Cerny R, Nitschke JR. J. Am. Chem. Soc. 2007;129:8774–8780. doi: 10.1021/ja070320j. [DOI] [PubMed] [Google Scholar]; (e) Langner A, Tait SL, Lin N, Rajadurai C, Ruben M, Kern K. Proc. Natl. Acad. Sci. USA. 2007;104:17927–17930. doi: 10.1073/pnas.0704882104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (9).(a) Chi K-W, Addicott C, Arif AM, Stang PJ. J. Am. Chem. Soc. 2004;126:16569–16574. doi: 10.1021/ja045542l. [DOI] [PubMed] [Google Scholar]; (b) Chi K-W, Addicott C, Moon M-E, Lee HJ, Yoon SC, Stang PJ. J. Org. Chem. 2006;71:6662–6665. doi: 10.1021/jo060971i. [DOI] [PubMed] [Google Scholar]
- (10).Ghosh S, Turner DR, Batten SR, Mukherjee PS. Dalton Trans. 2007:1869–1871. doi: 10.1039/b702353g. [DOI] [PubMed] [Google Scholar]
- (11).Some recent examples:Janzen DE, Patel KN, VanDerveer DG, Grant GJ. Chem. Commun. 2006:3540–3542. doi: 10.1039/b606753k.You C-C, Hippius C, Gruene M, Wuerthner F. Chem. Eur. J. 2006;12:7510–7519. doi: 10.1002/chem.200600413.Ferrer M, Gutierrez A, Mounir M, Rossell O, Ruiz E, Rang A, Engeser M. Inorg. Chem. 2007;46:3395–3406. doi: 10.1021/ic062373s.Ghosh S, Batten SR, Turner DR, Mukherjee PS. Organometallics. 2007;26:3252–3255.Blanco V, Chas M, Abella D, Peinador C, Quintela JM. J. Am. Chem. Soc. 2007;129:13978–13986. doi: 10.1021/ja074721a.Blanco V, Chas M, Abella D, Pía E, Platas-Iglesias C, Peinador C, Quintela JM. Org. Lett. 2008;10:409–412. doi: 10.1021/ol702701t.
- (12).(a) Fuss M, Siehl H-U, Olenyuk B, Stang PJ. Organometallics. 1999;18:758–769. [Google Scholar]; (b) Fan J, Whiteford JA, Olenyuk B, Levin MD, Stang PJ, Fleischer EB. J. Am. Chem. Soc. 1999;121:2741–2752. [Google Scholar]; (c) Yamamoto T, Arif AM, Stang PJ. J. Am. Chem. Soc. 2003;125:12309–12317. doi: 10.1021/ja0302984. [DOI] [PubMed] [Google Scholar]; (d) Chi K-W, Addicott C, Arif AM, Das N, Stang PJ. J. Org. Chem. 2003;68:9798–9801. doi: 10.1021/jo0353785. [DOI] [PubMed] [Google Scholar]; (e) Megyer T, Jude H, Grósz T, Bakó I, Radnai T, Tárkányi G, Pálinkás G, Stang PJ. J. Am. Chem. Soc. 2005;127:10731–10738. doi: 10.1021/ja0523690. [DOI] [PubMed] [Google Scholar]; (f) Tárkányi G, Jude H, Pálinkás G, Stang PJ. Org. Lett. 2005;7:4971–4973. doi: 10.1021/ol051910u. [DOI] [PubMed] [Google Scholar]; (g) Vacek J, Caskey DC, Horinek D, Shoemaker RK, Stang PJ, Michl J. J. Am. Chem. Soc. 2008;130:7629–7638. doi: 10.1021/ja801341m. [DOI] [PubMed] [Google Scholar]
- (13).(a) Yoshizawa M, Nagao M, Kumazawa K, Fujita M. J. Organomet. Chem. 2005;690:5383–5388. [Google Scholar]; (b) Rang A, Engeser M, Maier NM, Nieger M, Lindner W, Schalley CA. Chem. Eur. J. 2008;14:3855–3859. doi: 10.1002/chem.200800113. [DOI] [PubMed] [Google Scholar]
- (14).Stang PJ, Cao DH, Saito S, Arif AM. J. Am. Chem. Soc. 1995;117:6273–6283. [Google Scholar]
- (15).Evans RF, Brown HC. J. Org. Chem. 1962;27:1665–1667. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.