

Abstract
This article is concerned with statistical modeling of shotgun resequencing data and the use of such data for population genetic inference. We model data produced by sequencing-by-synthesis technologies such as the Solexa, 454, and polymerase colony (polony) systems, whose use is becoming increasingly widespread. We show how such data can be used to estimate evolutionary parameters (mutation and recombination rates), despite the fact that the data do not necessarily provide complete or aligned sequence information. We also present two refinements of our methods: one that is more robust to sequencing errors and another that can be used when no reference genome is available.
SANGER sequencing and fluorescence-based electrophoresis technologies are currently the standard methods for DNA sequencing. However, there is widespread interest in improving the efficiency with which sequence information can be obtained. Consequently, many novel sequencing technologies are being developed. Shendure et al. (2005a) reviews low-cost sequencing technologies, which can be classified into five groups: microelectrophoretic methods, sequencing by hybridization, cyclic-array sequencing on amplified molecules, cyclic-array sequencing on single molecules, and noncyclical, single-molecule, real-time methods.
Johnson and Slatkin (2006) developed methods for estimation of the mutation rate and the growth rate for Sanger shotgun sequencing data from metagenomic projects. In this article we develop tools that can be applied to the analysis of data resulting from resequencing technology. Such methods are rapidly growing in popularity and raise a variety of interesting practical and theoretical questions (e.g., Korbel et al. 2007; Brockman et al. 2008; Hillier et al. 2008). In this article we show how these data can be used to infer evolutionary parameters, such as mutation and recombination rates. We focus on three such systems: Solexa, 454, and polymerase colony (polony) sequencing (cf. Jarvie 2005). These methods are massively parallel and thereby offer the opportunity to improve, by several orders of magnitude, the speed with which sequence information is obtained. However, the technologies typically result in imperfect coverage of any given region of interest. While we develop inference methods for these three technologies in particular, our methods could easily be adapted to other similar techniques. In essence these methods first break genomic DNA into fragments from which library molecules are constructed. These library molecules are then clonally amplified in a highly parallel array and used as templates for sequencing by synthesis. Differences between the systems include the nature of the array surface and the type of sequencing chemistry that is used (Bentley 2006). These differences affect the details but not the spirit of the methods we introduce in this article. We now briefly summarize the three technologies (see also Figure 1). Each of them begins by randomly fragmenting the entire genome.
Figure 1.—

Illustration of Solexa reads in diploid individuals. Rectangles denote fragments, with the solid portion indicating the region that is sequenced for that fragment.
In the Solexa system (www.illumina.com) the fragments are then covalently attached to a planar surface and amplified in situ. Sequencing is performed in an iterative fashion using a mixture of four fluorescently labeled reversible chain terminators and DNA polymerases. The resulting data consist of regions of 20–50 bases at one end, or at both ends, of the fragment.
In the 454 system (www.454.com) (Margulies et al. 2005) specialized common adapters are added to the fragments. The fragments are then captured on beads and clonally amplified in aqueous-oil emulsion. Beads are placed in individual microfabricated picoliter wells for pyrosequencing. The resulting data typically consist of sequence information for regions of ∼200–300 bases at one end or at both ends of the fragment.
In polony sequencing (http://arep.med.harvard.edu/Polonator/) molecules are immobilized with a polyacrylamide gel on a microscope slide and amplified by forming polymerase colonies (Shendure et al. 2005b). A four-color sequencing-by-ligation scheme is used to generate reads and 13 bp at each end of a read are obtained.
We note that resequencing technology is a moving target in the sense that the details are evolving. For example, the number of bases in a typical read has changed over time. The methods in this article are robust to changes in parameters such as the exact number of bases per read or whether one or both ends of fragments are sequenced.
BACKGROUND
Each of the above technologies results in multiple short sequence fragments. We assume that the left ends of these fragments are sequenced and we refer to these sequenced portions as “reads” in this article. Finally, we initially assume that the reads have been uniquely mapped in the reference genome, regardless of the presence of polymorphism (but we relax this assumption later).
We wish to use these reads to infer population genetic parameters. Specifically, we explore the following question: When using data from reads, what degree of genome coverage is needed to estimate accurately population genetic parameters? Since fragments are created by a physical shearing process, it is a reasonable first approximation to assume that their left endpoints are randomly positioned along the genome. Specifically, we can view the start point of reads as occurring according to a Poisson process of rate λ along the genome. If several copies of the region of interest are fragmented according to independent shearing processes, the resulting process of left-hand ends is still a Poisson process but with a different value of λ.
