

Abstract
NR2B selective inhibitors show lower side-effects in preclinical pain models than non-selective NMDA receptor (NMDAR) antagonists, but it is unclear whether the improved safety of NR2B selective inhibitors is due to their subtype selectivity or to a unique mode of inhibition of the receptor. In the present study, the analgesic effects of intracerebral bolus injections of conantokin peptides with different NMDAR subunit selectivity were determined in mice by the standard hot-plate test, and following stimuli with acetic acid, formalin and complete Freund’s adjuvant (CFA). In the standard hot-plate model, con-G[S16Y], a NR2B–selective inhibitor, showed the highest analgesic activity among conantokin peptides tested. In the acetic acid- and CFA-induced pain models, con-G[S16Y] and, to a lesser extent, con-G exhibited higher analgesic activity compared to nonselective inhibitors, such as con-R[1-17]. In the formalin test, while all conantokin peptides could partially suppress the first phase response, only con-G[S16Y] and con-G significantly inhibited the second phase response and suppressed paw edema. Our results suggest that the antinociceptive action of the conantokins may be related to their NR2B selectivity and that these peptides may be useful as both neurobiological tools for probing mechanisms of nociception and as therapeutic agents for pain relief.
Keywords: NMDA receptor, NR2B subunit, pain, nociception, antinociception, conantokins
Introduction
Hyperactivation of the N-methyl-D-aspartate receptor (NMDAR) is generally recognized as a key factor in sustaining chronic inflammatory and neuropathic pain (Haley et al., 1990; Fisher et al., 2000; Hewitt, 2000). Hence, functional inhibitors of the NMDAR have garnered considerable attention for their analgesic potential. While uncompetitive NMDAR antagonists such as MK-801 and ketamine effectively relieve pain (Vaccarino et al., 1993) their clinical utility is seriously undermined by psychomimetic side-effects. NMDAR inhibitors selective for NR2B-containing receptors are effective and well-tolerated in rodent models of pain (Taniguchi et al., 1997; Boyce et al., 1999) suggesting the involvement of the NR2B subunit in mediating nociception. This hypothesis has been reinforced by the observations that overexpression of NR2B in mouse forebrain augments sensitivity to inflammatory pain (Wei et al., 2001) and that intrathecal administration of siRNA directed towards the NR2B subunit abrogates formalin-induced pain symptoms in rats (Tan et al., 2005). Furthermore, the contribution of NR2A- and NR2B-containing subunits in the induction of LTP and LTD, respectively (Liu et al., 2004), renders the use of NR2B-specific NMDAR antagonists, rather than non-selective agents, more desirable for the control of pain with minimal cognitive side effects.
The most commonly invoked NR2B-selective analgesics include ifenprodil and structurally related compounds (Parsons, 2001). These agents inhibit NMDAR activity through a high-affinity interaction with the N-terminal leucine/isoleucine/valine-binding protein (LIVBP)-like domain of the NR2B subunit (Perin-Dureau et al., 2002). This mode of binding, subunit-selective and peripheral to the ion channel pore, likely underlies the aforementioned effectiveness of these compounds in alleviating pain at doses that are well tolerated. However, enthusiasm for this class of compounds is mitigated by their capacity to interfere with the potassium channel activity of the human ether-a-go-go-related gene (HERG), the perturbation of which is linked to abnormal cardiac rhythms (McCauley et al., 2004). The conantokin family of peptides from the genus Conus represents an additional class of NMDAR antagonists that bind distal to the ion channel, at a location that either overlaps, or is tightly allosterically coupled to, the glutamate site (Klein et al., 2001; Wittekindt et al., 2001). In contrast to the ifenprodil class of compounds, the conantokins appear to be entirely specific for the NMDAR. In addition, one of the four known members of the conantokin family, viz., conantokin-G (con-G) from Conus geographus displays almost complete specificity for NR2B-containing receptor complexes (Donevan et al., 2000).
A previous investigation in mice has established that intrathecally administered con-G and conantokin-T (con-T) were effective in alleviating pain at doses that did not significantly interfere with motor function (Malmberg et al., 2003). Additionally, the therapeutic ratio for con-G was over 60% higher than that for con-T, which displays similar inhibitory potency for both NR2A- and NR2B-conatining NMDAR complexes. This observation strengthens the concept that NMDAR inhibitors that specifically target the NR2B subunit may be more desirable antinociceptives compared to their non-selective NMDAR counterparts. In the present study, the antinociceptive properties of the conantokins are further investigated using thermal, tissue injury, and inflammation models of pain. Included are conantokin variants having different NMDAR inhibitory potency and different subunit (NR2A and/or NR2B) selectivities. The results point to a contribution for the NR2B subunit in nociception and underscore the value of focusing on NR2B-selective agents for the treatment of pain.
Materials and methods
Peptide synthesis, purification and characterization
The methods for synthesis, purification, and characterization of conantokin variants were similar to those previously described (Dai et al., 2004). The primary sequences of the peptides used in this study are as follows:
| con-G: | GEγγLQγNQγLIRγKSN-NH2 |
| con-G[Q6A]: | GEγγLAγNQγLIRγKSN-NH2 |
| con-G[γ7K]: | GEγγLQKNQγLIRγKSN-NH2 |
| con-G[N8A]: | GEγγLQγAQγLIRγKSN-NH2 |
| con-G[γ14A]: | GEγγLQγNQγLIRAKSN-NH2 |
| con-G[S16Y]: | GEγγLQγNQγLIRAKYN-NH2 |
| Ala-con-G: | GEγγLGKAQALIRAAYA-NH2 |
| con-R[1-17]: | GEγγVAKMAAγLARγNI-NH2 |
| con-T: | GEγγYQKMLγNLRγAEVKKNA-NH2 |
| con-T[R13A]: | GEγγYQKMLγNLAγAEVKKNA-NH2 |
| con-T[γ14A]: | GEγγYQKMLγNLRAAEVKKNA-NH2 |
| con-T[γ10,14K]: | GEγγYQKMLKNLRKAEVKKNA-NH2 |
Animals and peptide administration
Male and female Kuming mice (20-24 g, Beijing Animal Center, China) were used in acetic acid, formalin and complete Freund’s adjuvant (CFA) stimulus experiments. Only female Kuming mice were used in the hot plate tests. Mice were housed in plastic boxes maintained between 24 ± 2 °C and a relative humidity of 50%. Food pellets and water were available ad libitum. All experiments were conducted in accordance with the guidelines of the Beijing Institutes for Biological Science Animal Research Advisory Committee and conformed to the European Community directives for the care and use of laboratory animals. Conantokin peptides were dissolved in 0.9% sterile saline and intracerebrally injected (i.c.v.) in a volume of 20 μl into each mouse. Saline (20 μl) was similarly administered to the control group.
