

Abstract
Under neuropathological conditions, reactive astrocytes release cytokines and chemokines, which act in an autocrine and/or paracrine fashion to modulate production of immunoregulatory factors from cells including microglia, astrocytes and neurons. In this way, astrocytes play an important role in orchestrating immune responses within the central nervous system (CNS). Suppressor Of Cytokine Signaling (SOCS) proteins are endogenous, negative regulators of the JAK/STAT signaling pathway, and function as attenuators of immune and inflammatory responses. As such, SOCS proteins may have critical roles in the CNS under neuroinflammatory conditions. In the inflamed CNS, expression of the IL-6 cytokine family member Oncostatin M (OSM) is elevated; however, its functional effects are not well understood. We demonstrate that OSM is a potent inducer of SOCS-3 in astrocytes. Analysis of the SOCS-3 promoter revealed that an AP-1 element, two IFN-γ activation sequence (GAS) elements, and a GC-rich region are crucial for SOCS-3 gene expression. Using small interfering RNA against STAT-3, as well as a STAT-3 dominant-negative construct, we demonstrate that STAT-3 activation is critical for OSM induction of SOCS-3 expression. The ERK1/2 and JNK pathways also contribute to OSM-induced SOCS-3 gene expression. OSM stimulation led to a time-dependent recruitment of the transcription factors STAT-3, c-Fos, c-Jun and Sp1, and the co-activators CREB-binding protein (CBP) and p300, to the endogenous SOCS-3 promoter. These data indicate that OSM-induced activation of STAT-3 and the ERK1/2 and JNK pathways are critical for astrocytic expression of SOCS-3, which provides for feedback inhibition of cytokine-induced inflammatory responses in the CNS.
Keywords: CNS, signal transduction, JAK/STAT pathway, cytokines, negative regulation
INTRODUCTION
Suppressor Of Cytokine Signaling (SOCS) proteins negatively regulate signaling pathways utilized by many cytokines, thereby modulating a wide range of cellular processes including innate and adaptive immune responses, inflammatory processes, growth and differentiation of lymphoid cells, and responses to bacterial and viral infections (Alexander and Hilton 2004; Ilangumaran et al. 2004; Lang et al. 2003; Marine et al. 1999; Seki et al. 2003; Yoshimura et al. 2007). The SOCS family is composed of eight members: CIS and SOCS-1 through SOCS-7. The structure of the SOCS proteins consists of a central SH2 domain, a C-terminal 40 amino acid motif termed the SOCS box, and an N-terminal region of variable length. Additionally, the N-terminus of SOCS-1 and SOCS-3 contains a kinase inhibitory region (KIR), which inhibits the kinase activity of JAK proteins (Yoshimura et al. 2007). The most well-characterized function of SOCS-3 is to negatively regulate signaling by IL-6 family cytokines through inhibition of STAT-3 activation in a classical feedback mechanism. Cells lacking SOCS-3 exhibit constitutive or enhanced STAT-3 activation and function (Cao et al. 2006; Croker et al. 2003; Lang et al. 2003; Miao et al. 2006; Okada et al. 2006; Yasukawa et al. 2003). Deletion of the Socs-3 gene is embryonic lethal and embryos exhibit dysregulated LIF signaling and extensive erythrocytosis (Marine et al. 1999). In addition, studies have shown that SOCS-3 inhibits signaling of other cytokines/stimuli including LPS, LIF, IL-2, -3, -4, IFN-γ and IFN-α (Elliott and Johnston 2004; Hanada et al. 2003; Kubo et al. 2003; Qin et al. 2006b). In general, SOCS-3 is thought to function as a negative regulator of inflammatory responses.
SOCS-3 is induced in a cell-type specific manner by a wide range of stimuli, including IL-2, -3, -4, -6, -10, -12, -21, -27, OSM, LIF, erythropoietin, growth hormone, LPS, IFN-γ, IFN-β, cAMP and CpG DNA (Barclay et al. 2007b; Dalpke et al. 2001; Gatto et al. 2004; Qin et al. 2007; Qin et al. 2006b). Induction of SOCS-3 by IL-10 in macrophages and by LIF in corticotroph AtT-20 cells occurs in a STAT-3-dependent manner (Auernhammer et al. 1999; Ding et al. 2003; Lang et al. 2003; Williams et al. 2004). However, IL-8-induced expression of SOCS-3 occurs in a STAT-independent manner in human myeloid cells (Stevenson et al. 2004). The peroxisome proliferator-activated receptor (PPAR)-γ agonists 15d-PGJ2 and rosiglitazone also induce SOCS-3 in a STAT-independent manner in primary astrocytes (Park et al. 2003a). In B cells, IL-4-induced SOCS-3 expression depends on activation of the p38 MAPK pathway, and in pro-B cells, growth hormone induction of SOCS-3 depends on STAT-5, p38 MAPK and JNK pathways (Barclay et al. 2007b). We recently demonstrated that induction of SOCS-3 by LPS in macrophages and microglia depends on the production of IL-10, STAT-3 activation, as well as activation of the ERK, JNK, and p38 MAPK pathways (Qin et al. 2007). Thus, the mechanism of SOCS-3 induction can vary greatly depending on the cell type and stimulus.
OSM is a member of the IL-6 family of cytokines which also includes IL-6, IL-11, LIF, CNTF, CT-1 and novel neurotrophin-1/B-cell stimulating factor-3 (NNT-BSF-3) (Heinrich et al. 2003). OSM strongly induces the expression of numerous immunoregulatory molecules, and has been implicated in a number of human neuropathologies such as Multiple Sclerosis (MS), HIV-Associated Dementia (HAD), and gliomas (Chen and Benveniste 2004). The main signaling pathways activated by OSM are the Janus Kinase/Signal Transducer and Activation of Transcription (JAK/STAT) and Mitogen-Activated Protein Kinase (MAPK) (Extracellular Signal Related Kinase (ERK), c-Jun N-terminal Kinase (JNK), and p38) pathways (Gomez-Lechon 1999; Korzus et al. 1997; Repovic et al. 2003a). OSM binds to the gp130 receptor subunit and recruits either the LIFR or OSMRβ subunit, resulting in dimerization of the receptor. Receptor-associated JAK1, JAK2, or TYK2 are then phosphorylated and activated (Heinrich et al. 2003). Activated JAK proteins in turn phosphorylate the cytoplasmic domain of the gp130 receptor, providing docking sites for STAT transcription factors (STAT-1, -3, -5 and -6), the protein tyrosine phosphatase SHP-2, and adaptor proteins such as SHC and Grb2 (Thoma et al. 1994). Association of such factors leads to activation of the JAK/STAT and MAPK pathways, which ultimately signal to the nucleus to regulate gene expression.
OSM has been implicated in the regulation of inflammatory responses, cell growth, proliferation and differentiation (Halfter et al. 2006; Hurst et al. 2002; Langdon et al. 2003; Repovic et al. 2003a; Repovic et al. 2003b; Weiss et al. 2006). The expression of OSM in normal brain is low, however, under neuroinflammatory conditions, activated microglia, reactive astrocytes and infiltrating leukocytes are significant producers of OSM. Elevated levels of OSM expression have been shown in inflammatory brain lesions, lesions of MS patients, HAD, and gliomas, as well as in mouse models of epilepsy and MS (Ensoli et al. 1999; Ensoli et al. 2002; Vecchiet et al. 2003). In addition, many cytokines and infectious compounds can induce the expression of OSM including GM-CSF, cisplatin, PMA, LPS, HIV-1, IL-3, and PGE2 (Chen and Benveniste 2004). The function of OSM in the CNS is unclear, however, due to seemingly disparate effects of OSM on pro- and anti-inflammatory processes (Chen and Benveniste 2004).
In this study, we demonstrate that OSM induces expression of SOCS-3 in primary astrocytes, which may serve an important regulatory role in the context of inflammation and neuroprotection. Because SOCS-3 has a non-redundant role in the inhibition of IL-6 family cytokines, we wished to characterize the molecular mechanism of OSM-induced SOCS-3 expression in astrocytes. We demonstrate that OSM activates a complex series of signaling pathways, leading to the recruitment of STAT-3, the AP-1 proteins c-Fos and c-Jun, and the histone acetyltransferases CBP and p300 to the SOCS-3 promoter to induce the expression of SOCS-3 in astrocytes.
MATERIALS AND METHODS
Recombinant proteins and reagents
Murine OSM, human soluble IL-6 receptor and human IL-11 were purchased from R&D Systems (Minneapolis, MN), and murine IL-6 was purchased from Endogen (Rockford, IL). Abs against phospho-Jak-1Tyr1022/Tyr1023, phospho-STAT-1αTyr701, phospho-STAT-3Tyr705, phospho-p44/42Thr202/Tyr204, phospho-SAPK/JNKThr183/Tyr185, phospho-p38 Thr180/Tyr182, and p44/42, were obtained from Cell Signaling Technology (Danvers, MA). Abs against STAT-3, SOCS-3, phospho-JAK-2Tyr1007/Tyr1008, phospho-c-JunSer63, JNK-1, actin, c-Fos, c-Jun, and Sp1 were obtained from Santa Cruz Biotechnology (Santa Cruz, CA). Abs against STAT-1α, JAK-1, JAK-2, AcH3 and AcH4 were purchased from Upstate Biotechnology (Lake Placid, NY). Ab against cyclophilin B was purchased from Abcam (Cambridge, MA) and the Ab against RNA Pol II was from Covance (Princeton, NJ). The pharmacological inhibitors for MEK-1/MEK-2 (U0126), JNK (SP600125), p38 (SB203580), JAK-2 (AG 490) and negative control compounds (U0124 and SB202474), were purchased from Calbiochem Biochemicals (San Diego, CA). The dominant-negative STAT-3 (STAT-3F) plasmid was a gift from Drs. M. Hibi, T. Hirano, and K. Nakajima, Osaka University Medical School, Osaka, Japan) (Nakajima et al. 1996). This construct has a phenylalanine substitution at Tyr705, which prevents it from being activated. The constitutively active STAT-3 construct (STAT-3C, with A662C and N664C mutations) (Bromberg et al. 1999) was generously provided by Dr. J. E. Darnell, Jr. (The Rockefeller University, New York, NY).
Cells
Primary astrocytes from C57BL/6J mice (The Jackson Laboratory, Bar Harbor, ME) and WT and STAT-1α deficient mice on the 129S6/SvEv background were prepared as described previously (Dong et al. 2001). After 2 weeks in culture, oligodendrocytes and microglia were separated from astrocytes by mechanical dislodgment. Astrocytes were monitored for purity by immunofluorescence, and were routinely > 97% positive for glial fibrillary acidic protein.