We now consider the estimation of genetic parameters from a random sample of diploid individuals who have been resequenced at a chromosomal segment of interest. It is well known that the number of segregating sites and the haplotype configuration at two-locus pairs in a sample are informative about the mutation parameter θ and recombination parameter ρ, respectively (Watterson 1975; Hudson and Kaplan 1985; Hudson 2001). In the present context we encounter a missing data problem. At any particular location in the genome, we have sequence data only for those individuals in the sample that have reads covering that position. As a result, some segregating sites are not detected due to lack of coverage, and some two-locus genotype configurations (which we use later as part of our method for estimating recombination rate) are not available because loci may not have complete sequence information for all individuals.
Hudson (2001) used two-locus sampling distributions to estimate recombination rates from SNP data. Hudson's method involves calculating a composite product likelihood and can be applied to diploid samples. Each term of this product corresponds to the probability of the observed SNP genotype configuration at a particular pair of loci, the product being taken across all pairs of loci. However, we note one complicating factor related to diploids: given that we observe data from reads at a particular locus for an individual, we cannot tell (using current applications of the technologies we study) whether one or both alleles have been sequenced.
We assume that the read length is constant and initially we assume that there are no sequencing errors. We focus primarily on results for single-end reads from the Solexa system and outline extensions to the other two systems in the discussion.
RESULTS
We now present results for estimating the mutation parameter θ and recombination parameter ρ for resequencing data from diploid individuals, assuming no sequencing errors. We then adapt our approach to deal with two types of sequencing errors. Finally, we address the issue of inference when there is no reference genome. For convenience, we list our notation in Table 1.
TABLE 1.
Notation for analysis of reads
| Symbol | Meaning |
|---|---|
| m | No. of diploid individuals in the sample |
| n | 2m, i.e., no. of chromosomes |
| G | Length of resequenced region in base pairs |
| l | Length of a read in base pairs |
| λ | Rate of Poisson process |
| X = λl | Expected coverage per haploid |
| θ | Mutation parameter |
| ρ | Recombination parameter |
| S | No. of SNPs in the sample |
| ST | No. of SNPs detected by reads |
| RMSE | Root mean square error |
Estimating θ:
We first derive a point estimator for θ and then give simulation results to assess its performance. The key to estimating θ from read data is to calculate the probability of detecting a segregating site, which enables us to apply Watterson's estimator. The probability of detecting a segregating site in a diploid sample of size m is the same as that in the corresponding haploid sample of size 2m, because it requires only that we observe two different types at the locus. Using the notation in Table 1, we have the following (proofs are deferred to the appendix):
- The probability q1 that a random point, in a single copy of a genome, is covered by at least one read is
The expected total length of covered regions is Gq1.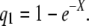
(1) - The probability q2(b) that a segregating site with b mutants in a sample of size n = 2m is detected by reads is
for 1 < b < n.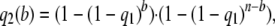
(2) - Under a constant population size coalescent model, the probability q3 that a segregating site is detected by reads is
where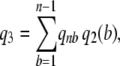
(3) 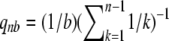 is the probability that a mutant is represented b times in the sample of size n (Griffiths and Tavaré 1998).
is the probability that a mutant is represented b times in the sample of size n (Griffiths and Tavaré 1998). - The expected number of detected SNPs is
Hence we obtain the point estimator of θ for read data,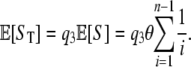
(4)
Note that these probabilities (e.g., q3) depend only on X, the expected coverage of reads per region. Figure 2 shows the proportion of the region covered for any given individual (q1) and the number of detected SNPs (ST) in graphical form, based on simulation of 25 diploid individuals over 1000 replicate data sets; circles refer to values obtained by the simulation and the solid line is based on the formulas above. Simulated values agree closely with the analytical predictions.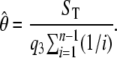
(5)
Figure 2.—

Properties of reads for a sample of 25 diploid individuals with θ = 100 and ρ = 20. Circles correspond to the average value of the statistic of interest in 1000 simulations. The solid line is the prediction based on the propositions in results. The horizontal dashed line in b indicates the expected number of segregating sites in the sample (Watterson 1975).
We now use the result in (5) to estimate θ. To assess performance we use standard coalescent simulations to generate 1000 samples of 25 diploid individuals (i.e., n = 2 × 25 = 50) with fixed values of θ and ρ, e.g., (100, 20) and (400, 20), for a 100-kb region. For each chromosome, we simulate the start point of reads as independent Poisson processes, and reads are assumed to be of length 36 bp. We record the number of segregating sites detected by reads, ST, and report point estimates of θ obtained using Equation 5. The mean and root mean square error (RMSE) are listed in Table 2. As we can see, the number of segregating sites detected by reads, ST, is highly informative for θ, with the variance increasing as the coverage decreases.
TABLE 2.