Hot plate test
In the hot plate model, a female mouse was placed on a plate maintained at 55 ± 0.1 °C and the time required to elicit jumping or licking of hind paw (latency) was recorded. A cutoff latency of 60 s was implemented to prevent tissue damage. Only those mice pretesting with a mean latency of less than 20 s were used in the study. The latency was measured at 30, 90, 150, and 240 min after peptide (300-600 pmol, i.c.v.), ifenprodil (25 nmol, i.c.v.), or saline injections.
Acid–induced writhing test
An 0.4 ml injection 1% (v/v) acetic acid in saline solution was administered intraperitoneally (i.p.) into each mouse 30 min after peptide injection (100-300 pmol/20 μl, i.c.v.). After 5 min, the number of abdominal contortions (writhes) that occurred was recorded over a 15 min period.
Formalin test
A 20 μl volume of 5% (v/v) formalin in saline was injected into the subplantar region of the right hind paw of each mouse 30 min after peptide administration (100 pmol, i.c.v.). The time spent licking or biting the affected paw was recorded in 5 min intervals for a total of 60 min. The inflammatory edematogenic response was evaluated 60 min after formalin injection by using a vernier caliper to measure paw diameter. The effects of peripherally administered conantokin peptides were also tested by subcutaneous injection of 10 ⌠l of peptide solution (100-300 pmol in saline) into the dorsal surface of the hind paw 30 min prior to formalin injection (20 μl of the above 5% formalin solution) at the same dorsal surface site.
Inflammation-evoked pain model
Inflammation was produced by subcutaneous injection of 20 ⌠l of a 1:1 mineral oil/saline emulsion of complete Freund’s adjuvant (CFA) into the plantar surface of the right hind paw. Conantokin peptides were administered (300 pmol, i.c.v.) approximately 72 h later. Thermal hyperalgesia was determined 2 h after peptide injection by measuring the latency of the afflicted paw of female mice to a radiant heat stimulus using the aforementioned hot plate test. As an index of edema, paw diameter was measured immediately after testing.
Assessment of motor function
Motor impairment was determined on an accelerating rotarod treadmill (Ugo Basile, Comerio, Italy) as previously described (Malmberg et al., 2003). The peptides, ifenprodil (25 nmol), or saline were administered i.c.v. to the mice (n = 5) in a volume of 20 μl. At 60 and 240 min following injection, the mice were placed on the rotating rod at a speed of 3 rpm. After 50 s, the rod was accelerated from 3 rpm to 30 rpm over a 5 min interval. The time that the mice remained balanced on the rod was recorded.
Statistical analysis
All data are expressed as the mean ± S.D., and were analyzed using either the unpaired t test or one-way analysis of variance (ANOVA) followed by the Student-Newman-Keul’s test. Differences with a p value < 0.05 were considered to be statistically significant. Indicated n values represent the number of animals tested a single time.
Results
The effects of the conantokins on thermal nociception
At a dose of 300 pmol (i.c.v.), several of the conantokin peptides significantly increased paw withdrawal latency compared with the saline control group (Table 1). The onset of antinociception occurred between 30 and 90 min following peptide administration and peaked near the 150 min time point for NR2B-selective (con-G-based) and non-selective (con-R[1-17] and con-T-based) peptides alike. Significant increases in latencies at 240 min were only observed with con-G[γ7K] and con-G[S16Y]. The highest antinociceptive activity was attained with con-G[S16Y], an NR2B selective inhibitor. This peptide not only significantly increased paw withdrawal latency compared to the saline control, but was the only conantokin whose effects differed significantly from those displayed by the other peptides. As determined previously through inhibition of [3H]MK-801 binding to crude rat membrane preparations (Klein et al., 2003) this variant manifested the highest NMDAR inhibitory activity of the con-G-based peptides examined in this study. However, potency in the radioligand binding assay does not strictly correlate with increased paw withdrawal latency since con-R[1-17], which is the most potent conantokin inhibitor of [3H]MK-801 binding examined in the current study, is a less effective antinociceptive than con-G[S16Y]. Similar observations apply to NR2B-selective con-G[Q6A], con-G[γ7K], con-G[N8A], con-G[γ14A], and Ala-con-G. These peptides inhibit [3H]MK-801 binding with IC50 values comparable to that of con-G[S16Y], but do not provide the same degree of antinociception. Peptides manifesting activity at NR2A- and NR2B-containing receptors, namely con-R[1-17], con-T[R13A], con-T[γ14A], and con-T[γ10,14K], were not especially effective in alleviating thermal nociception, while con-T was without effect in this regard. At some time points, higher doses (600 pmol, i.c.v.) of con-G, con-G[γ7K], con-G[S16Y] prolonged paw withdrawal latencies beyond that observed at 300 pmol. In the case of Ala-con-G and con-R[1-17], the 600 pmol injection failed to significantly increase latency at any time following peptide administration compared with the lower dose. Ifenprodil, at a dose approximately 80–fold greater than conantokins, had no effect in this assay.
Table 1.
Effects of conantokins on thermal nociception in the hot-plate testa
| Paw withdrawal latency before peptide administration (s) | Paw withdrawal latency after peptide administrationc (s)
|
|||||
|---|---|---|---|---|---|---|
| Peptide | IC50 (μM)b | 30 min | 90 min | 150 min | 240 min | |