RNA isolation, riboprobes, and RNase protection assay (RPA)
Total cellular RNA was isolated from unstimulated, OSM-treated, or OSM plus inhibitor-treated cells using TRIzol reagent (Invitrogen, Carlsbad, CA) as previously described (Qin et al. 2006b). The riboprobes for SOCS-1, SOCS-3 and GAPDH were prepared as described previously (Qin et al. 2006a; Qin et al. 2006b), and Ribonuclease Protection Assay (RPA) was performed on the RNA samples as described previously (Qin et al. 2006b). Briefly, ten μg of total RNA was hybridized with the riboprobes at 56°C overnight. The hybridized mixture was treated with RNase A/TI (1:200) and then separated by PAGE on a 5% denaturing (8 M urea) gel. Expression values for SOCS-1 and SOCS-3 mRNA expression were normalized to GAPDH mRNA levels for each experimental condition using ImageQuant Software.
Immunoblotting
Thirty μg of cell lysate was separated by electrophoresis on 10% polyacrylamide gels, transferred to nitrocellulose membranes, and probed with antibodies against phospho-Jak-1Tyr1022/Tyr1023, phospho-JAK-2Tyr1007/Tyr1008, phospho-STAT-1αTyr701, phospho-STAT-3Tyr705, phospho-p44/42Thr202/Tyr204, phospho-SAPK/JNKThr183/Tyr185, phospho-c-JunSer63, and phospho-p38 Thr180/Tyr182, as described previously (Qin et al. 2006b). SOCS-3 protein expression was analyzed using 50 μg of protein and separated on 12% SDS-PAGE. Membranes were stripped for 30 minutes at room temperature in buffer containing 100 mM 2-ME, 2% SDS, and 62.5 mM Tris-HCl (pH 6.7) with continuous shaking, and reprobed with antibodies against JAK-1, JAK-2, STAT-1α, STAT-3, p44/42, JNK1, c-Jun, p38, or actin.
SOCS-3 promoter constructs, plasmids, transient transfection, and luciferase assays
The 1556-bp (−1429- to +127) murine SOCS-3 promoter was cloned as described previously (Qin et al. 2006b). Serial deletion mutants were generated as previously described (Qin et al. 2007). The potential regulatory elements in the SOCS-3 promoter are three AP-1 elements, one p300 site, one C/EBP site, two GATA-1 elements, one NF-κB site, three Sp1 elements, and two IFN-γ activation sequence (GAS) elements (see Fig 8A). The deletion constructs are named as follows: Δ1, which lacks the distal AP-1 and C/EBP site; Δ2, which lacks two GATA-1 elements and a p300 site; Δ3, which lacks the medial AP-1 element; Δ4, which lacks two Sp1 sites and an NF-κB site; Δ5, which lacks the proximal AP-1 site; Δ6, which lacks the distal GAS element; and Δ7, which lacks the proximal GAS and Sp1 elements. The site-directed mutation constructs were generated on the SOCS-3 Δ4 (−120 bp) plasmid backbone using the QuikChange site-directed mutagenesis kit (Stratagene, La Jolla, CA) following the manufacturer’s instructions, and were confirmed by sequencing (Qin et al. 2007). The SOCS-3 promoter constructs were transiently transfected into 5 × 105 astrocytes in 6-well plates using the Lipofectamine Plus (Invitrogen, Carlsbad, CA) method as described previously (Qin et al. 2006b). Transfected cells recovered overnight and then were treated with medium or OSM for 18 h. Luciferase activity of each sample was measured using a luminometer and was normalized to the protein concentration of each well. Luciferase activity from the untreated sample was arbitrarily set to 1 for calculations of fold induction. In some experiments, cells were pre-treated with 0.2% DMSO, or with 5, 10 or 20 μM U0126 or U0124 for 1 hour before OSM treatment. The Lipofectamine Plus method was also used for co-transfection of the SOCS-3 promoter and STAT-3F. Cells recovered for 48 h to allow for expression of STAT-3F and then were treated with OSM for 18 h. Luciferase expression was analyzed as described above.
Figure 8. The Proximal AP-1 Site and the Two GAS Elements are Critical for OSM-induced SOCS-3 Promoter Activity.

A. Primary astrocytes were transiently transfected with 200 ng of the WT or Δ1–Δ7 SOCS-3 promoter constructs. Cells recovered for 24 h, and were treated with OSM for 18 h. Cell lysates were analyzed for luciferase expression. Mean +/− S.D. of three independent experiments. B. Site-directed mutagenesis was performed on the proximal AP-1 site and on each of the two GAS elements in the Δ4 SOCS-3 promoter. Primary astrocytes were transfected with either the Δ4, Δ4 GAS #1 SDM, Δ4 GAS #2 SDM, or Δ4 AP-1 SDM construct (200 ng). After recovering for 24 h, cells were treated with OSM for 18 h and cell lysates were analyzed for luciferase expression. Mean +/− S.D. of three independent experiments.
RNA interference
RNA interference was performed using a protocol provided by Dharmacon with modifications as described previously (Qin et al. 2006a). DharmaFECT 1 small interfering RNA (siRNA) transfection reagent, SMARTpool siRNAs specific for murine STAT-3, murine cyclophilin B (a housekeeping gene), and siCONTROL nontargeting siRNAs were purchased from Dharmacon (Lafayette, CO). Astrocytes (1 × 106) were plated in 6-well plates and incubated with DharmaFECT 1 reagent alone or with 100 nM cyclophilin B, siCONTROL or STAT-3 specific siRNAs following the manufacturer’s recommendations. After 72 h of transfection, 30 μg of cell lysate was subjected to immunoblotting for cyclophilin B, STAT-3 and actin as described above. In some experiments, immunoblotting for activated STAT-3Tyr705 and STAT-1Tyr701 was also performed. To analyze SOCS-1 and SOCS-3 mRNA expression, 10 μg of RNA was subjected to RPA as described above.
Chromatin immunoprecipitation (ChIP) assays
ChIP analysis was performed as described previously (Qin et al. 2006b). Primary astrocytes were unstimulated or incubated with OSM for 0.25, 0.5, 1 or 4 h. Cells were fixed with 1% formaldehyde for 15 min at room temperature to crosslink DNA and proteins, and nuclei were isolated. Chromatin was sheared by sonication and samples were aliquoted and precleared for 2 h at 4°C with salmon sperm DNA-saturated protein A/G Sepahrose beads. Chromatin solutions were incubated with 5 μg of Abs or isotype-matched control IgG overnight at 4°C. Input and immunoprecipitated chromatin were incubated with 200 mM NaCl at 65°C overnight to reverse cross-links. After digestion of protein with proteinase K, DNA was purified over a Qiagen column and analyzed by PCR with GoTaq polymerase. The primer pair 5′-GCTTTGTCTCCCTCTCGGTGAGT-3′ and 5′-AGTGTAGAGTCAGAGTTAGAGCC-3′ was used to amplify a 230-bp region in the mouse SOCS-3 promoter containing the proximal AP-1 element, two GAS elements, and an Sp1 element.
Statistical analysis
All experiments were repeated a minimum of three times. Levels of significance between samples were determined by the student’s t test distribution. A value of p ≤ 0.05 was considered to be statistically significant.
RESULTS
Characterization of SOCS-3 gene expression by OSM
Cytokines of the IL-6 family have been shown to induce the expression of SOCS-1 and SOCS-3 with varying intensities and kinetics in many cell types (Endo et al. 1997; Heinrich et al. 2003; Masuhara et al. 1997; Nakajima et al. 1996; Park et al. 2003b; Suzuki et al. 1998). In primary astrocytes, OSM, IL-6, in conjunction with the sIL-6R, and IL-11 all induced SOCS-3 expression (data not shown). Of these, OSM induced stronger and more sustained SOCS-3 expression compared to IL-6 and IL-11. To gain a better understanding of the extent of SOCS-3 expression by OSM, astrocytes were treated with OSM from 0–48 h and analyzed for SOCS-3 mRNA by RPA. Basal expression levels of SOCS-3 mRNA were very low in astrocytes as demonstrated in the untreated (Un) condition (Fig. 1A). OSM-induced expression of SOCS-3 mRNA began at 15 min, peaked at 1 h, and was still upregulated at 48 h post-OSM treatment (Fig. 1A). Expression of SOCS-3 protein by OSM was detected at 30 min, peaked between 1 and 2 h, and was still upregulated at 24 h (Fig. 1B). To determine if induction of SOCS-3 expression was transcriptionally regulated, a SOCS-3 promoter assay was utilized. Astrocytes were transiently transfected with a pGL3 firefly luciferase expression vector containing a 1.5 kb portion of the SOCS-3 promoter (Qin et al. 2007). Treatment of these cells with OSM led to a 10.9-fold increase in SOCS-3 promoter activation compared to unstimulated cells (Fig. 1C). Therefore, OSM induces robust and sustained expression of SOCS-3, and this expression is regulated at the transcriptional level.
Figure 1. Induction of SOCS-3 Expression by OSM.

A, B. Primary astrocytes were treated with OSM (5 ng/ml) for 0–48 h. A. 10 μg of RNA extracts was analyzed for SOCS-3 mRNA expression by RPA. GAPDH mRNA expression serves as a control. B. Protein lysates were collected and separated by SDS-PAGE, immunoblotted for SOCS-3 protein, and for actin as a loading control. C. Primary astrocytes were transiently transfected with the pGL3 firefly luciferase-expression vector containing the murine SOCS-3 promoter. Cells recovered overnight and were then treated with OSM for 16 h. Lysates were then analyzed for luciferase activity. Values were normalized to total protein, and fold induction calculated by dividing the OSM treatment values by untreated levels. Data represent the mean +/− S.D. of three experiments.
OSM activates the JAK/STAT pathway in primary astrocytes
OSM utilizes several signaling pathways to regulate gene expression, including the JAK/STAT pathway (Gomez-Lechon 1999; Korzus et al. 1997; Van Wagoner et al. 2000). Several studies have demonstrated a requirement for activation of this pathway to induce SOCS-3 expression in response to a variety of stimuli in different cell types (Brender et al. 2001; Campbell 2005; Gatto et al. 2004; Qin et al. 2007; Ramana et al. 2005). We therefore analyzed activation of the JAK/STAT pathway following exposure of astrocytes to OSM. Treatment with OSM led to rapid JAK-1 and JAK-2 tyrosine phosphorylation. Activation of JAK-1 was evident at 15 min, peaked at 30 min and diminished by 1 h (Fig. 2A). JAK-2 activation was evident at 15 min and decreased after 30 min (Fig. 2B). Tyrosine phosphorylation of STAT-1 and STAT-3 was detected at 15 min, which persisted for 1 h in the case of STAT-1 (Fig. 2C), and out to 24 h for STAT-3 (Fig. 2D). OSM-induced activation of STAT-3 was much more robust and sustained compared to that of STAT-1 in astrocytes.
Figure 2. OSM-induced Activation of the JAK/STAT Pathway in Astrocytes.