Estimating θ using resequencing data
| X | θ0 = 100 | θ0 = 400 |
|---|---|---|
| 16× | 99.5 (14.5) | 400.6 (57.4) |
| 8× | 99.3 (15.3) | 399.5 (58.7) |
| 4× | 101.6 (15.5) | 404.1 (58.3) |
| 2× | 105.1 (16.8) | 400.6 (62.9) |
The recombination parameter is fixed at ρ = 20. We report the mean (and RMSE) of the estimates over 1000 simulations.
Application to data:
It is, of course, of interest to apply our methods to existing data sets. Given the new nature of the technology, and the fact that the aim of our article is to anticipate the need for analysis methodologies for forthcoming data, we find ourselves in a situation in which there are very few publicly available resequencing data. For this reason we use a data set that was analyzed in Hellmann et al. (2008), an article that appeared during the revision process for this article and that presented an independently derived method for estimating mutation rate on the basis of resequencing data. The article included an analysis of the Celera shotgun sequence data from Venter et al. (2001). Those data consist of contigs from seven human individuals. The average number of reads per base, across the sample, is about five, leading to an expected coverage of 0.36 per haploid. Consequently, we apply our method for estimation of mutation rate to these data and compare results to those of Hellmann et al. (2008). However, we note that the depth of coverage in these data does not allow for application of the estimator for recombination rates that we present later in this article.
In the method of Hellmann et al. (2008), each chromosome is treated as a collection of segments, each segment being defined as the maximal continuous region over which the observed coverage is constant. This raises a key issue: when coverage is low, or varies significantly over the genome due to the existence of repetitive regions, for example, the use of an estimator, such as ours, that conditions on theoretical expectation of coverage rather than the coverage actually observed may be suboptimal. To assess this issue, when analyzing these data we also present results for a version of our method that conditions on the observed coverage. We now detail this modification.
The key to our method for estimating θ is the calculation of the probability that a segregating site is detected. To calculate this probability conditional on the observed coverage within a segment, where segments are defined as regions of maximal length over which coverage is constant, we consider a particular segregating site, in a particular segment v, and let r denote the number of reads observed to cover that site. We first condition on the number of mutant alleles at the segregating site (i.e., b) and calculate the probability of detecting this segregating site as
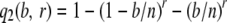 |
(6) |
(cf. Equation 2), where n is the number of (haploid) chromosomes. We then substitute into Equation 3 to obtain the conditional probability of detecting a segregating site, given r observed reads as
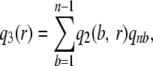 |
(7) |
where, as before, qnb is the probability of there being b mutants at a segregating site. The point estimator of θ per base for the region covered by r reads is then
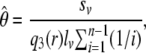 |
(8) |
where sv is the number of detected segregating sites and lv is the length of segment v.
We then write the overall estimate of θ as
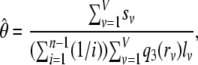 |
(9) |
where V is the total number of segments (where r > 0).
Results are listed in Table 3. As well as analyzing the data of Venter et al. (2001) (the first row of results), we present analyses of a set of 1000 simulated data sets (the second row of results) designed to assess the generality of our conclusions. We report both the mean and the RMSE of the resulting θ-estimates for the 1000 analyses. We simulated 10-kb regions for n = 20 haplotypes using a mutation rate of θ = 1/kb and further simulated short reads of length 100 bp with an expected coverage of 0.5, without sequencing error. In column a we show the results of applying the method of Hellmann et al. (2008), in column b we give results for the method presented in this article, and in column c we report results for the extension of our approach to perform calculations conditional on observed rather than expected coverage. Existing estimates for θ in the region corresponding to the data of Hellmann et al. (2008) are ∼1.0/kb (Halushka et al. 1999, reported as 1.5/kb for silent substitutions and 1.05/kb for introns), suggesting that our method may perform somewhat better than that of Hellmann et al. (2008). The analyses of the simulated data suggest that this might be true in general, since we obtain both a smaller bias and a smaller RMSE. Finally, we note that, across the scenarios considered in this article, as is suggested by the results in Table 3, there appears to be little measurable difference in performance between the versions of our estimator based on expected and observed coverage (results not shown). However, when coverage levels are very low, it may be the case that conditioning on observed coverage would show an improvement in performance.
TABLE 3.
Comparisons of estimates of θ (per kb) for the data of Venter et al. (2001) and for a set of 1000 simulated data sets
| Data | a | b | c |
|---|---|---|---|
| Celera data | 1.67 | 1.13 | 1.21 |
| Simulated data | 1.68 (0.9) | 1.36 (0.6) | 1.39 (0.6) |
Columns indicate estimates resulting from (a) the method of Hellmann et al. (2008), (b) the method presented in this article, and (c) the extension of our approach to perform calculations conditional on observed rather than expected coverage. We show the mean estimate for the data of Venter et al. (2001) and the mean (and RMSE) for the simulated data.