| con-G | 0.48 | 11.6 ± 2.1 | 12.2 ± 4.5 | *ˆ25.5 ± 7.9 | 19.4 ± 9.8 | ˆ19.5 ± 8.0 |
| con-G | 11.6 ± 2.5 | 12.6 ± 12.5 | *#ˆ47.1 ± 18.1 | *#ˆ37.6 ± 20.0 | *30.1 ± 14.8 | |
| con-G[Q6A] | 0.51 | 16.1 ± 3.6 | 12.0 ± 5.0 | 24.2 ± 17.6 | *ˆ34.8 ± 19.4 | 26.4 ± 15.6 |
| con-G[γ7K] | 0.22 | 14.9 ± 3.5 | 11.9 ± 4.3 | 16.1 ± 4.8 | 20.3 ± 4.3 | *ˆ23.6 ± 7.3 |
| con-G[γ7K] | 15.1 ± 3.9 | 21.5 ± 12.5 | *#ˆ41.8 ±12.4 | *#ˆ52.0 ± 12.6 | *30.1 ± 14.8 | |
| con-G[N8A] | 0.49 | 11.1 ± 2.5 | 11.6 ± 4.2 | 13.5 ± 4.8 | 12.6 ± 2.5 | 13.8 ± 3.1 |
| con-G[γ14A] | 0.23 | 11.3 ± 1.5 | 10.0 ± 3.3 | 20.4 ± 14.2 | ˆ25.4 ± 9.3 | ˆ22.8 ± 8.6 |
| con-G[S16Y] | 0.14 | 13.4 ± 3.1 | 18.8 ± 11.3 | *¶ˆ43.2 ± 20.0 | *ˆ46.4 ± 17.5 | *¶ˆ45.7 ± 20.0 |
| con-G[S16Y] | 9.0 ± 2.1 | *#¶ˆ48.6 ± 16.8 | *ˆ54.0 ±11.1 | *ˆ56.0 ± 11.3 | *ˆ47.0 ± 21.0 | |
| Ala-con-G | 0.18 | 15.3 ± 3.0 | 20.3 ± 8.6 | 20.9 ± 5.7 | 24.6 ± 10.4 | 16.1 ± 5.0 |
| Ala-con-G | 11.3 ± 1.9 | 11.9 ± 3.4 | 22.4 ± 9.5 | 17.1 ± 7.5 | 20.0 ± 16.1 | |
| con-R[1-17] | 0.09 | 15.8 ± 3.6 | 14.6 ± 7.4 | *22.9 ± 6.7 | *ˆ28.4 ± 9.5 | 22.3 ± 9.7 |
| con-R[1-17] | 10.9 ± 2.5 | 10.8 ± 4.3 | *ˆ27.5 ± 8.8 | *ˆ29.9 ± 12.9 | ˆ25.0 ±16.2 | |
| con-T | 0.40 | 11.9 ± 2.9 | 13.1 ± 3.1 | 15.4 ± 4.8 | 16.5 ± 4.6 | 16.8 ± 4.5 |
| con-T [R13A] | 0.43 | 14.1 ± 5.1 | 15.1 ± 6.3 | 22.7 ± 13.0 | 28.1 ± 14.9 | 28.1 ± 15.6 |
| con-T[γ14A] | 0.20 | 14.2 ± 2.5 | 21.9 ± 14.7 | 25.9 ± 14.6 | *25.4 ± 4.8 | 19.3 ± 7.6 |
| con-T[γ10,14K] | 0.75 | 10.6 ± 1.9 | 12.5 ± 2.5 | 18.9 ± 6.1 | *ˆ30.4 ± 11.7 | ˆ20.1 ± 6.5 |
| ifenprodil | 10.8 ± 2.4 | 11.1 ± 3.7 | 11.1 ± 4.6 | 11.2 ± 3.0 | 12.8 ± 3.0 | |
| saline | 13.4 ± 4.9 | 14.5 ± 3.7 | 16.5 ± 3.7 | 17.8 ± 6.5 | 16.7 ± 3.4 | |
Peptides (300 pmol) or ifenprodil (25 nmol) were injected intracerebrally (i.c.v.) in a volume of 20 μl. Table entries in bold face correspond to values obtained with 600 pmol (i.c.v.) of peptides.
Values represent previously determined peptide concentrations required to achieve 50% inhibition of the spermine potentiation of [3H]MK-801 binding to rat brain membranes [25-27].
Values for paw withdrawal latency are presented as mean ± S.D. (n = 8).
Significantly different from the saline control (p < 0.05, unpaired t test).
Significantly different from the value obtained at 300 pmol for the indicated peptide (p < 0.05, unpaired t test).
Significantly different from the baseline value (prior to peptide administration) (p < 0.05, unpaired t test).
Significantly different from all other peptides at the designated dose and time (p < 0.05, Student-Newman-Keul’s test).
The effects of the conantokins on acid-induced contortion behavior
Intraperitoneal injections of acetic acid typically induce writhing responses that can be quantified and equated with visceral pain. Several peptides displaying varying degrees of effectiveness in reducing thermal nociception were examined in this model. As shown in Figure 1, a 300 pmol dose (i.c.v.) of either con-G or con-G[S16Y] resulted in a significant reduction of abdominal contortions (8.3 ± 6.0 and 6.4 ± 4.6, respectively) compared to the control experiment (17.6 ± 4.8). In contrast, con-G[γ7K], con-R[1-17] and Ala-con-G failed to attenuate writhing responses at the same dosage. A 100 pmol dose of con-G[S16Y] was also significantly effective in modulating responses to the acid stimulus.
Figure 1.
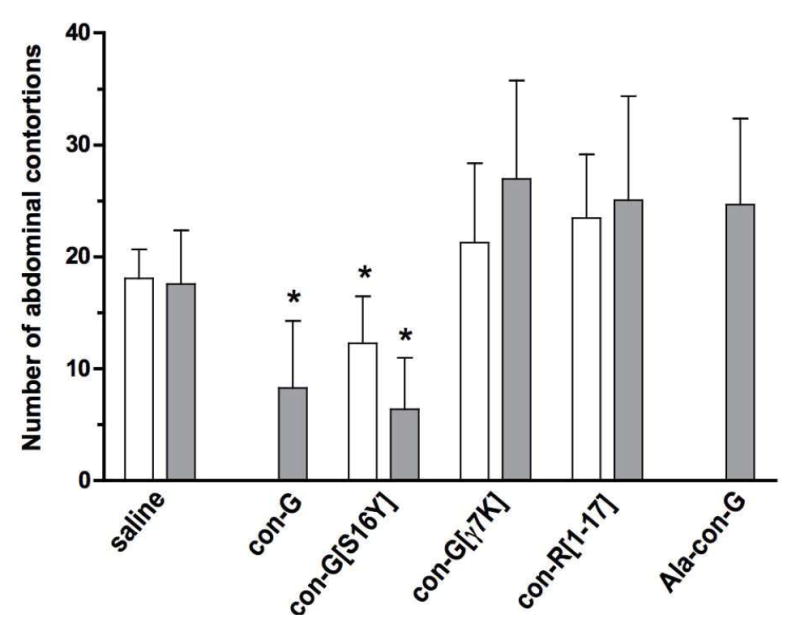
Effects of conantokin peptides on the number of abdominal contortions induced by an 0.4 ml i.p. injection of 1% (v/v) acetic acid in saline solution 30 min after i.c.v. 100 pmol (white bars) and 300 pmol (gray bars) conantokin administration. Counting of contortions began 5 min after acetic acid injection and was continued for a 15 min period. The data are presented as mean ± S.D (n = 8). *Significantly different from control (p < 0.05, unpaired t test).
Antinociceptive effects of the conantokins in the formalin test
Two distinct phases of nociceptive behaviors (Tjolsen et al., 1993) were produced after intraplantar (i.pl.) injection of formalin. The first phase, which stems from primary afferent fiber activation, started immediately after formalin injection and lasted for 3~5 min. The second phase, ascribed to peripheral tissue inflammation, developed 10-15 min following cessation of the first phase and lasted for 20-40 min. Both phases are characterized by the flinching, lifting, shaking, licking and biting of the injected paw. As shown in Figure 2, all of the conantokin peptides tested (100 pmol, i.c.v.) were capable of partially suppressing the first phase response. During the second phase, con-G[S16Y] and con-G were highly effective in reducing the responses to the afflicted paw, whereas neither Ala-con-G nor con-R[1-17] provided antinociception compared to the saline group. Ifenprodil, an NR2B-selective antagonist, was without effect in both phases. At the end of the experiment (i.e., 60 min post-formalin injection) paw width, as a measure of edema, was evaluated. Both con-G and con-G[S16Y] reduced paw swelling to a significant extent (Table 2). Consistent with their ineffectiveness in alleviating second phase responses to formalin, neither Ala-con-G, con-R[1-17], nor ifenprodil had any effect on paw edema. Despite the high degree of antinociception afforded by i.c.v. delivery of con-G and con-G[S16Y] in both phases of the formalin test, subcutaneous injections of these two peptides (100 pmol) 30 min prior to formalin administration did not significantly diminish paw biting/licking behaviors, nor did they reduce the edematogenic response (data not shown).