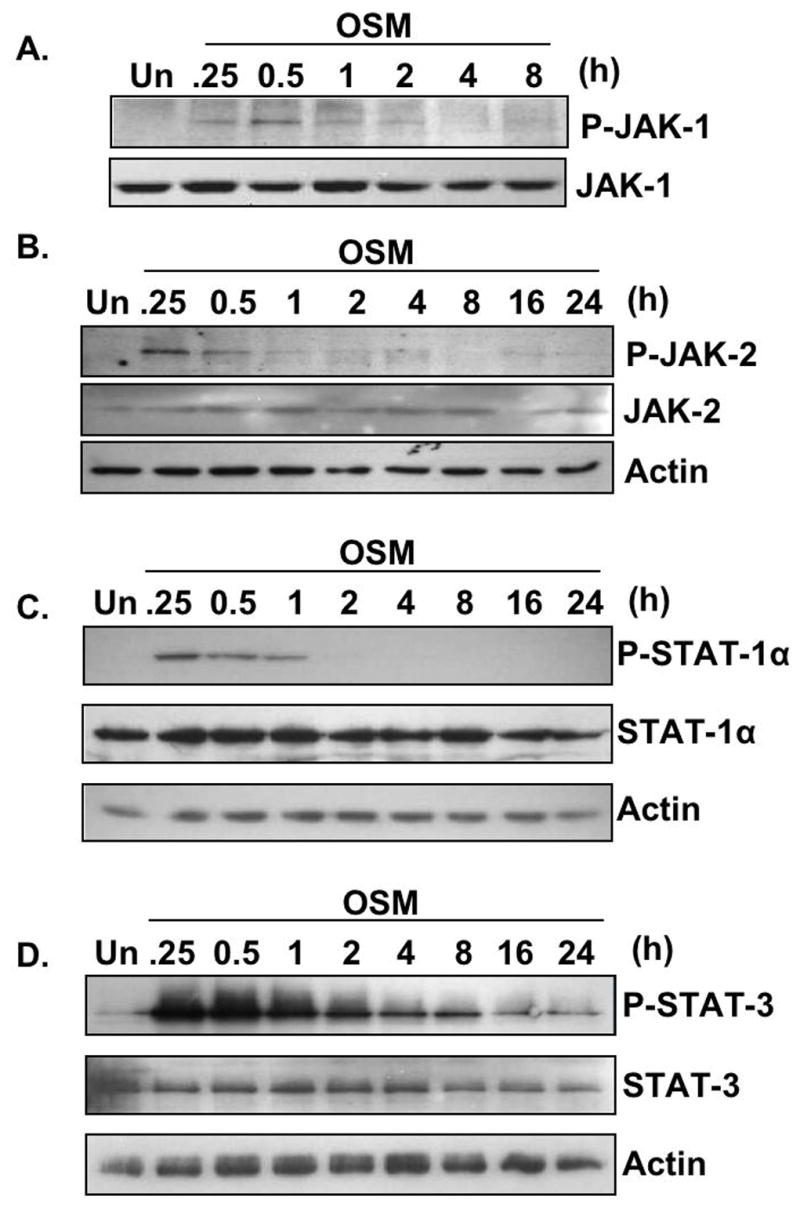
A. Primary astrocytes were treated with OSM (5 ng/ml) for 0–8 h. 500 μg of protein lysates were immunoprecipitated with an anti-JAK-1 antibody, separated by SDS-PAGE, and immunoblotted for phosphorylated JAK-1Tyr1022/1023. The membrane was stripped and reprobed with an antibody against total JAK-1. Representative of three experiments. B–D. Primary astrocytes were treated with OSM (5 ng/ml) for 0–24 h. Protein lysates were prepared and subjected to immunoblot analysis with antibodies against phosphorylated JAK-2Tyr1007/1008, STAT-1Tyr701, or STAT-3Tyr705. Membranes were stripped and reprobed for total JAK-2, STAT-1, and STAT-3, respectively, and actin as a loading control. Representative of four experiments.
STAT-3 activation is necessary for SOCS-3 expression by OSM
OSM induced strong STAT-3 activation, therefore we determined the contribution of STAT-3 to SOCS-3 expression in astrocytes. STAT-3 knockout mice are embryonic lethal (Takeda et al. 1997), so siRNA against STAT-3 was utilized to inhibit its expression. Primary astrocytes were transfected with siRNA against STAT-3 or with a positive control siRNA against cyclophilin B. Non-target siRNA was also utilized as a negative control. Specific knock-down of STAT-3 and cyclophilin B was confirmed by analyzing STAT-3 and cyclophilin B protein expression, respectively (Fig. 3A). As expected, the non-target siRNA did not have an effect on protein expression. The knockdown of STAT-3 was then analyzed over a time-course of OSM treatment, from 0–4 h (Fig. 3B). Tranfection of STAT-3 siRNA resulted in varying levels of STAT-3 knockdown when compared to the control conditions: 59.7% (Un), 61.0% (0.25 h), 55.2% (0.5 h), 71.7% (1 h), 46.3% (2 h), and 59.8% (4 h). In the transfection reagent only conditions, addition of OSM led to phosphorylation of STAT-3 in a time-dependent manner, beginning at 0.25 h and lasting through 4 h (Fig. 3B). We did not detect activated STAT-3 in response to OSM in the STAT-3 siRNA conditions (Fig. 3B). While OSM treatment led to slight activation of STAT-1 in the transfection reagent only conditions, activation of STAT-1 in the STAT-3 siRNA cells was more pronounced (Fig. 3B). With respect to SOCS-3 expression, knock-down of STAT-3 resulted in decreased OSM-induced SOCS-3 mRNA expression at all time points tested (Fig. 3C). Inhibition of SOCS-3 mRNA at different time-points was as follows: 50.1% (Un), 74.6% (0.25 h), 71.8% (0.5 h), 41.0% (1 h), 41.7% (2 h), and 63.0% (4 h). We also tested the ability of OSM to induce SOCS-1 mRNA expression in these cells. Modest induction of SOCS-1 mRNA was observed upon treatment with OSM, which was elevated in the STAT-3 siRNA cells at 1 h (2-fold), 2 h (2.2-fold), and 4 h (6.2-fold) (Fig. 3C). Several studies have demonstrated a requirement of STAT-1 for SOCS-1 expression in response to different stimuli (O’Keefe et al. 2001; Zimmerer et al. 2007). Also, compensatory roles of STAT-1 and STAT-3 have been proposed, such that enhanced activation and signaling of one STAT protein occurs when the other is absent or downregulated (Costa-Pereira et al. 2002). These results indeed support such a hypothesis.
Figure 3. Inhibition of STAT-3 Expression or Activation Inhibits OSM-induced SOCS-3 Expression and Promoter Activity.

A. Astrocytes were transiently transfected with 100 nM siRNA against STAT-3, cyclophilin B as a positive control, or non-target siRNA as a negative control, and incubated for 72 h. Specific STAT-3 knock-down was confirmed by immunobloting for total STAT-3 protein, cyclophilin B and actin. B, C. Primary astrocytes were incubated with transfection reagent alone or with transfection reagent plus 100 nM siRNA against STAT-3, and incubated for 72 h. Cells were then treated with OSM (5 ng/ml) for the indicated times (0–4 h). B. Protein lysates were prepared and subjected to immunoblot analysis with antibodies against phosphorylated STAT-3Tyr705 or STAT-1Tyr701. Membranes were stripped and reprobed for total STAT-3 and actin as loading controls. Representative of three independent experiments. C. 10 μg of RNA was analyzed for SOCS-3 and SOCS-1 mRNA expression by RPA. GAPDH mRNA expression serves as a loading control. Representative of three independent experiments. D. Primary astrocytes were transiently co-transfected with increasing concentrations of a STAT-3 dominant-negative vector (STAT-3F), along with the mSOCS-3 promoter (200 ng). The total amount of cDNA was kept constant using an empty pcDNA3 vector. Following transfection, cells recovered for 48 h to allow for expression of the STAT-3F construct. The cells were then treated with OSM for 24 h, and analyzed for luciferase expression as a measure of SOCS-3 promoter activity. Mean +/− S.D. of three independent experiments. *, p ≤ 0.05. E. Astrocytes were transiently co-transfected with the mSOCS-3 promoter (200 ng) and either 200 ng of pcDNA3 or 200 ng of STAT-3C. Cells recovered for 48 h and then were analyzed for luciferase expression. Mean +/− S.D. of three experiments. **, p ≤ 0.001.
To further investigate the role of STAT-3 in SOCS-3 expression, the effect of a dominant-negative STAT-3 construct (STAT-3F), which contains an inactivating phenylalanine substitution at a critical tyrosine residue (Nakajima et al. 1996), on OSM-induced SOCS-3 promoter activity was tested. Inclusion of STAT-3F significantly inhibited OSM-induced activation of the SOCS-3 promoter at concentrations of 100 ng and above (Fig. 3D). Similarly, the inclusion of a constitutively active form of STAT-3 (STAT-3C) (Bromberg et al. 1999) was capable of activating the SOCS-3 promoter in the absence of OSM (Fig. 3E). Together, these results demonstrate the requirement of STAT-3 for SOCS-3 expression in response to OSM in primary astrocytes.
STAT-1 expression is not necessary for OSM-induced SOCS-3 expression
Unlike STAT-3 deficient mice, STAT-1 deficient mice are viable (Meraz et al. 1996). Because other studies have implicated STAT-1 in SOCS-3 expression (Gatto et al. 2004), and also because OSM induced its activation in astrocytes (Fig. 2C), we analyzed SOCS-3 mRNA expression in STAT-1 deficient astrocytes. Absence of STAT-1 phosphorylation and protein expression in STAT-1−/− cells was confirmed by immunoblotting (Fig. 4A). Comparative levels of STAT-3 activation were observed between WT and STAT-1−/− astrocytes. Consistent with this result, induction of SOCS-3 in response to OSM treatment did not differ between wild-type and STAT-1−/− primary astrocytes (Fig. 4B). As expected, induction of SOCS-1 was completely absent in STAT-1−/− cells. These data indicate that STAT-1 is not necessary for the induction of SOCS-3 by OSM in primary astrocytes.
Figure 4. OSM-induced Expression of SOCS-1, but not SOCS-3, Depends on STAT-1.

A, B. Astrocytes isolated from STAT-1+/+ (WT) or STAT-1−/− mice were incubated with OSM (5 ng/ml) for the indicated times. A. Protein lysates were subjected to immunoblotting for phospho-STAT-1αTyr701 and phospho-STAT-3Tyr705. Blots were stripped and reprobed for total STAT-1, total STAT-3 and actin. B. 10 μg of RNA was analyzed for SOCS-3 and SOCS-1 mRNA expression by RPA. GAPDH mRNA expression serves as a loading control. Representative of three independent experiments.
OSM activates the MAPK pathways in astrocytes
In addition to the JAK/STAT pathway, OSM regulates gene expression through other pathways including the p38 MAPK, ERK1/2, and JNK pathways (Korzus et al. 1997; Repovic et al. 2003b; Van Wagoner et al. 2000). We therefore analyzed the activation of these pathways in astrocytes. Treatment with OSM led to the activation of p38 MAPK at 15 min, which was maximal at 1 h, and remained elevated above basal levels out to 24 h (Fig. 5A). OSM treatment also increased activation of SAPK/JNK at 15 min, which remained elevated for at least 24 h (Fig. 5B). Activation of p44/42 ERK was also detected in response to OSM from 15 min to 4 h (Fig. 5C). These results demonstrate that OSM activates the p38 MAPK, ERK1/2, and JNK pathways in astrocytes.