Estimating ρ:
When estimating ρ we adapt the approximate product-likelihood approach of Hudson (2001). The major difficulty here is that we cannot determine the genotypes of individuals with certainty. Suppose two alleles exist at a given polymorphic locus. For now, we assume that bases are called correctly. Consequently, for any given individual, if we observe two types at that locus, we know that we have sequenced both alleles and that the individual is a heterozygote. However, if only one type is observed, two explanations are possible: (i) the individual is homozygous at that position and we have sequenced one or both alleles or (ii) the individual is heterozygous but we have only sequenced one allele. The probability of the second of these two situations decreases as the number of reads at that locus for that individual increases (and can be calculated directly). Thus, we propose a pragmatic approach in which data are included in the analysis if either of the following “read criteria” are satisfied: (1) we observe two different sequence types or (2) we observe one type and there are at least NT reads, for that individual at that locus. We vary the value of NT to find a good choice for a given coverage X.
In addition to the above, we make a further adjustment to the approximate product-likelihood approach of Hudson (2001) to deal with the imperfect coverage that results from read data. Hudson's method relies upon lookup tables that contain empirical estimates of the probability of observing each possible two-locus genotype configuration for a given ρ and sample size. Each lookup table gives results for a range of ρ-values and a particular sample size. In traditional applications a single lookup table is used, corresponding to the appropriate sample size n. In our context, due to (a) incomplete read coverage and (b) the fact that we discard genotypes not meeting the read criteria given earlier, we do not have full data. For example, if for the jth two-locus pair, only Nj individuals meet either of the two read criteria given above at both loci, we then use the lookup table of size nj = 2Nj to find the sample probability for this diploid configuration. Since Nj varies between locus pairs, the lookup table that we use will also vary from pair to pair.
We now present a more formal description. Let mj denote a two-locus genotype configuration obtained from full sequence data over n chromosomes. Let pn(mj; ρ) denote the probability of this configuration given ρ. The composite likelihood of the full sample is defined as
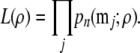 |
(10) |
In the present setting we let  be the configuration of the jth two-locus pair we observe from reads meeting the read criteria. Suppose there are Nj individuals that meet the read criteria for both loci of this locus pair. We then use the tabulated two-locus sampling distribution of size nj = 2Nj to look up the value of the likelihood of
be the configuration of the jth two-locus pair we observe from reads meeting the read criteria. Suppose there are Nj individuals that meet the read criteria for both loci of this locus pair. We then use the tabulated two-locus sampling distribution of size nj = 2Nj to look up the value of the likelihood of  given ρ, denoted by
given ρ, denoted by 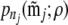 , which we then use as a surrogate for pn(mj; ρ) in the above expression. Therefore, we write the approximate product likelihood as
, which we then use as a surrogate for pn(mj; ρ) in the above expression. Therefore, we write the approximate product likelihood as
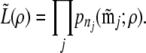 |
(11) |
Finally, we compute the approximate product likelihood 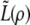 for ρ-values ranging from 0 to 120 and record the MLE as the estimate of ρ for each sample.
for ρ-values ranging from 0 to 120 and record the MLE as the estimate of ρ for each sample.
In our simulation study we generate 1000 coalescent samples of 25 diploid individuals with fixed θ but differing ρ, e.g., (100, 20), (100, 40), and (100, 60), for a 100-kb region, and report results for estimating ρ in Table 4. First, we note that the mean number of reads, nr, covering a locus increases as the coverage X increases. That is, for a threshold NT, the larger the coverage is, the larger nr becomes on average, and the more two-locus pairs are included in the approximate product-likelihood calculation, which improves the estimates. On the other hand, for large NT and relatively small X, we do not obtain estimates of ρ because there are too few two-locus pairs meeting the read criteria.
TABLE 4.
Estimating ρ for resequencing data where θ = 100 and NT = number of reads required to use homozygous genotype calls in the inference
| X | NT | ρ0 = 20 | ρ0 = 40 | ρ0 = 60 |
|---|---|---|---|---|
| 16× | 3 | 21.6 (7.4) | 42.2 (12.1) | 63.9 (16.3) |
| 5 | 21.9 (7.6) | 43.3 (12.9) | 64.2 (16.7) | |
| 7 | 21.9 (7.6) | 42.3 (12.5) | 64.2 (16.7) | |
| 8× | 3 | 21.6 (7.6) | 42.2 (12.1) | 63.9 (16.6) |
| 5 | 21.6 (7.3) | 43.3 (12.9) | 63.2 (15.9) | |
| 7 | 21.9 (7.5) | 42.3 (12.5) | 62.7 (16.3) | |
| 4× | 3 | 21.4 (7.6) | 42.7 (12.9) | 62.1 (15.8) |
| 5 | 21.2 (7.3) | 40.9 (11.4) | 61.5 (15.5) | |
| 7 | 19.8 (6.3) | 37.6 (10.8) | 54.6 (14.6) | |
| 2× | 3 | 19.8 (6.6) | 38.0 (11.1) | 55.9 (14.3) |
| 5 | 13.4 (7.7) | 22.7 (18.5) | 30.4 (30.5) | |
| 7 | NA | NA | NA |
We report mean (and RMSE) of the estimates over 1000 simulations. NA, not applicable due to too few two-locus pairs meeting the read criteria.