Figure 2.
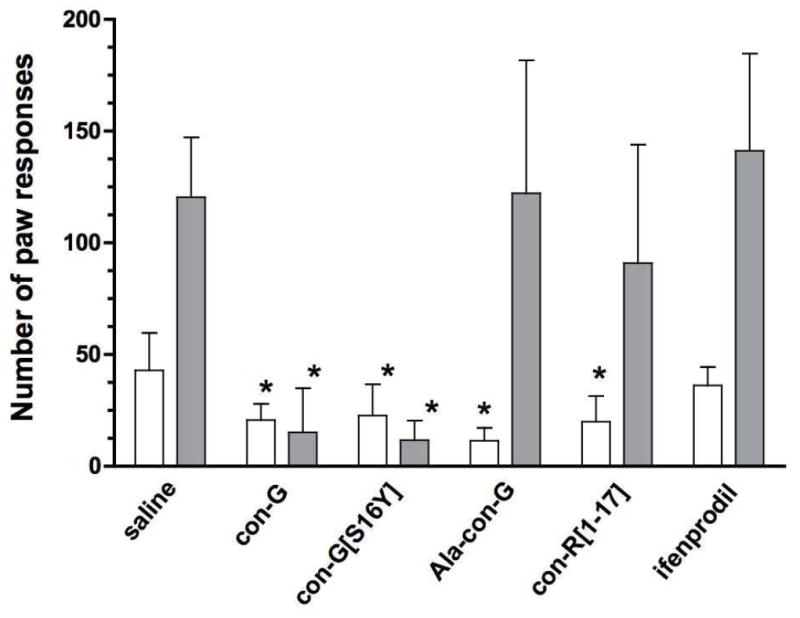
Ability of the conantokin peptides to modulate the first and second phases of nociceptive behavior produced by an injection of formalin into the paw. Conantokins (100 pmol), ifenprodil (25 nmol), or saline, were administered (i.c.v.) 30 min prior to formalin injection. The total time spent responding to the affected paw (licking and/or biting) in the first phase (0-10 min after injection, white bars) and second phase (10-60 min, gray bars). The data are presented as mean ± S.D. (n = 8). *Significantly different from the control (p < 0.05, unpaired t test).
Table 2.
Effects of the conantokins on formalin-induced edemaa
| Peptides b | Injected paw width a (mm)
|
||
|---|---|---|---|
| Before formalin injection | After formalin injectionc | Increase in paw width | |
| con-G* | 1.55 ± 0.02 | 2.38 ± 0.03 | 0.83 ± 0.03 |
| con-G[S16Y]* | 1.57 ± 0.03 | 2.33 ± 0.05 | 0.76 ± 0.05 |
| Ala-con-G | 1.60 ± 0.02 | 2.56 ± 0.05 | 0.97 ± 0.05 |
| con-R[1-17] | 1.60 ± 0.04 | 2.60 ± 0.05 | 1.00 ± 0.08 |
| ifenprodil | 1.59 ± 0.04 | 2.70 ± 0.04 | 1.10 ± 0.05 |
| saline | 1.57 ± 0.03 | 2.53 ± 0.03 | 0.97 ± 0.05 |
Edema was elicited through injection of 20 μl of 5% formalin in saline into the subplantar region of the right hind paw. Data are presented as mean ± S.D. (n = 8).
Peptides (100 pmol) or ifenprodil (25 nmol) were injected intracerebrally (i.c.v.) in a volume of 20 μl.
Measurements were taken 60 min after formalin injection.
Significantly diminished paw swelling compared to the saline control group (p < 0.05, unpaired t test).
Antinociceptive properties of the conantokins in a model of chronic inflammation
Intraplantar injection of 1:1 CFA is known to elicit hyperalgesia and edema that can last for several days. Using the hot plate test, a decrease in paw withdrawal latencies confirmed a hyperalgesic response on days 1 and 3 (p < 0.001 for all day 1 and day 3 measurements compared to their respective baseline values). following CFA administration (Figure 3). The ability of the conantokins (300 pmol i.c.v.) to reduce thermal sensitivity was evaluated 2 h post-injection using the hot plate test. In this experiment, con-G and con-G[S16Y] increased the paw withdrawal latency approximately 1 s and 2 s, respectively, compared to the day 3 controls, although the differences between these latencies and each peptide’s baseline withdrawal threshold value were still significant (p ≤ 0.002). Ala-con-G and con-R[1-17] failed to alter withdrawal latencies. Prior to CFA delivery, average paw width was 1.47 ± 0.04 mm and increased to a post-CFA diameter of 2.35 ± 0.04 mm on day 3. Only those mice injected with con-G[S16Y] showed a small but significant (p = 0.0012; unpaired t test) decrease in paw swelling upon comparison of the day 3 paw diameter before (2.36 ± 0.039 mm) and after (2.26 ± 0.058 mm) peptide injection (measured up to 4 h after peptide injections)
Figure 3.
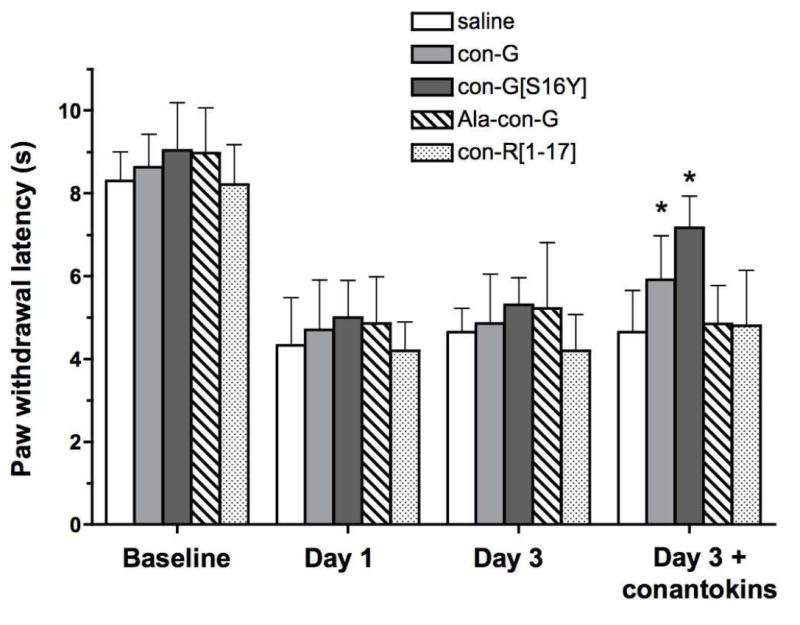
Conantokin effects on paw withdrawal latencies in response to thermal stimulus in the CFA model of chronic inflammation. Mice were grouped and tested before (“Baseline”) and after (“Day 1” and “Day 3”) intraplantar CFA injection. The last grouping of bars represents latencies recorded 2 h after i.c.v. injection of the specific peptide designated for the group. The data are presented as mean ± S.D. (n=8). *Significantly different from the control (i.e., day 3 prior to conantokin administration; p < 0.05, unpaired t test).