Figure 5. OSM-induced Activation of the MAPK Pathways in Primary Astrocytes.
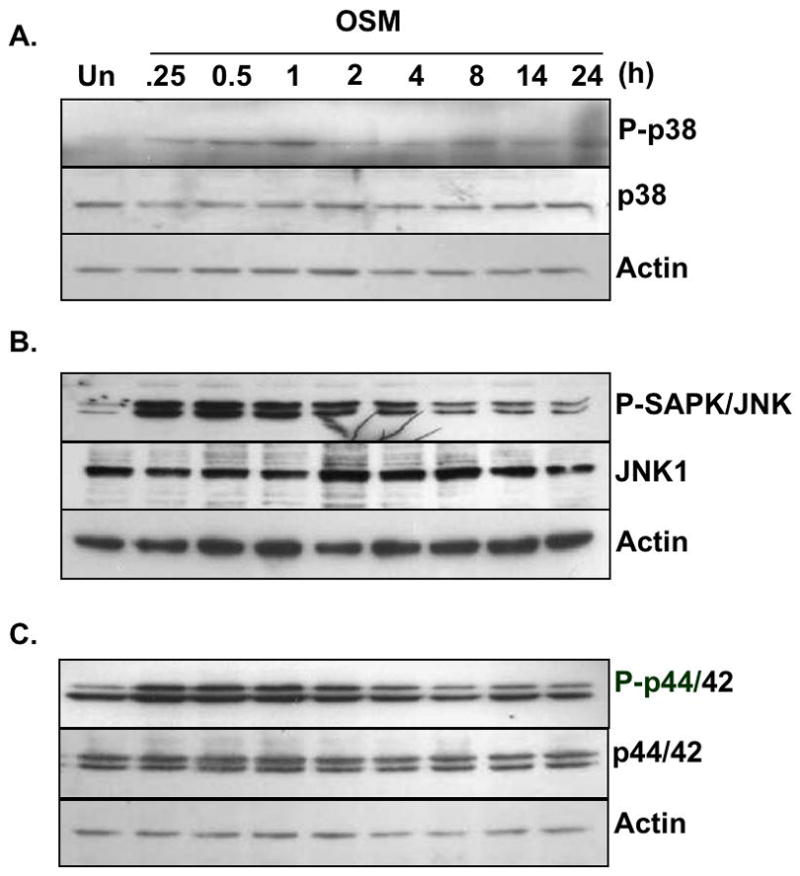
A–C. Primary astrocytes were treated with OSM (5 ng/ml) for 0–24 h. Protein lysates were prepared, separated by SDS-PAGE, and subjected to immunoblot analysis with antibodies against (A) phosphorylated p38Thr180/Tyr182, (B) SAPK/JNKThr183/Tyr182 or (C) p44/42 ERKThr202/Tyr204. Membranes were stripped and reprobed for total p38, SAPK/JNK, and p44/42 ERK, respectively, and actin as a loading control. Representative of three experiments.
Activation of the ERK1/2 pathway contributes to OSM-induced SOCS-3 expression in astrocytes
SOCS-3 expression was not completely inhibited in the absence of STAT-3 (Fig. 3), suggesting that other signaling pathways may be involved in regulating OSM-induced SOCS-3 expression. We have recently demonstrated that the MAPK pathways are involved in LPS-induced expression of SOCS-3 in macrophages and microglia (Qin et al. 2007). Therefore, the potential involvement of the p38 MAPK, ERK1/2, and JNK pathways in OSM-induced SOCS-3 expression was examined. Pretreatment of astrocytes for 1 h with the p38 MAPK inhibitor SB203580 (Frantz et al. 1998) modestly decreased OSM-induced SOCS-3 mRNA expression at 4, 8 and 18 h following treatment, but not at earlier timepoints (data not shown). To determine the involvement of the ERK1/2 pathway, a specific inhibitor of MEK1 and MEK2, U0126, was utilized (Favata et al. 1998). Pretreatment of astrocytes for 1 h with 5, 10 or 20 μM U0126 revealed a dose-dependent inhibition of OSM-induced SOCS-3 expression (19% at 5 μM, 29% at 10 μM, and 38% at 20 μM), while the control compound U0124 had no effect (Fig. 6A). U0126, but not U0124, inhibited OSM-induced phosphorylation of p44/42 ERK, demonstrating the efficacy and specificity of the compound (Fig. 6B). To further examine the importance of the ERK1/2 pathway in SOCS-3 expression, we extended the use of the inhibitor compound to the SOCS-3 promoter assay. Pretreatment of astrocytes with 10 and 20 μM of U0126 significantly inhibited OSM-induced SOCS-3 promoter activity, while pretreatment with DMSO or U0124 had no effect (Fig. 6C). Extended kinetic analysis of SOCS-3 mRNA expression revealed that inhibition of the ERK pathway significantly decreased OSM-induced SOCS-3 expression by 44.7% at 15 min, 44.9% at 30 min, 22.6% at 1 h, 31.3% at 2 h, and 17.8% at 4 h, compared to DMSO pretreatment or pretreatment with U0124 (Figs. 6D and 6E). SOCS-3 expression was also inhibited at 8 and 18 h following OSM treatment, but did not reach significance. OSM-induced STAT-3 activation was not affected by U0126 at all the concentrations tested (Fig. 6F), indicating that the inhibition of SOCS-3 expression was not due to non-specific inhibition of STAT-3. These results indicate that OSM-induced activation of the ERK1/2 pathway is involved in transcriptional regulation of SOCS-3 gene expression.
Figure 6. OSM-induced SOCS-3 Expression Partially Depends on Activation of the ERK Pathway.

A, B. Primary astrocytes were pretreated with 0.2% DMSO or with increasing concentrations (0–20 μM) of U0126, a specific inhibitor of MEK1 and MEK2, or U0124, a negative control, for 1 h. Cells were then incubated with OSM (5 ng/ml) for 30 min. A. 10 μg of RNA was analyzed for SOCS-3 mRNA expression by RPA. GAPDH mRNA expression serves as a control. Representative of three independent experiments. B. Protein lystes were prepared, separated by SDS-PAGE and immunoblotted for phosphorylated p44/42 ERKThr202/Tyr204. Lysates were also probed for total p44/42 ERK as loading control. Representative of three independent experiments. C. Primary astrocytes were transiently transfected with the SOCS-3 promoter (200 ng) and allowed to recover for 24 h. The cells were then pretreated with 0.2% DMSO or with increasing concentrations of U0126 or U0124 for 1 h. Cells were then incubated in the absence or presence of OSM (5 ng/ml) for 8 h, and analyzed for luciferase expression as a measure of SOCS-3 promoter activity. Mean +/− S.D. of three independent experiments. *, p ≤ 0.05; **, p ≤ 0.001 compared to DMSO condition. D. Primary astrocytes were pretreated with 0.2% DMSO or with 20 μM of U0126 for 1 h. Cells were then incubated with OSM (5 ng/ml) for the indicated times. 10 μg of RNA was analyzed for SOCS-3 mRNA expression by RPA. GAPDH mRNA expression serves as a control. Representative of three independent experiments. E. Average fraction of SOCS-3 mRNA expression induced by OSM in the presence of 20 μM U0126. Data represent the mean +/− S.D. of three experiments. *, p ≤ 0.05 compared to DMSO condition. F. Primary astrocytes were pretreated with 0.2% DMSO or with increasing concentrations (0–20 μM) of U0126 for 1 h. Cells were then incubated with OSM (5 ng/ml) for 30 min. Protein lysates were subjected to immunoblotting for phospho-STAT-3Tyr705 and actin.
Activation of the JNK pathway contributes to SOCS-3 gene expression
To characterize the potential contribution of the JNK pathway, a specific inhibitor of the JNK pathway, SP600125, was used (Bennett et al. 2001). Astrocytes were pretreated with various concentrations of SP600125 (0–50 μM) or with 0.5% DMSO for 1 h, and then incubated with OSM (5 ng/ml) for 30 min. Concentrations of 1 μM and above significantly inhibited SOCS-3 mRNA expression, and maximal inhibition was seen at 20 μM (Figs. 7A and 7B). SP600125 inhibited the phosphorylation of c-Jun in a dose-dependent manner, demonstrating the efficacy of this compound (Fig. 7C). Inclusion of 20 μM of SP600125 significantly inhibited OSM-induced SOCS-3 mRNA expression by 80.3% at 15 min, 55.2% at 30 min, 36.2% at 1 h, 39.6% at 2 h, 36.4% at 4 h, 43.6% at 8 h, and 26.6% at 18 h compared to DMSO pretreatment (Figs. 7D and 7E). The phosphorylation status of STAT-3 in response to OSM was not affected by inclusion of SP600125 at all concentrations tested (Fig. 7F). Together, these data indicate an involvement of the JNK pathway in SOCS-3 expression induced by OSM.
Figure 7. OSM-induced SOCS-3 Expression Involves the JNK Pathway.

A–C. Primary astrocytes were incubated with 0.5% DMSO or with increasing concentrations (0–50 μM) of SP600125, a specific JNK inhibitor, for 1 h. Cells were then stimulated with OSM (5 ng/ml) for 30 min. A. 10 μg of RNA was analyzed for SOCS-3 mRNA expression by RPA. GAPDH mRNA expression serves as a control. Representative of three independent experiments. B. Average SOCS-3 mRNA fold inductions were calculated from three independent experiments. Data represent the mean +/− S.D. of three experiments. *, p ≤ 0.05 compared to DMSO condition. C. Protein lystes were prepared, separated by SDS-PAGE and immunoblotted for phosphorylated c-Jun (Ser 63). Lysates were also probed for total c-Jun as loading control. Representative of three independent experiments. D. Primary astrocytes were pretreated with 0.2% DMSO or with 20 μM of SP600125 for 1 h. Cells were then incubated with OSM (5 ng/ml) for the indicated times. 10 μg of RNA was analyzed for SOCS-3 and GAPDH mRNA expression by RPA. Representative of three independent experiments. E. Average fraction of SOCS-3 mRNA expression was calculated from three independent experiments. Data represent the mean +/− S.D. of three experiments. *, p ≤ 0.05 compared to DMSO condition. F. Primary astrocytes were pretreated with 0.2% DMSO or with increasing concentrations (0–20 μM) of SP600125 for 1 h. Cells were then incubated with OSM (5 ng/ml) for 30 min. Protein lysates were subjected to immunoblotting for phospho-STAT-3Tyr705 and actin.