An important observation from Table 4 is that we tend to significantly underestimate ρ when there is low coverage and a stringent threshold for NT, e.g., twofold coverage with NT = 5. We speculate that this is caused by the increasing tendency to misinterpret an observed homozygote as an actual homozygote as coverage decreases. Clearly, if resequencing data are to be used to estimate recombination rates, adequate coverage must be used.
Robustness:
So far we have assumed that there are no sequencing errors in read data; in reality, of course, sequencing errors do exist. We focus on two types of errors in this article: traditional sequencing error and biased amplification.
We first allow for traditional base-calling sequencing errors. In Solexa data, for example, the error rate is ∼1% per base per read. Motivated by the fact that a given base will be sequenced more than once if it is contained within more than one read, we provide a simple heuristic procedure to deal with sequencing errors. For the read data at any given locus, for any given individual, we proceed as follows:
Treat the data as missing if the number of reads is less than NT.
Find the most frequent and second most frequent alleles, denoted by A and B; denote their counts by nA and nB, respectively.
Set the genotype as AA if nB = 0 or BB if nA = 0; i.e., we observe only one type at this locus.
Set the genotype as AB if nA ≥ 2 and nB ≥ 2; i.e., we require at least two observations of both alleles to call it as a heterozygote (so, if nA = 1 or nB = 1, we treat the data as missing for that individual).
The rationale for the last condition is that sequencing errors at a homozygous locus will most often result in a single instance of the erroneous call (assuming, as is realistic, that error rates are relatively low). Therefore, we exclude such occurrences from the analysis since this pattern cannot be distinguished from a situation in which one of two different alleles has been read once only. Clearly, other schemes are possible.
In following this procedure the probability of making an error in calling the genotype is a decreasing function of NT, but, conversely, the number of called genotypes will decrease as NT increases. For example, when the error rate is 1% per base per read, and when using 1× coverage with NT = 3 (i.e., we require at least three reads present to call a locus), we make calls for ∼32% of homozygotes (with 100% accuracy), while calling ∼13% of heterozygotes (with 54% accuracy). As we increase the coverage and the threshold NT, we make better calls. For example, with 4× coverage and NT = 5, we make calls for 81.2% heterozygotes, with 98.2% accuracy. The results in Table 5 show that we still successfully estimate θ, but there is some impact on performance when estimating ρ (Table 6) at low coverage levels.
TABLE 5.
Estimating θ in the presence of sequencing errors where ρ is fixed at 20 and NT = 3
| X | θ0 = 100 | θ0 = 400 |
|---|---|---|
| 16× | 99.8 (11.4) | 404.2 (59.6) |
| 8× | 99.6 (11.4) | 403.6 (60.5) |
| 4× | 104.8 (12.9) | 406.5 (61.0) |
| 2× | 103.2 (12.0) | 415.6 (64.7) |
We report the mean (and RMSE) of the estimates over 1000 simulations.
TABLE 6.
Estimating ρ in the presence of sequencing errors
| X | NT | ρ0 = 20 | ρ0 = 40 | ρ0 = 60 |
|---|---|---|---|---|
| 16× | 3 | 21.2 (6.1) | 41.6 (11.1) | 62.4 (15.3) |
| 5 | 21.2 (6.2) | 41.8 (11.0) | 62.9 (15.8) | |
| 7 | 21.1 (6.2) | 41.9 (11.0) | 62.9 (15.8) | |
| 8× | 3 | 21.4 (6.6) | 41.2 (10.2) | 61.5 (14.1) |
| 5 | 19.9 (6.3) | 39.3 (11.2) | 58.9 (14.9) | |
| 7 | 20.3 (6.7) | 39.6 (11.4) | 57.7 (14.2) | |
| 4× | 3 | 20.7 (6.9) | 41.9 (12.0) | 60.6 (15.7) |
| 5 | 19.9 (6.6) | 39.5 (11.3) | 58.2 (14.6) | |
| 7 | 18.8 (5.9) | 35.0 (11.2) | 51.4 (15.2) | |
| 2× | 3 | 19.0 (6.1) | 37.0 (10.6) | 53.6 (15.4) |
| 5 | 12.5 (8.4) | 22.8 (19.9) | 27.6 (23.2) | |
| 7 | NA | NA | NA |
Here, θ = 100 and NT = number of reads required to use homozygous genotype calls in the inference. NA, not applicable, due to there being too few two-locus pairs for which calls are made. We report mean (and RMSE) of the estimates over 1000 simulations.