Effects of the conantokins on motor function
As the most effective peptide in ameliorating voluntary responses (i.e., paw latencies and paw responses) to painful stimuli, the possibility that con-G[S16Y] might merely be acting through suppression of motor function was tested using the rotarod test. For comparison, the less effective con-R[1-17], as well as ifenprodil, were also examined. From the data summarized in Figure 4, it can be concluded that, relative to the saline control, some toxicity is associated with both con-G[S16Y] and con-R[1-17] at the doses used in our studies. After 60 min, 100 pmol of con-R[1-17] actually impaired motor function to a greater extent than the same dose of con-G[S16Y]. The reverse effect was observed at the 600 pmol does of con-R[1-17] and con-G[S16Y]. No difference between the two conantokin peptides was observed at the 300 pmol dose. Hence, because con-R[1-17] compromised motor function to a degree similar to that observed with con-G[S16Y], the mitigated responses to stimuli of those mice treated with con-G[S16Y] are likely ascribable to improved antinociception rather than peptide-induced impairment of coordination. Also, for the 300 and 600 pmol doses of con-G [S16Y], no significant differences were noted between the 60 and 240 min test times.
Figure 4.
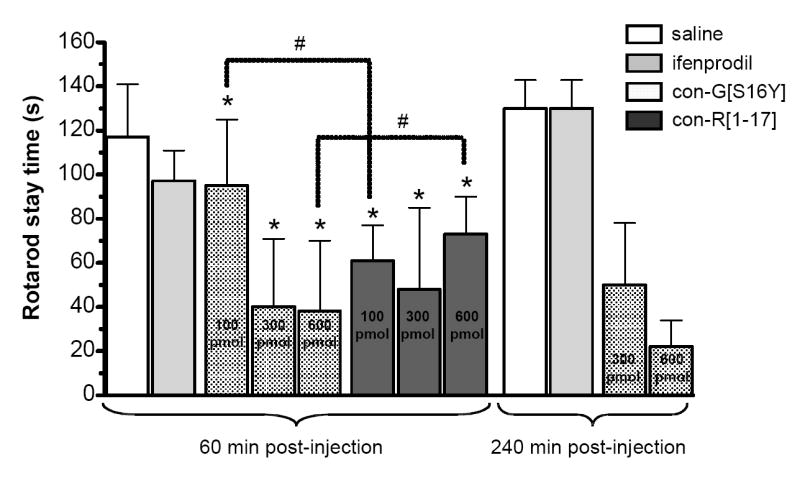
Conantokin effects on motor function using the rotarod test. Con-G[S16Y] or con-R[1-17] at the indicated doses, ifenprodil (25 nmol), or saline were administered (i.c.v.) to the mice prior to placement on the rotarod at 60 min or 240 min post-injection. The stay times are presented as mean ± S.D. (n=8). *Significantly different from the saline control; #indicated pairs are significantly different (p < 0.05, unpaired t test).
Discussion
The aim of the present study was to evaluate the antinociceptive properties of the conantokins in an array of mouse pain models, with emphasis on the contribution of non- and NR2B-selective peptides to antinociception. Excepting the first phase of the formalin test, the ability of con-G[S16Y] and, to a lesser extent con-G, to curtail responses to painful stimuli and formalin-induced edema to a greater extent than the non-selective conantokins suggests that the action of these peptides may be related to their NR2B selectivity. However, as demonstrated through the screening of numerous con-G-based peptides in the hot-plate test, NR2B-selectivity alone is insufficient for antinociceptive character. For example, con-G[N8A], con-G[γ14A] and Ala-con-G are ineffective at antinociception at all time points examined, despite exhibiting NR2B-selectivity. Few clues concerning the molecular features of con-G[S16Y] that underlie its high antinociceptive effectiveness are provided from the results of in vitro pharmacology since IC50 values obtained from previous radioligand binding studies (Blandl et al., 1998; Blandl et al., 2001; Warder et al., 2001; Klein et al., 2003) do not strictly parallel the results of this in vivo study. For example, while con-G[S16Y] manifests a 3.5-fold increase in potency in the [3H]MK-801 binding assay compared to con-G, (IC50 con-G = 0.48 μM; IC50 con-G[S16Y] = 0.18 μM), a similar increase in NMDAR inhibitory activity displayed by Ala-con-G (IC50 Ala-con-G = 0.18 μM) is not reflected in the antinociceptive properties of this derivative relative to the con-G parent in the hot-plate test. Despite the improvement in in vitro and in vivo effectiveness associated with the Ser→Tyr replacement at position 16, it is unlikely that this derives from a direct involvement of the Tyr side-chain with the cognate binding site for conantokins on the NMDAR. This is concluded from previous truncation studies with con-G that revealed that residues 13-17 are not especially critical to NMDAR activity (Blandl et al., 1998). Hence, the Ser→Tyr substitution augments NMDAR inhibitory activity in an indirect fashion, perhaps by rendering the resultant peptide less susceptible to proteolytic degradation. Based on the identical circular dichroism-derived helical contents of con-G and con-G[S16Y] variant in both apo- and Ca2+-bound states (5% and 50%, respectively) (Blandl et al., 1998; Klein et al., 2003), this putative resistance to biodegradation cannot be ascribed to an alteration in secondary structure. Furthermore, the observed inactivity of con-T despite its high apo- and Ca2+-saturated helicities (55% and 82%, respectively) fails to support any correlation between conantokin secondary structure and antinociception with respect to thermal stimuli (Warder et al., 2001). It is conceivable that the superior antinociceptive properties of con-G[S16Y] are related to the increase in lipophilicity imparted by the Ser→Tyr replacement. This sequence element may facilitate peptide accumulation on the neuronal membrane, raising the effective concentration in the vicinity of the NMDAR target and shielding the peptide from proteolytic enzymes. Support for this is provided by direct binding studies of radiolabeled con-G[S16Y] to adult rat brain sections (Klein et al., 2003). In this investigation, approximately 80% of peptide was associated with a low-affinity, non-receptor site, whereas the remainder was tightly and specifically bound to the receptor. In contrast, Ala-con-G manifested only the high-affinity NMDAR-specific component of binding and therefore does not pre-adsorb to the membrane surface.