OSM-induced SOCS-3 promoter activity requires AP-1 and GAS elements
The SOCS-3 promoter contains a number of cis-regulatory elements including three AP-1 sites, one NF-κB element, two GAS elements, and others (Auernhammer et al. 1999; Gatto et al. 2004; Qin et al. 2007). To determine the regulatory elements necessary for SOCS-3 expression, a series of deletion constructs were generated from the 5′ end of the SOCS-3 promoter (Qin et al. 2007). The full length (WT) SOCS-3 promoter and various deletion constructs were transiently transfected into primary astrocytes, which were treated with OSM for 18 h and then analyzed for luciferase activity. OSM stimulation resulted in a 7.9-fold increase in WT SOCS-3 promoter activity (Fig. 8A). Deletion of the distal AP-1 element and C/EBP element (Δ1) did not significantly affect OSM-induced SOCS-3 promoter activity. Deletion of a p300 element and two GATA-1 sites (Δ2) led to a slight reduction (18%) in promoter activity. Deletion of the region containing the medial AP-1 element (Δ3), and an NF-κB and Sp1 site (Δ4) had no significant effect on OSM-induced SOCS-3 promoter activity compared with the WT construct. In construct Δ5, deletion of the proximal AP-1 element reduced OSM-induced SOCS-3 promoter activity by ~48% compared with Δ4. Deletion of the distal GAS element (GAS #1) (Δ6) did not significantly affect OSM-induced SOCS-3 promoter activity compared to Δ5. However, in construct Δ7, deletion of both GAS elements, GAS #1 and #2, rendered the SOCS-3 promoter inactive (Fig. 8A). As well, the proximal Sp1 element is deleted in construct Δ7.
Activated STAT protein complexes bind to GAS elements in the promoters of STAT-responsive genes (Bromberg and Darnell 2000). Therefore, site-directed mutagenesis was used to generate individual mutations in each of the GAS elements to better define their contributions in SOCS-3 promoter activity (Fig. 8B). Mutation of the distal GAS element (GAS #1) decreased OSM-induced SOCS-3 promoter activity by ~48%, whereas mutation of the proximal GAS #2 element reduced activity by 40%. In addition, site-directed mutagenesis was performed on the proximal AP-1 site, as its deletion in Δ5 reduced OSM-induced SOCS-3 promoter activity. Mutation of the distal AP-1 element decreased OSM-induced SOCS-3 promoter activity by 22% compared to Δ4. These results indicate that the proximal AP-1 site and both GAS elements have pivotal roles in OSM-induced SOCS-3 promoter activity.
Recruitment of STAT-3, c-Fos, c-Jun and Sp1, and the coactivatos CBP and p300, to the SOCS-3 promoter in response to OSM
We have shown that STAT-3, ERK1/2, and JNK activation are involved in OSM-induced SOCS-3 gene expression. As well, the proximal AP-1 element and two GAS elements are important for SOCS-3 transcription. To analyze transcription factor binding to the endogenous SOCS-3 promoter, primary astrocytes were incubated in the absence or presence of OSM for up to 4 h, and ChIP assays performed using Abs against STAT-3, c-Fos, c-Jun and Sp1, or normal rabbit IgG as a negative control. PCR analysis of the positive control (input) samples indicated that the soluble chromatin in each sample contained equal amounts of the SOCS-3 promoter (Fig. 9). STAT-3 was associated with the SOCS-3 promoter at low levels in untreated cells. Treatment with OSM induced a strong and rapid recruitment of STAT-3 to the SOCS-3 promoter from 15 min to 1 h after treatment (Fig 9). c-Fos and c-Jun are AP-1 transcription factors downstream of the MAPK pathways (Turjanski et al. 2007). Low levels of c-Fos and c-Jun were associated with the SOCS-3 promoter in unstimulated cells; however, treatment with OSM resulted in robust recruitment of these factors from 15 min to 1 h. At 4 h following OSM treatment, recruitment of STAT-3, c-Fos and c-Jun to the SOCS-3 promoter was diminished. Recruitment of Sp1 to the SOCS-3 promoter has been demonstrated in response to PGE2 in T47D breast cancer cells (Barclay et al. 2007a). This study also showed the requirement for the Sp1/Sp3 element in the SOCS-3 promoter for SOCS-3 gene expression. Sp1 was recruited to the SOCS-3 promoter in response to OSM in primary astrocytes, but with somewhat later kinetics compared to the other transcription factors investigated (Fig. 9). This result would suggest that Sp1 is not required for early SOCS-3 gene expression, but may contribute to its expression at later timepoints.
Figure 9. Characterization of OSM-induced Effects at the SOCS-3 Promoter In Vivo.
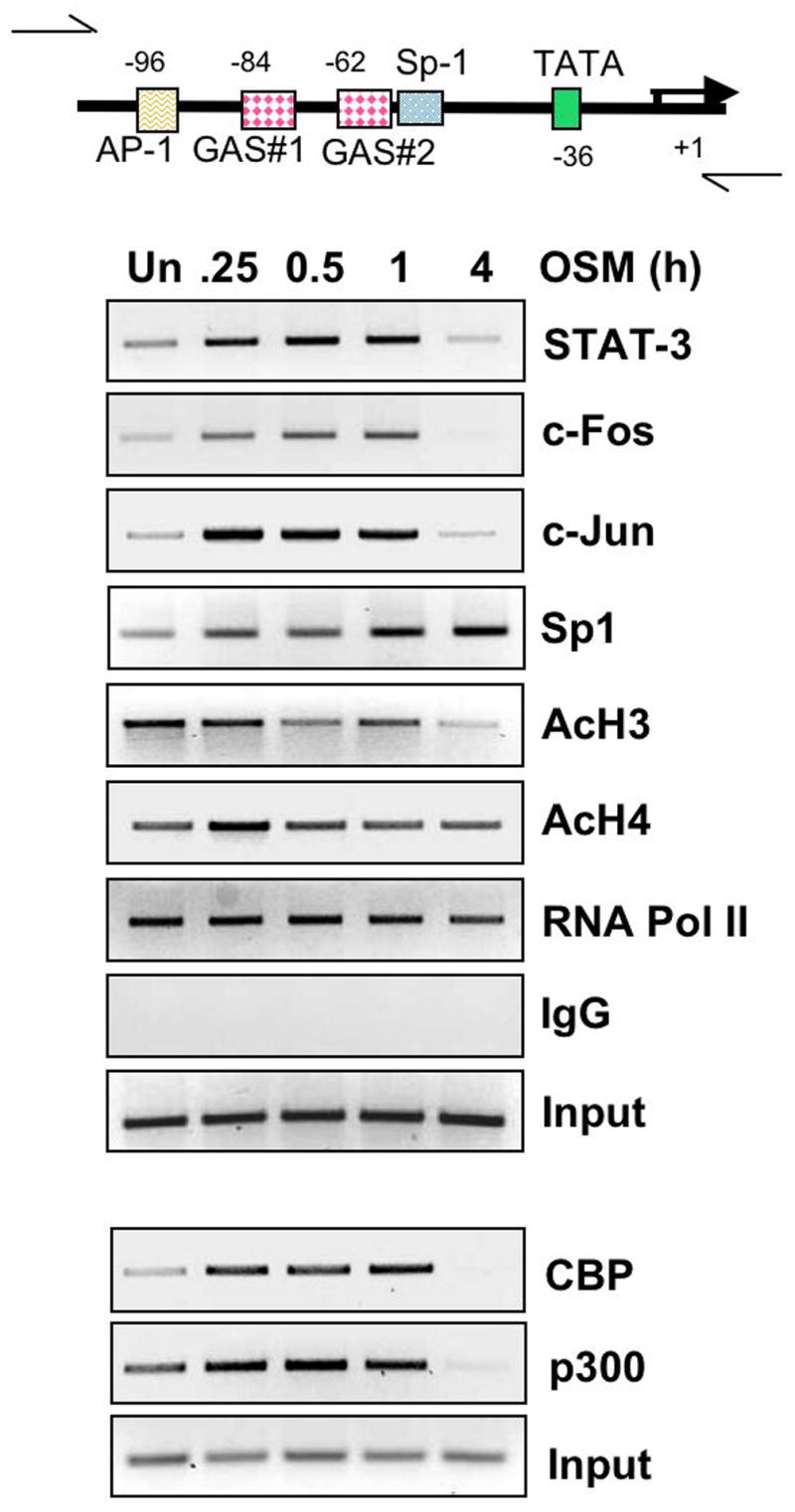
Primary astrocytes were treated with OSM (5 ng/ml) for the indicated times, and subjected to chromatin immunoprecipitation (ChIP) assay. Briefly, formaldehyde was used to cross-link proteins to DNA and the soluble chromatin was subjected to immunoprecipitation with IgG control serum or 5 μg of antibody against STAT-3, c-Fos, c-Jun, Sp1, acetylated histone H3, acetylated histone H4 and RNA Polymerase II. In a separate experiment, immunoprecipitation was performed with 5 μg of antibody against CBP and p300. Primers specific for the indicated region of the SOCS-3 promoter were used to amplify the DNA bound to particular proteins at different times. Input serves as a control for total DNA. Representative of three independent experiments.
It is well established that covalent histone modifications alter the structure of chromatin to influence gene transcription. The N-terminal tails of histones may be modified by acetylation, methylation, phosphorylation, and ubiquitination, resulting in altered chromatin conformation (Barski et al. 2007). To determine histone modifications in response to OSM, ChIP assays were performed using Abs against the acetylated forms of histones H3 and H4. Histone H3 was constitutively acetylated but acetylation was diminished after 4 h of OSM treatment. Histone H4 was also constitutively acetylated and this pattern was not affected by OSM treatment. These data demonstrate varying degrees of constitutive acetylation of histones H3 and H4 in primary astrocytes (Fig. 9). Histone acetylation requires the activities of histone acetyltransferases (HATs) such as CBP and p300, which tend to promote open chromatin conformations (Verdone et al. 2005). In addition to their roles as HATs, CBP and p300 are also important for recruitment of the transcriptional machinery and can act as adaptor molecules in transcriptional complexes (Blobel 2002; Chan and La Thangue 2001). CBP and p300 were present at low levels at the SOCS-3 promoter under basal conditions. Treatment with OSM enhanced the recruitment of CBP and p300 by 15 min, which lasted out to 1 h (Fig. 9). By 4 h following OSM treatment, the presence of CBP and p300 at the SOCS-3 promoter was diminished. ChIP assays were also performed for the recruitment of RNA Pol II to the SOCS-3 promoter. Pol II was constitutively present at the SOCS-3 promoter and its presence slightly diminished by 4 h following OSM treatment. These results suggest that under basal conditions, the SOCS-3 promoter is in a “ready” state that would allow for extremely fast initiation of gene transcription upon recruitment of the necessary factors.