We also note that the method for dealing with sequencing errors inherent in the method of Hellmann et al. (2008) is not applicable in this scenario. In that article it was assumed that each sequencing error leads to a unique new segregating site or SNP. Their method works in the context of the data of Venter et al. (2001) because of the extremely small error rate of ∼0.003% per base per read, but fails to produce accurate estimates in the present context (results not shown).
Note that, for convenience, we assume that sequencing errors are distributed uniformly along a read. In fact, error rates typically increase toward the growing end of each read and may be influenced by composition of neighboring bases (e.g., in homopolymer tracts), but we expect this to have little impact on the results presented here.
We now consider a second type of error: biased amplification. Here there are different success rates in sequencing, depending on the actual type being sequenced. Clearly a range of scenarios is possible, but here we illustrate how to adapt our approach in a situation in which we are able sequence the major allele with 100% success but fail to sequence a percentage of minor alleles at each locus. We now recalculate the probability that a segregating site is detected in this context. We let pe denote the probability that a minor allele is not sequenced and then recalculate q2(b) and q3 as follows:
- (
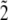 ): The probability q2e(b) that a segregating site with b mutants in a sample of size n is detected by reads is
): The probability q2e(b) that a segregating site with b mutants in a sample of size n is detected by reads is
for 1 < b < n, where q1 is defined as before.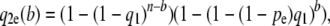
(12) - (
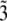 ): Under a constant population size coalescent model, the probability q3e that a segregating site is detected by reads is
): Under a constant population size coalescent model, the probability q3e that a segregating site is detected by reads is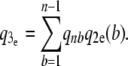
(13)
The estimate of θ is given by 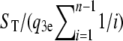 , where ST is the number of detected segregating sites. We simulate 1000 data sets using the coalescent model to assess performance of this estimator. Results for estimating θ are listed in Table 7, where we see that we remain able to infer θ from the adjusted formulas. There is no straightforward way to adapt the procedure for estimating ρ in the presence of biased amplification. However, in Table 8 we show results obtained by applying the method we used for “traditional” sequencing errors earlier in this article to this new context. We see the same trends as in Table 4; i.e., if coverage is low we tend to underestimate ρ, but as coverage increases, the estimator begins to perform well. Note that, with onefold coverage, performance is relatively poor when estimating θ or ρ since relatively few data meet the threshold NT (results not shown).
, where ST is the number of detected segregating sites. We simulate 1000 data sets using the coalescent model to assess performance of this estimator. Results for estimating θ are listed in Table 7, where we see that we remain able to infer θ from the adjusted formulas. There is no straightforward way to adapt the procedure for estimating ρ in the presence of biased amplification. However, in Table 8 we show results obtained by applying the method we used for “traditional” sequencing errors earlier in this article to this new context. We see the same trends as in Table 4; i.e., if coverage is low we tend to underestimate ρ, but as coverage increases, the estimator begins to perform well. Note that, with onefold coverage, performance is relatively poor when estimating θ or ρ since relatively few data meet the threshold NT (results not shown).
TABLE 7.
Estimating θ in the presence of biased amplification
| X | pe = 0.0 | pe = 0.01 | pe = 0.05 | pe = 0.1 | pe = 0.15 |
|---|---|---|---|---|---|
| 16× | 100.1 (14.4) | 100.0 (14.7) | 101.8 (15.2) | 102.4 (15.1) | 102.6 (15.6) |
| 8× | 100.1 (14.4) | 100.1 (15.4) | 101.9 (15.5) | 102.3 (15.4) | 103.0 (15.9) |
| 4× | 100.2 (14.9) | 101.5 (15.6) | 102.8 (16.5) | 100.6 (15.8) | 103.7 (15.9) |
| 2× | 99.6 (15.7) | 103.7 (16.3) | 105.1 (16.7) | 105.6 (16.5) | 107.3 (18.7) |
Here we set θ0 (the generating value of θ) = 100 and use ρ0 = 20. pe is the probability that a minor allele is not sequenced. We report the mean (and RMSE) of the estimates over 1000 simulations.
TABLE 8.