As observed in the thermal nociception paradigm, con-G and conG[S16Y] were effective in reducing acetic acid-induced acute visceral pain whereas other NR2B-selective peptides, viz., con-G[γ7K] and Ala-con-G, as well as the non-selective con-R[1-17], had no effect in this assay. This is in contrast to the first phase of nociceptive behavior following formalin injection into the paw, in which all tested peptides, including con-G, conG[S16Y], con-G[γ7K], Ala-con-G, and con-R[1-17] significantly reduced the time spent responding to the afflicted paw. It thus appears that a reduction in nociceptive behaviors in the short-lived first phase can be achieved with subunit-selective and non-selective conantokins alike. These observations are consistent with those previously reported for 100 pmol doses of con-G and con-T in the first phase of the formalin test (Malmberg et al., 2003) although the percent inhibition of responses was lesser (25-35%) than those associated with the current work (50-75%). This may be attributable to the different modes of peptide administration (intrathecal versus i.c.v.) and time of agent delivery (10 min prior versus 30 min prior to formalin injection). Based on previous reports that estimated a median toxic dose of ca. 300 pmol for the con-G, -T (i.t. delivery), and –R (i.c.v. delivery) as determined by the rotarod test (White et al., 2000; Malmberg et al., 2003), it is possible that the general nature of conantokin effects on the first phase of the formalin test reflects a slight diminution of motor function rather than true antinociception. This conclusion is supported by the rotarod results presented in this study, which reveal that 100 pmol doses (i.c.v.) of both con-G[S16Y] and con-R[1-17] effect similar decreases in motor coordination. In contrast to the results of the first phase, suppression of second phase formalin responses and a reduction of paw swelling was limited to con-G and conG[S16Y]. These outcomes are consistent with the involvement of NMDARs in mediating the tonic phase following formalin injection (Coderre and Melzack, 1992) and, more specifically, are in support of evidence that couples NR2B-containing subunits in the central nervous system to formalin-induced tonic pain (Gaunitz et al., 2002; Tan et al., 2005). As established in a previous study (Coderre, 1993), the NR2B-selective small-molecule, ifenprodil, was ineffective in both the early and late phases of the test, a possible consequence of the different NMDAR binding sites targeted by ifenprodil and the conantokins (Gaunitz et al., 2002; Perin-Dureau et al., 2002; Tsai et al., 2005). Subcutaneous injections of con-G and con-G[S16Y] failed to diminish formalin-evoked behaviors and did not reduce paw swelling. Despite the expression of NR2B subunits on primary afferent neurons (Ma and Hargreaves, 2000) these null results indicate that conantokin antinociception involves inhibition of NR2B-containing NMDARs through action at central, rather than peripheral, sites. In the CFA model of chronic inflammation, con-G[S16Y] and con-G were capable of reversing thermal hyperalgesia, and increased paw withdrawal latencies to 80% of pre-administration values.
The mechanism(s) by which i.c.v. administered conantokins slightly reduce edema in both the formalin and CFA models are not obvious, since NMDAR antagonists such as ifenprodil or MK-801 are not known to decrease swelling in inflammatory pain models. However, it is well accepted that such peripheral inflammatory stimuli can induce pain sensitization through enhanced activity of spinal NMDARs and the concomitant increase in glutamate and aspartate release in the dorsal horn (Ren et al., 1992; Lawand et al.,1997; Hama et al., 2003). In particular, the expression and activity of NR2B-containing receptors appears to be upregulated under such conditions (Tan et al., 2005; Iwata et al., 2007). Increased expression of NR2B receptors in the anterior cingulate cortex during peripheral inflammation has also been noted (Wu et al., 2005). The modulation of hyperalgesia herein observed following central administration of the conantokins is consistent with these observations. In a rat model of arthritis it has been demonstrated that i.t. injection of CNQX, an AMPA/kainate receptor antagonist, was effective in reducing knee joint swelling, whereas treatment with AP7, a competitive antagonist of the glutamate site on the NMDAR, had no effect (Sluka et al., 1994). This observation supports glutamatergic transmission as one component of the edematogenic response. Hence, it is possible that centrally-delivered NR2B-specific conantokins can uniquely alter glutamate-evoked NMDAR activity in a manner that leads to modest reductions in paw swelling observed in this study.
Our results raise an obvious question as to why NR2B-selective con-G[S16Y] and, to a lesser extent, con-G are superior analgesics to non-selective conantokins such as con-R[1-17], which interact with similar potency at both NR2A- and NR2B-containing receptor complexes (White et al., 2000). Presumably, as in the case of the non-selective competitive glutamate antagonist AP5, an agent that inhibits NR2B-containing NMDARs should demonstrate analgesia irrespective of the level of NR2A interaction (McKenna and Melzack, 2001). However, it is possible that the aforementioned ability of con-G[S16Y] to pre-adsorb to the neuronal membrane facilitates NMDAR binding in a mechanism reminiscent of that proposed for the interaction of neuropeptide Y with its G protein-coupled receptor targets (Bader et al., 2001). A longer physiological half-life of con-G and con-G[S16Y], possibly related to the membrane partitioning mechanism--including resistance to proteolysis--may also be operating to increase their effectiveness. Another possible explanation for the higher antinociceptive activity of NR2B-selective versus non-selective conantokins may be related to the extrasynaptic/synaptic distribution of NR2A and NR2B subunits. In adult animals, NR2B represents the major NR2 component or extrasynaptic NMDARs, while the NR2A subunit is confined to the synaptic receptor pool (Li et al., 1998, Thomas et al., 2006). If opposing roles for pain mediation exist between the synaptic and extrasynaptic populations, as has been demonstrated for synaptic plasticity and extracellular signal-regulated kinases (ERK) signalling (Massey et al., 2004; Ivanov et al., 2006), this could account for the enhanced antinociceptive properties of the NR2B-selective (extrasynaptically targeted) versus the non-selective (synaptically and extrasynaptically targeted) conantokins. Support for this possibility is provided by studies that have demonstrated that a potent phosphatidylinositol 3-kinase inhibitor reduces inflammatory pain and is linked to a reduction in both ERK signalling and NR2B phosphorylation (Pezet et al., 2008). When considering these data in tandem with the observation that con-G reduces NR2B phosphorylation in hippocampal cultures (Waxman and Lynch, 2005), it is reasonable to infer that the conantokins may be involved in altering ERK signalling. As such, non-selective conantokins may exert opposite effects in the ERK pathway (based on the targeting of synaptic and extrasynaptic NMDARs alike), whereas the NR2B-selective conantokins do not produce a cancellation effect, as these peptides are primarily directed to the extrasynaptic NMDAR pool. Futhermore, the antinociceptive effectiveness of con-G and con-G[S16Y] following the i.c.v. route of administration suggests that cortical NR2B-containing NMDARs contribute to nociception, although diffusion to spinal sites cannot be discounted. Taken together, our results suggest that further studies of con-G and the S16Y variant are warranted, not only for their utility as neurobiological tools, but as potential templates for the design of compounds that can effectively alleviate NMDAR-mediated nociception.