DISCUSSION
In astrocytes and cerebral endothelial cells, OSM has been shown to induce the expression of pro-inflammatory molecules such as matrix metalloproteinases (MMPs) -1 and -3, ICAM-1 and MCP-1, which may contribute to CNS inflammation by promoting immune cell migration and invasion (Korzus et al. 1997). Plasminogen Activator Inhibitor-1 and urokinase-type Plasminogen Activator, other proteins involved in tissue remodeling and cell invasion, are also induced by OSM in astrocytes (Kasza et al. 2002). In astrocytes and astroglioma cells, IL-1β synergizes with OSM to enhance PGE2 and COX-2 production (Repovic et al. 2003b). Because these factors contribute to CNS inflammation, OSM may promote an inflammatory environment in the brain. However, OSM also induces TIMP-1 expression in astrocytes and in brain microvascular endothelial cells (Bugno et al. 1999; Kasza et al. 2002). TIMP proteins inhibit the actions of MMPs and have been shown to inhibit tumor growth and invasion as well as inflammatory processes (Bugno et al. 1999). Also, OSM treatment protected mice in a mouse model of MS, and limited inflammatory cell infiltration into the CNS compared to untreated animals (Wallace et al. 1999). Finally, OSM was shown to protect neurons from NMDA-induced excitotoxicity in vitro and in vivo (Weiss et al. 2006). Therefore, the function of OSM on cells of the CNS is complex, with inflammatory, anti-inflammatory and neuroprotective activities.
In this study we demonstrate that OSM induces rapid and strong SOCS-3 expression in primary astrocytes (Fig. 1). This expression is induced at the level of transcription as evidenced by activation of the SOCS-3 promoter in response to OSM (Fig. 1C). Induction of SOCS-3 is 71-fold above basal at the mRNA level, and 10.9-fold at the promoter level. The difference between these values is likely due to the experimental approach used in each case. SOCS-3 mRNA expression occurs due to activation of the endogenous SOCS-3 promoter, which potentially contains many regulatory elements that are not present on the 1500 bp fragment used in the promoter assay. OSM induced rapid and transient activation of both JAK-1 and JAK-2 (Figs. 2A and 2B). STAT-1 and STAT-3 were both activated by 15 min following OSM treatment; however, STAT-3 activation was more pronounced and prolonged compared to STAT-1 (Figs. 2C and 2D). Targeted knock down of STAT-3 by siRNA inhibited OSM-induced SOCS-3 expression at all timepoints, with an average inhibition of 58.4% (Figure 3C). On the other hand, SOCS-1 mRNA was enhanced under STAT-3 siRNA conditions, supporting an inhibitory role of STAT-3 in the expression of SOCS-1. The dominant-negative STAT-3F inhibited OSM-induced SOCS-3 promoter activity, while STAT-3C alone was capable of activating the SOCS-3 promoter (Figs. 3D and 3E). Exploration of STAT-1 involvement in SOCS-3 expression using STAT-1 deficient primary astrocytes revealed that STAT-1 is not necessary for OSM-induced SOCS-3 expression (Figs. 4A and 4B). However, STAT-1 was necessary for expression of SOCS-1 in astrocytes by OSM.
Activation of the MAPK pathways by OSM has been demonstrated for a number of cells types (Korzus et al. 1997; Van Wagoner et al. 2000). Treatment of primary astrocytes with OSM induced activation of the three main MAPK pathways: the p38, ERK1/2 and JNK pathways (Figs. 5A, 5B and 5C). Although the p38 pathway was determined not to be involved in OSM-induced SOCS-3 expression (data not shown), both the ERK1/2 and JNK pathways contributed to SOCS-3 expression (Figs. 6 and 7). The kinetic analysis of SOCS-3 mRNA expression demonstrated that inhibition of the JNK pathway using SP600125 led to a greater reduction in SOCS-3 expression than when the ERK1/2 pathway was inhibited by U0126. These results suggest that OSM-induced SOCS-3 expression in astrocytes relies more on activation of the JNK pathway than the ERK1/2 pathway.
The SOCS-3 promoter contains many regulatory elements including three AP-1 sites and two GAS elements (Gatto et al. 2004; Qin et al. 2007). AP-1 transcription factors are activated downstream of the MAPK pathways and GAS elements bind activated STAT proteins. Removal of the proximal AP-1 site (Δ5) decreased OSM-induced SOCS-3 promoter activity by 48%, and deletion of both the distal and proximal GAS elements (GAS #1 and #2) and the proximal Sp1 site (Δ7) completely eliminated activation of the SOCS-3 promoter by OSM (Fig. 8A). Within the Δ4 construct, mutation of the proximal AP-1 site led to a 22% decrease in OSM-induced SOCS-3 promoter activity, whereas mutation of GAS #1 caused a 48% decrease, and mutation of GAS #2 led to a 40% decrease (Fig. 8B). These results indicate that all three elements, proximal AP-1, GAS #1, and GAS #2, are important for optimal OSM-induced SOCS-3 promoter activity.
The ChIP assay was utilized to characterize the recruitment of relevant transcription factors and cofactors to the endogenous SOCS-3 promoter in astrocytes under basal and OSM-treatment conditions. While low levels of STAT-3 were present on the SOCS-3 promoter under basal condition, OSM treatment induced robust recruitment of STAT-3 by 15 min, which lasted to 1 h (Fig. 9). A similar pattern of recruitment was seen for the AP-1 transcription factors c-Fos and c-Jun, as well as for the co-factors CBP and p300 (Fig. 9). The identical kinetics of recruitment for these factors, and the proximity of the AP-1 and GAS elements suggest that they may form a transcriptional complex to activate SOCS-3 gene expression. Recruitment of Sp1 occurred with delayed kinetics, thus Sp1 may participate in later activation of SOCS-3 transcription. RNA Pol II is present on the SOCS-3 promoter under basal conditions and histones H3 and H4 are constitutively acetylated to varying degrees. These data suggest that the open reading frame of the SOCS-3 promoter is accessible in resting cells, but cannot be activated until the formation of an appropriate transcriptional complex.
Recently, in vivo studies have attempted to define the role of SOCS-3 in cells of the CNS. Conditional deletion of SOCS-3 in astrocytes improved functional recovery in a mouse model of spinal cord injury and promoted formation of glial scars, prevented excessive inflammatory infiltrates and enhanced survival of oligodendrocytes (Okada et al. 2006). In contrast, conditional deletion of STAT-3 in astrocytes inhibited astrocyte migration, glial scar formation, and delayed functional recovery (Okada et al. 2006). A rat model of neurite outgrowth in primary sensory neurons demonstrated that lentivirus-mediated delivery of SOCS-3 inhibited neurite growth. Lentivirus delivery of STAT-3, however, significantly increased the length of neurites (Miao et al. 2006). Targeted expression of SOCS-3 in oligodendrocytes limited the protective effect of LIF, an IL-6 cytokine, on cuprizone-induced demyelination (Emery et al. 2006). Finally, one group demonstrated that overexpression of SOCS-3 in neural stem cells decreased STAT-3 activation, promoted the maintenance of the neural stem cell phenotype and inhibited astrogliogenesis (Cao et al. 2006). SOCS-3 has been shown to modulate several important cellular processes within the CNS including cell migration, growth, cell survival, and differentiation, in part by inhibiting STAT-3 signaling. However, it is important to consider that SOCS-3 has also been shown to inhibit signaling by the growth hormone receptor, insulin receptor, focal adhesion kinases and src family kinases, among others (Baetz et al. 2004; Cacalano et al. 2001; Johnston 2004; Liu et al. 2003). In addition to the inhibitory actions of SOCS-3, one study demonstrated that SOCS-3 can prolong growth-factor-induced ERK signaling by binding to p120 RasGAP, a Ras inhibitor (Cacalano et al. 2001). This function of SOCS-3 has important implications for cell proliferation, migration, and cell-cycle progression which depend, in part, on the intensity and duration of ERK signaling. Therefore, the effects of SOCS-3 within the CNS could be due to regulation of a number of signaling cascades. Because SOCS-3 has the potential to regulate responses in astrocytes, oligodendrocytes and neurons, developing a better understanding of its regulation will be important clinically.
Since the discovery of the SOCS proteins in the late 1990s, there have been many studies directed towards determining the regulation and actions of these proteins both in vitro and in vivo. Also, researchers have attempted to elucidate the beneficial versus harmful effects of OSM within the CNS, but many questions remain. SOCS-3 may play an important role in regulating the balance between the protective and deleterious effects of OSM in the CNS, specifically in astrocytes. Therefore, understanding the relationship between OSM regulation of SOCS-3 and subsequent SOCS-3 regulation of OSM cellular effects will be important.
Acknowledgments
This work was supported in part by NIH grants NS45290 and NS36765 to E.N.B., and a grant from the National Multiple Sclerosis Society, RG-3892-A-12, to E.N.B.. B.J.B. is supported by the NIH Training Grant T32-NS48039.