Estimating ρ in the presence of biased amplification
| X | NT | pe = 0.01 | pe = 0.05 | pe = 0.1 | pe = 0.15 |
|---|---|---|---|---|---|
| 16× | 3 | 21.7 (7.8) | 21.9 (7.8) | 21.8 (7.6) | 21.7 (7.5) |
| 5 | 21.9 (7.6) | 21.9 (7.7) | 21.7 (7.5) | 21.6 (7.5) | |
| 8× | 3 | 21.8 (7.6) | 22.4 (8.2) | 20.9 (7.6) | 21.2 (7.3) |
| 5 | 21.7 (7.7) | 22.1 (7.6) | 22.1 (7.8) | 21.1 (7.2) | |
| 4× | 3 | 21.5 (7.7) | 21.6 (7.5) | 22.0 (7.8) | 21.1 (7.7) |
| 5 | 21.0 (7.4) | 20.5 (7.1) | 21.0 (7.5) | 20.5 (7.2) | |
| 2× | 3 | 19.5 (6.7) | 20.1 (6.9) | 19.6 (6.6) | 19.5 (6.5) |
| 5 | 13.4 (7.8) | 12.7 (8.22) | 12.5 (8.28) | 12.1 (8.8) |
Here we set the generating values, θ0 = 100 and ρ0 = 20. NT is the number of reads required to use homozygous genotype calls in the inference and pe is the probability that a minor allele is not sequenced. We report mean (and RMSE) of the estimates over 1000 simulations.
Inference with no reference genome:
Thus far we have assumed that there is a reference genome to which all reads can be uniquely mapped. However, reference genomes are not always available, e.g., in bacterial genome resequencing projects. The common fragment assembly programs such as Phrap (Ewing and Green 1998) work well on 500- to 700-bp reads produced by Sanger sequencing, but their overlap-layout-consensus approach becomes more difficult when applied to shorter reads produced by next-generation sequencing techniques. Chaisson et al. (2004) demonstrated that while it is feasible to assemble short reads (of length 80–200 bp), it requires significantly higher coverage (≥30×).
Addressing the lack of a reference genome in diploid data is a difficult problem, and it is not clear how one should proceed. As a first step in that direction, we demonstrate a simple but effective algorithm that is usable in the context where individuals are haploid:
Assume R reads (length l bp) are obtained for each of the haploid individuals.
Assume that we can merge adjacent reads into a larger fragment if they share at least k bases. Do this for each individual and call the resulting collections of reads “islands.”
Examine the set of islands across all individuals and create “superislands” by joining islands that overlap by at least k bases.
This algorithm results in a set of superislands. The relative ordering of the superislands is unknown. We now present results for inferring the mutation and recombination parameters on the basis of the superislands. We show results for a context in which there are no sequencing errors in the reads. (Sequencing errors can be dealt with using the heuristic procedures employed earlier.) We explore a range of different values for k.
In Table 9 we list the estimates of θ and ρ, respectively, from simulated data. For each combination of generating values (θ, ρ), we simulate 1000 coalescent samples of 50 haploid individuals over a 100-kb region and generate reads according to a Poisson process, as before, with intensity specified by the coverage X for each chromosome. We then construct superislands. For both analyses we consider only the SNPs detected by reads in the superislands. When estimating ρ, we include only the covered pairs for which both loci fall within the same superisland (since only for such pairs is the distance between them known). We then use composite likelihood as before and report the maximum-likelihood estimate (MLE). We present mean and RMSE of the estimates from the 1000 samples. Results for θ are generated from data simulated with θ0 = 100 and ρ0 = 20. Results for ρ use data simulated with θ0 = 20 and ρ0 = 20, 40 and 60 (per column headings).
TABLE 9.
Estimating θ and ρ in the absence of a reference genome
| X | l | k | θ0 = 100 | ρ0 = 20 | ρ0 = 40 | ρ0 = 60 |
|---|---|---|---|---|---|---|
| 1× | 30 | 20 | 100.5 (16.7) | 21.3 (7.5) | 42.2 (11.4) | 63.2 (15.7) |
| 1× | 50 | 40 | 100.3 (16.3) | 21.5 (7.7) | 42.2 (13.0) | 61.8 (15.1) |
| 0.5× | 30 | 20 | 99.3 (18.0) | 22.2 (11.1) | 44.2 (16.1) | 62.8 (19.2) |
| 0.5× | 50 | 40 | 101.2 (18.2) | 32.8 (34.9) | 47.7 (34.3) | 61.5 (30.9) |
We report the mean (and RMSE) of the estimates over 1000 simulations.
As we can see from Table 9, in general we obtain good results for estimating both θ and ρ. When estimating ρ we lose some efficiency when coverage is low and large overlap is required for alignment (last row of Table 9). This is because, in such a case, the number of superislands becomes very large, with each being of relatively short length. For example, for data corresponding to the last row of Table 9, there are an average of ∼250 superislands with a mean length ∼440 bp in the 100-kb region for each data set (see Figure 3). Consequently there are far fewer SNPs within each superisland, the number of covered pairs becomes much lower, and those pairs will be relatively close together. This increases the variance of our estimates of ρ, and the asymmetry of those errors at low ρ-values leads to an apparent upward bias in the final MLE.