Footnotes
This work was supported by Grant 30470388 from the Natural Science Foundation of China (to Q.Y.D.) and Grant 2006AA09Z404 from the High Technology Program of Oceans in China (to Q.Y.D.) and Grant HL-019982 from the National Institutes of Health (to F.J.C.)
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Bader R, Bettio A, Beck-Sickinger AG, Zerbe O. Sturcture and dynamics of micelle-bound neuropeptide Y: Comparison with unligated NPY and implications for receptor selection. J Mol Biol. 2001;305:307–329. doi: 10.1006/jmbi.2000.4264. [DOI] [PubMed] [Google Scholar]
- Blandl T, Prorok M, Castellino FJ. NMDA-receptor antagonist requirements in conantokin-G. FEBS Lett. 1998;435:257–262. doi: 10.1016/s0014-5793(98)01077-1. [DOI] [PubMed] [Google Scholar]
- Blandl T, Zajicek J, Prorok M, Castellino FJ. Sequence requirements for the N-methyl-D-aspartate receptor antagonist activity of conantokin-R. J Biol Chem. 2001;276:7391–7396. doi: 10.1074/jbc.M006648200. [DOI] [PubMed] [Google Scholar]
- Boyce S, Wyatt A, Webb JK, O’Donnell R, Mason G, Rigby M, Sirinathsinghji D, Hill RG, Rupniak NM. Selective NMDA NR2B antagonists induce antinociception without motor dysfunction: correlation with restricted localisation of NR2B subunit in dorsal horn. Neuropharmacology. 1999;38:611–23. doi: 10.1016/s0028-3908(98)00218-4. [DOI] [PubMed] [Google Scholar]
- Coderre T, Melzack R. The contribution of excitatory amino acids to central sensitization and persistent nociception after formalin-induced tissue injury. J Neurosci. 1992;12:3665–3670. doi: 10.1523/JNEUROSCI.12-09-03665.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Coderre TJ. Potent analgesia induced in rats by combined action at PCP and polyamine recognition sites of the NMDA receptor complex. Eur J Neurosci. 1993;5:390–393. doi: 10.1111/j.1460-9568.1993.tb00506.x. [DOI] [PubMed] [Google Scholar]
- Dai QY, Prorok M, Castellino FJ. A new mechanism for metal ion-assisted interchain helix assembly in a naturally occurring peptide mediated by optimally spaced γ-carboxyglutamic acid residues. J Mol Biol. 2004;336:731–734. doi: 10.1016/j.jmb.2003.12.027. [DOI] [PubMed] [Google Scholar]
- Donevan SD, McCabe RT. Conantokin G is an NR2B-selective competitive antagonist of N-methyl-D-aspartate receptors. Mol Pharmacol. 2000;58:614–623. doi: 10.1124/mol.58.3.614. [DOI] [PubMed] [Google Scholar]
- Fisher K, Coderre TJ, Hagen NA. Targeting the N-methyl-D-aspartate receptor for chronic pain management: preclinical animal studies, recent clinical experience and future research directions. J Pain Symptom Manage. 2000;20:358–373. doi: 10.1016/s0885-3924(00)00213-x. [DOI] [PubMed] [Google Scholar]
- Gaunitz C, Schüttler A, Gillen C, Allgaier C. Formalin-induced changes of NMDA receptor subunit expression in the spinal cord of the rat. Amino Acids. 2002;23:177–182. doi: 10.1007/s00726-001-0125-3. [DOI] [PubMed] [Google Scholar]
- Haley JE, Sullivan AF, Dickenson AH. Evidence for spinal N-methyl-D-aspartate receptor involvement in prolonged chemical nociception in the rat. Brain Res. 1990;518:218–226. doi: 10.1016/0006-8993(90)90975-h. [DOI] [PubMed] [Google Scholar]
- Hama A, Woon Lee J, Sagen J. Differential efficacy of intrathecal NMDA receptor antagonists on inflammatory mechanical and thermal hyperalgesia in rats. Eur J Pharmacol. 2003;459:49–58. doi: 10.1016/s0014-2999(02)02828-5. [DOI] [PubMed] [Google Scholar]
- Hewitt DJ. The use of NMDA-receptor antagonists in the treatment of chronic pain. Clin J Pain. 2000;16:S73–79. doi: 10.1097/00002508-200006001-00013. [DOI] [PubMed] [Google Scholar]
- Ivanov A, Pellegrino C, Rama S, Dumalska I, Salyha Y, Ben-Ari Y, Medina I. Opposing role of synaptic and extrasynaptic NMDA receptors in regulation of the extracellular signal-regulated kinases (ERK) activity in cultured rat hippocampal neurons. J Physiol. 2006;572:789–798. doi: 10.1113/jphysiol.2006.105510. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Iwata H, Takasusuki T, Yamaguchi S, Hori Y. NMDA receptor 2B subunit-mediated synaptic transmission in the superficial dorsal horn of peripheral nerve-injured neuropathic mice. Brain Res. 2007;1135:92–101. doi: 10.1016/j.brainres.2006.12.014. [DOI] [PubMed] [Google Scholar]
- Klein RC, Warder SE, Galdzicki Z, Castellino FJ, Prorok M. Kinetic and Mechanistic Characterization of NMDA Receptor Antagonism by Replacement and Truncation Variants of the Conantokin Peptides. Neuropharmacology. 2001;41:801–810. doi: 10.1016/s0028-3908(01)00119-8. [DOI] [PubMed] [Google Scholar]
- Klein RC, Prorok M, Castellino FJ. Direct binding properties of conantokins to native N-methyl-D-aspartate receptors. J Pept Res. 2003;61:307–317. doi: 10.1034/j.1399-3011.2003.00059.x. [DOI] [PubMed] [Google Scholar]
- Lawand NB, Willis WD, Westlund KN. Excitatory amino acid receptor involvement in peripheral nociceptive transmission in rats. Eur J Pharmacol. 1997;324:169–177. doi: 10.1016/s0014-2999(97)00072-1. [DOI] [PubMed] [Google Scholar]
- Liu L, Wong TP, Pozza MF, Lingenhoehl K, Wang Y, Sheng M, Auberson YP, Wang YT. Role of NMDA receptor subtypes in governing the direction of hippocampal synaptic plasticity. Science. 2004;304:1021–1024. doi: 10.1126/science.1096615. [DOI] [PubMed] [Google Scholar]
- Ma QP, Hargreaves RJ. Localization of N-methyl-D-aspartate NR2B subunits on primary sensory neurons that give rise to small-caliber sciatic nerve fibers in rats. Neuroscience. 2000;101:699–707. doi: 10.1016/s0306-4522(00)00419-x. [DOI] [PubMed] [Google Scholar]
- Malmberg AB, Gilbert H, McCabe RT, Basbaum AI. Powerful antinociceptive effects of the cone snail venom-derived subtype-selective NMDA receptor antagonists conantokins G and T. Pain. 2003;101:109–116. doi: 10.1016/s0304-3959(02)00303-2. [DOI] [PubMed] [Google Scholar]