References
- Alexander WS, Hilton DJ. The role of suppressors of cytokine signaling (SOCS) proteins in regulation of the immune response. Annu Rev Immunol. 2004;22:503–529. doi: 10.1146/annurev.immunol.22.091003.090312. [DOI] [PubMed] [Google Scholar]
- Auernhammer CJ, Bousquet C, Melmed S. Autoregulation of pituitary corticotroph SOCS-3 expression: characterization of the murine SOCS-3 promoter. Proc Natl Acad Sci U S A. 1999;96:6964–6969. doi: 10.1073/pnas.96.12.6964. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baetz A, Frey M, Heeg K, Dalpke AH. Suppressor of cytokine signaling (SOCS) proteins indirectly regulate toll-like receptor signaling in innate immune cells. J Biol Chem. 2004;279:54708–54715. doi: 10.1074/jbc.M410992200. [DOI] [PubMed] [Google Scholar]
- Barclay JL, Anderson ST, Waters MJ, Curlewis JD. Characterization of the SOCS3 promoter response to prostaglandin E2 in T47D cells. Mol Endocrinol. 2007a;21:2516–2528. doi: 10.1210/me.2007-0030. [DOI] [PubMed] [Google Scholar]
- Barclay JL, Anderson ST, Waters MJ, Curlewis JD. Regulation of suppressor of cytokine signaling 3 (SOC3) by growth hormone in pro-B cells. Mol Endocrinol. 2007b;21:2503–2515. doi: 10.1210/me.2006-0498. [DOI] [PubMed] [Google Scholar]
- Barski A, Cuddapah S, Cui K, Roh TY, Schones DE, Wang Z, Wei G, Chepelev I, Zhao K. High-resolution profiling of histone methylations in the human genome. Cell. 2007;129:823–837. doi: 10.1016/j.cell.2007.05.009. [DOI] [PubMed] [Google Scholar]
- Bennett BL, Sasaki DT, Murray BW, O’Leary EC, Sakata ST, Xu W, Leisten JC, Motiwala A, Pierce S, Satoh Y, et al. SP600125, an anthrapyrazolone inhibitor of Jun N-terminal kinase. Proc Natl Acad Sci U S A. 2001;98:13681–13686. doi: 10.1073/pnas.251194298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blobel GA. CBP and p300: versatile coregulators with important roles in hematopoietic gene expression. J Leukoc Biol. 2002;71:545–556. [PubMed] [Google Scholar]
- Brender C, Nielsen M, Kaltoft K, Mikkelsen G, Zhang Q, Wasik M, Billestrup N, Odum N. STAT3-mediated constitutive expression of SOCS-3 in cutaneous T-cell lymphoma. Blood. 2001;97:1056–1062. doi: 10.1182/blood.v97.4.1056. [DOI] [PubMed] [Google Scholar]
- Bromberg J, Darnell JE., Jr The role of STATs in transcriptional control and their impact on cellular function. Oncogene. 2000;19:2468–2473. doi: 10.1038/sj.onc.1203476. [DOI] [PubMed] [Google Scholar]
- Bromberg JF, Wrzeszczynska MH, Devgan G, Zhao Y, Pestell RG, Albanese C, Darnell JE., Jr Stat3 as an oncogene. Cell. 1999;98:295–303. doi: 10.1016/s0092-8674(00)81959-5. [DOI] [PubMed] [Google Scholar]
- Bugno M, Witek B, Bereta J, Bereta M, Edwards DR, Kordula T. Reprogramming of TIMP-1 and TIMP-3 expression profiles in brain microvascular endothelial cells and astrocytes in response to proinflammatory cytokines. FEBS Lett. 1999;448:9–14. doi: 10.1016/s0014-5793(99)00323-3. [DOI] [PubMed] [Google Scholar]
- Cacalano NA, Sanden D, Johnston JA. Tyrosine-phosphorylated SOCS-3 inhibits STAT activation but binds to p120 RasGAP and activates Ras. Nat Cell Biol. 2001;3:460–465. doi: 10.1038/35074525. [DOI] [PubMed] [Google Scholar]
- Campbell IL. Cytokine-mediated inflammation, tumorigenesis, and disease-associated JAK/STAT/SOCS signaling circuits in the CNS. Brain Res Brain Res Rev. 2005;48:166–177. doi: 10.1016/j.brainresrev.2004.12.006. [DOI] [PubMed] [Google Scholar]
- Cao F, Hata R, Zhu P, Ma YJ, Tanaka J, Hanakawa Y, Hashimoto K, Niinobe M, Yoshikawa K, Sakanaka M. Overexpression of SOCS3 inhibits astrogliogenesis and promotes maintenance of neural stem cells. J Neurochem. 2006;98:459–470. doi: 10.1111/j.1471-4159.2006.03890.x. [DOI] [PubMed] [Google Scholar]
- Chan HM, La Thangue NB. p300/CBP proteins: HATs for transcriptional bridges and scaffolds. J Cell Sci. 2001;114:2363–2373. doi: 10.1242/jcs.114.13.2363. [DOI] [PubMed] [Google Scholar]
- Chen SH, Benveniste EN. Oncostatin M: a pleiotropic cytokine in the central nervous system. Cytokine Growth Factor Rev. 2004;15:379–391. doi: 10.1016/j.cytogfr.2004.06.002. [DOI] [PubMed] [Google Scholar]
- Costa-Pereira AP, Tininini S, Strobl B, Alonzi T, Schlaak JF, Is’harc H, Gesualdo I, Newman SJ, Kerr IM, Poli V. Mutational switch of an IL-6 response to an interferon-gamma-like response. Proc Natl Acad Sci U S A. 2002;99:8043–8047. doi: 10.1073/pnas.122236099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Croker BA, Krebs DL, Zhang JG, Wormald S, Willson TA, Stanley EG, Robb L, Greenhalgh CJ, Forster I, Clausen BE, et al. SOCS3 negatively regulates IL-6 signaling in vivo. Nat Immunol. 2003;4:540–545. doi: 10.1038/ni931. [DOI] [PubMed] [Google Scholar]
- Dalpke AH, Opper S, Zimmermann S, Heeg K. Suppressors of cytokine signaling (SOCS)-1 and SOCS-3 are induced by CpG-DNA and modulate cytokine responses in APCs. J Immunol. 2001;166:7082–7089. doi: 10.4049/jimmunol.166.12.7082. [DOI] [PubMed] [Google Scholar]
- Ding Y, Chen D, Tarcsafalvi A, Su R, Qin L, Bromberg JS. Suppressor of cytokine signaling 1 inhibits IL-10-mediated immune responses. J Immunol. 2003;170:1383–1391. doi: 10.4049/jimmunol.170.3.1383. [DOI] [PubMed] [Google Scholar]
- Dong Y, Tang L, Letterio JJ, Benveniste EN. The Smad3 protein is involved in TGF-beta inhibition of class II transactivator and class II MHC expression. J Immunol. 2001;167:311–319. doi: 10.4049/jimmunol.167.1.311. [DOI] [PubMed] [Google Scholar]
- Elliott J, Johnston JA. SOCS: role in inflammation, allergy and homeostasis. Trends Immunol. 2004;25:434–440. doi: 10.1016/j.it.2004.05.012. [DOI] [PubMed] [Google Scholar]
- Emery B, Cate HS, Marriott M, Merson T, Binder MD, Snell C, Soo PY, Murray S, Croker B, Zhang JG, et al. Suppressor of cytokine signaling 3 limits protection of leukemia inhibitory factor receptor signaling against central demyelination. Proc Natl Acad Sci U S A. 2006;103:7859–7864. doi: 10.1073/pnas.0602574103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Endo TA, Masuhara M, Yokouchi M, Suzuki R, Sakamoto H, Mitsui K, Matsumoto A, Tanimura S, Ohtsubo M, Misawa H, et al. A new protein containing an SH2 domain that inhibits JAK kinases. Nature. 1997;387:921–924. doi: 10.1038/43213. [DOI] [PubMed] [Google Scholar]
- Ensoli F, Fiorelli V, DeCristofaro M, Santini Muratori D, Novi A, Vannelli B, Thiele CJ, Luzi G, Aiuti F. Inflammatory cytokines and HIV-1-associated neurodegeneration: oncostatin-M produced by mononuclear cells from HIV-1-infected individuals induces apoptosis of primary neurons. J Immunol. 1999;162:6268–6277. [PubMed] [Google Scholar]
- Ensoli F, Fiorelli V, Lugaresi A, Farina D, De Cristofaro M, Collacchi B, Muratori DS, Scala E, Di Gioacchino M, Paganelli R, et al. Lymphomononuclear cells from multiple sclerosis patients spontaneously produce high levels of oncostatin M, tumor necrosis factors alpha and beta, and interferon gamma. Mult Scler. 2002;8:284–288. doi: 10.1191/1352458502ms817oa. [DOI] [PubMed] [Google Scholar]
- Favata MF, Horiuchi KY, Manos EJ, Daulerio AJ, Stradley DA, Feeser WS, Van Dyk DE, Pitts WJ, Earl RA, Hobbs F, et al. Identification of a novel inhibitor of mitogen-activated protein kinase kinase. J Biol Chem. 1998;273:18623–18632. doi: 10.1074/jbc.273.29.18623. [DOI] [PubMed] [Google Scholar]
- Frantz B, Klatt T, Pang M, Parsons J, Rolando A, Williams H, Tocci MJ, O’Keefe SJ, O’Neill EA. The activation state of p38 mitogen-activated protein kinase determines the efficiency of ATP competition for pyridinylimidazole inhibitor binding. Biochemistry. 1998;37:13846–13853. doi: 10.1021/bi980832y. [DOI] [PubMed] [Google Scholar]
- Gatto L, Berlato C, Poli V, Tininini S, Kinjyo I, Yoshimura A, Cassatella MA, Bazzoni F. Analysis of SOCS-3 promoter responses to interferon gamma. J Biol Chem. 2004;279:13746–13754. doi: 10.1074/jbc.M308999200. [DOI] [PubMed] [Google Scholar]
- Gomez-Lechon MJ. Oncostatin M: signal transduction and biological activity. Life Sci. 1999;65:2019–2030. doi: 10.1016/s0024-3205(99)00296-9. [DOI] [PubMed] [Google Scholar]
- Halfter H, Friedrich M, Resch A, Kullmann M, Stogbauer F, Ringelstein EB, Hengst L. Oncostatin M induces growth arrest by inhibition of Skp2, Cks1, and cyclin A expression and induced p21 expression. Cancer Res. 2006;66:6530–6539. doi: 10.1158/0008-5472.CAN-04-3734. [DOI] [PubMed] [Google Scholar]
- Hanada T, Kinjyo I, Inagaki-Ohara K, Yoshimura A. Negative regulation of cytokine signaling by CIS/SOCS family proteins and their roles in inflammatory diseases. Rev Physiol Biochem Pharmacol. 2003;149:72–86. doi: 10.1007/s10254-003-0015-z. [DOI] [PubMed] [Google Scholar]
- Heinrich PC, Behrmann I, Haan S, Hermanns HM, Muller-Newen G, Schaper F. Principles of interleukin (IL)-6-type cytokine signalling and its regulation. Biochem J. 2003;374:1–20. doi: 10.1042/BJ20030407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hurst SM, McLoughlin RM, Monslow J, Owens S, Morgan L, Fuller GM, Topley N, Jones SA. Secretion of oncostatin M by infiltrating neutrophils: regulation of IL-6 and chemokine expression in human mesothelial cells. J Immunol. 2002;169:5244–5251. doi: 10.4049/jimmunol.169.9.5244. [DOI] [PubMed] [Google Scholar]
- Ilangumaran S, Ramanathan S, Rottapel R. Regulation of the immune system by SOCS family adaptor proteins. Semin Immunol. 2004;16:351–365. doi: 10.1016/j.smim.2004.08.015. [DOI] [PubMed] [Google Scholar]
- Johnston JA. Are SOCS suppressors, regulators, and degraders? J Leukoc Biol. 2004;75:743–748. doi: 10.1189/jlb.1003507. [DOI] [PubMed] [Google Scholar]
- Kasza A, Kiss DL, Gopalan S, Xu W, Rydel RE, Koj A, Kordula T. Mechanism of plasminogen activator inhibitor-1 regulation by oncostatin M and interleukin-1 in human astrocytes. J Neurochem. 2002;83:696–703. doi: 10.1046/j.1471-4159.2002.01163.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Korzus E, Nagase H, Rydell R, Travis J. The mitogen-activated protein kinase and JAK-STAT signaling pathways are required for an oncostatin M-responsive element-mediated activation of matrix metalloproteinase 1 gene expression. J Biol Chem. 1997;272:1188–1196. doi: 10.1074/jbc.272.2.1188. [DOI] [PubMed] [Google Scholar]
- Kubo M, Hanada T, Yoshimura A. Suppressors of cytokine signaling and immunity. Nat Immunol. 2003;4:1169–1176. doi: 10.1038/ni1012. [DOI] [PubMed] [Google Scholar]
- Lang R, Pauleau AL, Parganas E, Takahashi Y, Mages J, Ihle JN, Rutschman R, Murray PJ. SOCS3 regulates the plasticity of gp130 signaling. Nat Immunol. 2003;4:546–550. doi: 10.1038/ni932. [DOI] [PubMed] [Google Scholar]
- Langdon C, Kerr C, Tong L, Richards CD. Oncostatin M regulates eotaxin expression in fibroblasts and eosinophilic inflammation in C57BL/6 mice. J Immunol. 2003;170:548–555. doi: 10.4049/jimmunol.170.1.548. [DOI] [PubMed] [Google Scholar]
- Liu E, Cote JF, Vuori K. Negative regulation of FAK signaling by SOCS proteins. Embo J. 2003;22:5036–5046. doi: 10.1093/emboj/cdg503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marine JC, McKay C, Wang D, Topham DJ, Parganas E, Nakajima H, Pendeville H, Yasukawa H, Sasaki A, Yoshimura A, et al. SOCS3 is essential in the regulation of fetal liver erythropoiesis. Cell. 1999;98:617–627. doi: 10.1016/s0092-8674(00)80049-5. [DOI] [PubMed] [Google Scholar]
- Masuhara M, Sakamoto H, Matsumoto A, Suzuki R, Yasukawa H, Mitsui K, Wakioka T, Tanimura S, Sasaki A, Misawa H, et al. Cloning and characterization of novel CIS family genes. Biochem Biophys Res Commun. 1997;239:439–446. doi: 10.1006/bbrc.1997.7484. [DOI] [PubMed] [Google Scholar]
- Meraz MA, White JM, Sheehan KC, Bach EA, Rodig SJ, Dighe AS, Kaplan DH, Riley JK, Greenlund AC, Campbell D, et al. Targeted disruption of the Stat1 gene in mice reveals unexpected physiologic specificity in the JAK-STAT signaling pathway. Cell. 1996;84:431–442. doi: 10.1016/s0092-8674(00)81288-x. [DOI] [PubMed] [Google Scholar]
- Miao T, Wu D, Zhang Y, Bo X, Subang MC, Wang P, Richardson PM. Suppressor of cytokine signaling-3 suppresses the ability of activated signal transducer and activator of transcription-3 to stimulate neurite growth in rat primary sensory neurons. J Neurosci. 2006;26:9512–9519. doi: 10.1523/JNEUROSCI.2160-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nakajima K, Yamanaka Y, Nakae K, Kojima H, Ichiba M, Kiuchi N, Kitaoka T, Fukada T, Hibi M, Hirano T. A central role for Stat3 in IL-6-induced regulation of growth and differentiation in M1 leukemia cells. Embo J. 1996;15:3651–3658. [PMC free article] [PubMed] [Google Scholar]
- O’Keefe GM, Nguyen VT, Ping Tang LL, Benveniste EN. IFN-gamma regulation of class II transactivator promoter IV in macrophages and microglia: involvement of the suppressors of cytokine signaling-1 protein. J Immunol. 2001;166:2260–2269. doi: 10.4049/jimmunol.166.4.2260. [DOI] [PubMed] [Google Scholar]
- Okada S, Nakamura M, Katoh H, Miyao T, Shimazaki T, Ishii K, Yamane J, Yoshimura A, Iwamoto Y, Toyama Y, et al. Conditional ablation of Stat3 or Socs3 discloses a dual role for reactive astrocytes after spinal cord injury. Nat Med. 2006;12:829–834. doi: 10.1038/nm1425. [DOI] [PubMed] [Google Scholar]
- Park EJ, Park SY, Joe EH, Jou I. 15d-PGJ2 and rosiglitazone suppress Janus kinase-STAT inflammatory signaling through induction of suppressor of cytokine signaling 1 (SOCS1) and SOCS3 in glia. J Biol Chem. 2003a;278:14747–14752. doi: 10.1074/jbc.M210819200. [DOI] [PubMed] [Google Scholar]
- Park SH, Kim KE, Hwang HY, Kim TY. Regulatory effect of SOCS on NF-kappaB activity in murine monocytes/macrophages. DNA Cell Biol. 2003b;22:131–139. doi: 10.1089/104454903321515931. [DOI] [PubMed] [Google Scholar]
- Qin H, Roberts KL, Niyongere SA, Cong Y, Elson CO, Benveniste EN. Molecular mechanism of lipopolysaccharide-induced SOCS-3 gene expression in macrophages and microglia. J Immunol. 2007;179:5966–5976. doi: 10.4049/jimmunol.179.9.5966. [DOI] [PubMed] [Google Scholar]
- Qin H, Wilson CA, Lee SJ, Benveniste EN. IFN-beta-induced SOCS-1 negatively regulates CD40 gene expression in macrophages and microglia. Faseb J. 2006a;20:985–987. doi: 10.1096/fj.05-5493fje. [DOI] [PubMed] [Google Scholar]
- Qin H, Wilson CA, Roberts KL, Baker BJ, Zhao X, Benveniste EN. IL-10 inhibits lipopolysaccharide-induced CD40 gene expression through induction of suppressor of cytokine signaling-3. J Immunol. 2006b;177:7761–7771. doi: 10.4049/jimmunol.177.11.7761. [DOI] [PubMed] [Google Scholar]
- Ramana CV, Kumar A, Enelow R. Stat1-independent induction of SOCS-3 by interferon-gamma is mediated by sustained activation of Stat3 in mouse embryonic fibroblasts. Biochem Biophys Res Commun. 2005;327:727–733. doi: 10.1016/j.bbrc.2004.12.074. [DOI] [PubMed] [Google Scholar]
- Repovic P, Fears CY, Gladson CL, Benveniste EN. Oncostatin-M induction of vascular endothelial growth factor expression in astroglioma cells. Oncogene. 2003a;22:8117–8124. doi: 10.1038/sj.onc.1206922. [DOI] [PubMed] [Google Scholar]
- Repovic P, Mi K, Benveniste EN. Oncostatin M enhances the expression of prostaglandin E2 and cyclooxygenase-2 in astrocytes: synergy with interleukin-1beta, tumor necrosis factor-alpha, and bacterial lipopolysaccharide. Glia. 2003b;42:433–446. doi: 10.1002/glia.10182. [DOI] [PubMed] [Google Scholar]
- Seki Y, Inoue H, Nagata N, Hayashi K, Fukuyama S, Matsumoto K, Komine O, Hamano S, Himeno K, Inagaki-Ohara K, et al. SOCS-3 regulates onset and maintenance of T(H)2-mediated allergic responses. Nat Med. 2003;9:1047–1054. doi: 10.1038/nm896. [DOI] [PubMed] [Google Scholar]
- Stevenson NJ, Haan S, McClurg AE, McGrattan MJ, Armstrong MA, Heinrich PC, Johnston JA. The chemoattractants, IL-8 and formyl-methionyl-leucyl-phenylalanine, regulate granulocyte colony-stimulating factor signaling by inducing suppressor of cytokine signaling-1 expression. J Immunol. 2004;173:3243–3249. doi: 10.4049/jimmunol.173.5.3243. [DOI] [PubMed] [Google Scholar]
- Suzuki R, Sakamoto H, Yasukawa H, Masuhara M, Wakioka T, Sasaki A, Yuge K, Komiya S, Inoue A, Yoshimura A. CIS3 and JAB have different regulatory roles in interleukin-6 mediated differentiation and STAT3 activation in M1 leukemia cells. Oncogene. 1998;17:2271–2278. doi: 10.1038/sj.onc.1202143. [DOI] [PubMed] [Google Scholar]
- Takeda K, Noguchi K, Shi W, Tanaka T, Matsumoto M, Yoshida N, Kishimoto T, Akira S. Targeted disruption of the mouse Stat3 gene leads to early embryonic lethality. Proc Natl Acad Sci U S A. 1997;94:3801–3804. doi: 10.1073/pnas.94.8.3801. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thoma B, Bird TA, Friend DJ, Gearing DP, Dower SK. Oncostatin M and leukemia inhibitory factor trigger overlapping and different signals through partially shared receptor complexes. J Biol Chem. 1994;269:6215–6222. [PubMed] [Google Scholar]
- Turjanski AG, Vaque JP, Gutkind JS. MAP kinases and the control of nuclear events. Oncogene. 2007;26:3240–3253. doi: 10.1038/sj.onc.1210415. [DOI] [PubMed] [Google Scholar]
- Van Wagoner NJ, Choi C, Repovic P, Benveniste EN. Oncostatin M regulation of interleukin-6 expression in astrocytes: biphasic regulation involving the mitogen-activated protein kinases ERK1/2 and p38. J Neurochem. 2000;75:563–575. doi: 10.1046/j.1471-4159.2000.0750563.x. [DOI] [PubMed] [Google Scholar]
- Vecchiet J, Dalessandro M, Falasca K, Di Iorio A, Travasi F, Zingariello P, Schiavone C, Ensoli F, Pizzigallo E, Paganelli R. Increased production of oncostatin-M by lymphomononuclear cells from HIV-1-infected patients with neuroAIDS. J Acquir Immune Defic Syndr. 2003;32:464–465. doi: 10.1097/00126334-200304010-00019. [DOI] [PubMed] [Google Scholar]
- Verdone L, Caserta M, Di Mauro E. Role of histone acetylation in the control of gene expression. Biochem Cell Biol. 2005;83:344–353. doi: 10.1139/o05-041. [DOI] [PubMed] [Google Scholar]
- Wallace PM, MacMaster JF, Rouleau KA, Brown TJ, Loy JK, Donaldson KL, Wahl AF. Regulation of inflammatory responses by oncostatin M. J Immunol. 1999;162:5547–5555. [PubMed] [Google Scholar]
- Weiss TW, Samson AL, Niego B, Daniel PB, Medcalf RL. Oncostatin M is a neuroprotective cytokine that inhibits excitotoxic injury in vitro and in vivo. Faseb J. 2006;20:2369–2371. doi: 10.1096/fj.06-5850fje. [DOI] [PubMed] [Google Scholar]
- Williams L, Bradley L, Smith A, Foxwell B. Signal transducer and activator of transcription 3 is the dominant mediator of the anti-inflammatory effects of IL-10 in human macrophages. J Immunol. 2004;172:567–576. doi: 10.4049/jimmunol.172.1.567. [DOI] [PubMed] [Google Scholar]
- Yasukawa H, Ohishi M, Mori H, Murakami M, Chinen T, Aki D, Hanada T, Takeda K, Akira S, Hoshijima M, et al. IL-6 induces an anti-inflammatory response in the absence of SOCS3 in macrophages. Nat Immunol. 2003;4:551–556. doi: 10.1038/ni938. [DOI] [PubMed] [Google Scholar]
- Yoshimura A, Naka T, Kubo M. SOCS proteins, cytokine signalling and immune regulation. Nat Rev Immunol. 2007;7:454–465. doi: 10.1038/nri2093. [DOI] [PubMed] [Google Scholar]
- Zimmerer JM, Lesinski GB, Kondadasula SV, Karpa VI, Lehman A, Raychaudhury A, Becknell B, Carson WE., 3rd IFN-alpha-induced signal transduction, gene expression, and antitumor activity of immune effector cells are negatively regulated by suppressor of cytokine signaling proteins. J Immunol. 2007;178:4832–4845. doi: 10.4049/jimmunol.178.8.4832. [DOI] [PubMed] [Google Scholar]