- Massey PV, Johnson BE, Moult PR, Auberson YP, Brown MW, Molnar E, Collingridge GL, Bashir ZI. Differential roles of NR2A and NR2B-containing NMDA receptors in cortical long-term potentiation and long-term depression. J Neurosci. 2004;24:7821–7828. doi: 10.1523/JNEUROSCI.1697-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McCauley JA, Theberge CR, Romano JJ, Billings SB, Anderson KD, Claremon DA, Freidinger RM, Bednar RA, Mosser SD, Gaul SL, Connolly TM, Condra CL, Xia M, Cunningham ME, Bednar B, Stump GL, Lynch JJ, Macaulay A, Wafford KA, Koblan KS, Liverton NJ. NR2B-selective N-methyl-D-aspartate antagonists: synthesis and evaluation of 5-substituted benzimidazoles. J Med Chem. 2004;47:2089–2096. doi: 10.1021/jm030483s. [DOI] [PubMed] [Google Scholar]
- McKenna JE, Melzack R. Blocking NMDA receptors in the hippocampal dentate gyrus with AP5 produces analgesia in the formalin pain test. Exp Neurol. 2001;172:92–99. doi: 10.1006/exnr.2001.7777. [DOI] [PubMed] [Google Scholar]
- Parsons CG. NMDA receptors as targets for drug action in neuropathic pain. Eur J Pharm. 2001;429:71–78. doi: 10.1016/s0014-2999(01)01307-3. [DOI] [PubMed] [Google Scholar]
- Perin-Dureau F, Rachline J, Neyton J, Paoletti P. Mapping the binding site of the neuroprotectant ifenprodil on NMDA receptors. J Neurosci. 2002;22:5955–5965. doi: 10.1523/JNEUROSCI.22-14-05955.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pezet S, Marchand F, D’Mello R, Grist J, Clark AK, Malcangio M, Dickenson AH, Williams RJ, McMahon SB. Phosphatidylinositol 3-kinase is a key mediator of central sensitization in painful inflammatory conditions. J Neurosci. 2008;28:4261–4270. doi: 10.1523/JNEUROSCI.5392-07.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ren K, Williams GM, Hylden JL, Ruda MA, Dubner R. The intrathecal administration of excitatory amino acid receptor antagonists selectively attenuated carrageenan-induced behavioral hyperalgesia in rats. Eur J Pharmacol. 1993;219:235–243. doi: 10.1016/0014-2999(92)90301-j. [DOI] [PubMed] [Google Scholar]
- Sheng Z, Dai Q, Prorok M, Castellino FJ. Subtype-selective antagonism of N-methyl-D-aspartate receptor ion channels by synthetic conantokin peptides. Neuropharmacology. 2007;53:145–156. doi: 10.1016/j.neuropharm.2007.04.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sluka KA, Jordan HH, Willis WD, Westlund KN. Differential effects of N-methyl-D-aspartate (NMDA) and non-NMDA receptor antagonists on spinal release of amino acids after development of acute arthritis in rats. Brain Res. 1994;664:77–84. doi: 10.1016/0006-8993(94)91956-9. [DOI] [PubMed] [Google Scholar]
- Tan PH, Yang LC, Shih HC, Lan KC, Cheng JT. Gene knockdown with intrathecal siRNA of NMDA receptor NR2B subunit reduces formalin-induced nociception in the rat. Gene Ther. 2005;12:59–66. doi: 10.1038/sj.gt.3302376. [DOI] [PubMed] [Google Scholar]
- Taniguchi K, Shinjo K, Mizutani M, Shimada K, Ishikawa T, Menniti FS, Nagahisa A. Antinociceptive activity of CP-101,606, an NMDA receptor NR2B subunit antagonist. Br J Pharmacol. 1997;122:809–812. doi: 10.1038/sj.bjp.0701445. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tjolsen A, Berge OG, Hunskaar S, Rosland JH, Hole K. The formalin test: an evaluation of method. Pain. 1993;51:5–17. doi: 10.1016/0304-3959(92)90003-T. [DOI] [PubMed] [Google Scholar]
- Tsai VW, Dodd PR, Lewis RJ. The effects of alanine-substituted conantokin-G and ifenprodil on the human spermine-activated N-methyl-D-aspartate receptor. Neuroscience. 2005;130:457–464. doi: 10.1016/j.neuroscience.2004.09.039. [DOI] [PubMed] [Google Scholar]
- Vaccarino AL, Marek P, Kest B, Weber E, Keana JF, Liebeskind JC. NMDA receptor antagonists, MK-801 and ACEA-1011, prevent the development of tonic pain following subcutaneous formalin. Brain Res. 1993;615:331–334. doi: 10.1016/0006-8993(93)90045-o. [DOI] [PubMed] [Google Scholar]
- Warder SE, Blandl T, Klein RC, Castellino FJ, Prorok M. Amino acid determinants for NMDA receptor inhibition by conantokin-T. J Neurochem. 2001;77:812–822. doi: 10.1046/j.1471-4159.2001.00281.x. [DOI] [PubMed] [Google Scholar]
- Waxman EA, Lynch DR. N-methyl-D-aspartate receptor subtype mediated bidirectional control of p38 mitogen-activated protein kinase. J Biol Chem. 2005;280:29322–29233. doi: 10.1074/jbc.M502080200. [DOI] [PubMed] [Google Scholar]
- Wei F, Wang G-D, Kerchner GA, Kim SJ, Xu H-M, Chen Z-C, Zhuo M. Genetic enhancement of inflammatory pain by forebrain NR2B overexpression. Nat Neurosci. 2001;4:164–169. doi: 10.1038/83993. [DOI] [PubMed] [Google Scholar]
- White HS, McCabe RT, Armstrong H, Donevan SD, Cruz LJ, Abogadie FC, Torres J, Rivier JE, Paarmann I, Hollmann M, Olivera BM. In vitro and in vivo characterization of conantokin-R, a selective NMDA receptor antagonist isolated from the venom of the fish-hunting snail Conus radiatus. J Pharmacol Exp Ther. 2000;292:425–432. [PubMed] [Google Scholar]
- Wittekindt B, Malany S, Schemm R, Otvos L, Maccecchini ML, Laube B, Betz H. Point mutations identify the glutamate binding pocket of the N-methyl-D-aspartate receptor as major site of conantokin-G inhibition. Neuropharmacology. 2001;41:753–761. doi: 10.1016/s0028-3908(01)00112-5. [DOI] [PubMed] [Google Scholar]
- Wu LJ, Toyoda H, Zhao MG, Lee YS, Tang J, Ko SW, Jia YH, Shum FW, Zerbinatti CV, Bu G, Wei F, Xu TL, Muglia LJ, Chen ZF, Auberson YP, Kaang BK, Zhuo M. Upregulation of forebrain NMDA NR2B receptors contributes to behavioral sensitization after inflammation. J Neurosci. 2005;25:11107–11116. doi: 10.1523/JNEUROSCI.1678-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]