Figure 3.—

Histograms of the number of SNPs detected within superislands for (a) X = 0.5× and l = 50 and (b) X = 0.5× and l = 30.
Note that the total length of the combined superislands, 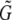 , will often be longer than the length of the region from which they were generated, G. This is because if two superislands (A and B, say) overlap by fewer base pairs than are required to align them, they are not combined to form a single superisland. Thus the region of overlap between the two is considered twice in the analysis: once for superisland A and once for superisland B. For example, with X = 0.5, l = 50, and k = 40, the total length of superislands is, on average
, will often be longer than the length of the region from which they were generated, G. This is because if two superislands (A and B, say) overlap by fewer base pairs than are required to align them, they are not combined to form a single superisland. Thus the region of overlap between the two is considered twice in the analysis: once for superisland A and once for superisland B. For example, with X = 0.5, l = 50, and k = 40, the total length of superislands is, on average 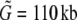 . Because of this, the estimate we obtain for θ is what would be appropriate for a region of length
. Because of this, the estimate we obtain for θ is what would be appropriate for a region of length 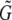 . Consequently, we report a “corrected” θ-estimate that multiplies the original estimate by a factor of
. Consequently, we report a “corrected” θ-estimate that multiplies the original estimate by a factor of 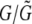 .
.
DISCUSSION
Our model and analysis show how read data from the Solexa (and similar) systems can be used to estimate mutation and recombination parameters. Inference accuracy increases as coverage increases, but 2× coverage provides good estimates for inferring mutation rate, while 4× coverage is sufficient for recombination rate. One of the primary advantages of sequencing by synthesis is that it is (in principle) fast and economic, and it is therefore relatively easy to increase coverage to whatever level is necessary to attain the desired accuracy.
In this article we have presented results for the Solexa system (with an assumed read length of 36 bp). However, our methods generalize naturally to longer read lengths and to the 454 and polony methods, since our theoretical results depend only on the expected coverage X. With respect to 454 technology, we simply have to change the read length to be 200 bp, say, rather than the 36 bp used here.
For polony data, the situation is slightly less clear since reads now occur in pairs (one read at each end of each fragment). It has been experimentally determined that the distance between each pair of reads is reasonably modeled by a Gaussian distribution with a mean of ∼950 bp and standard deviation of ∼97 bp (Shendure et al. 2005b). The theoretical results we have presented here still apply in this case.
It is also of interest to explore our ability to estimate decay of linkage disequilibrium (LD) using resequencing data. Recall that, for each pair of SNPs, the number of haplotypes that are covered (i.e., have data) at both SNPs will be less than or equal to the total number of haplotypes. In particular, the number of covered SNP pairs will vary from pair to pair. This leads to an interesting phenomenon, which we illustrate in the context of haploid data. In Figure 4a, we plot the decay of LD, measured in terms of r2, for differing levels of coverage. The r2 estimates are based on averages over 1000 simulations. We see that the estimated r2 decreases as distance increases, as it should, but also increases as coverage decreases. This is a simple consequence of the known tendency of r2 to be overestimated for small sample sizes (Terwilliger and Hiekkalinna 2006). For reference, Figure 4a also shows the decay of r2 when calculated from the full SNP data using samples of 30 and 50 chromosomes. One way of avoiding the observed bias is to condition on having a reasonably large number of covered chromosomes for each pair of loci. As an example of this, in Figure 4b we show results in which we include only pairs of loci for which have at least 30 covered chromosomes. We see that the bias now largely disappears. The results for X = 0.25× are not shown because there are no two-locus pairs that meet the conditioning criteria in this case.
Figure 4.—

LD decay of r2 based on SNPs observed in a sample of 50 chromosomes, presented as a function of coverage in a 100-kb region. (a) Plot of the r2 estimates obtained from all reads; (b) plot of those estimates when we condition on the number of covered chromosomes being at least 30. “SNP (50)” presents results when using full SNP data and “SNP (30)” is the same as SNP (50) but using only a subset of 30 chromosomes from the sample.
Throughout this article we have assumed that reads are randomly distributed and that read locations are independent across chromosomes. Since fragments are created by physical shearing processes, this assumption appears to be a reasonable approximation. However, there is evidence suggesting that in short-read resequencing data the distribution of the number of reads covering a base pair depends on the location of that base pair. The effects of this lack of homogeneity, thought to be due to underlying variation in GC content across the genome, can be modeled in a straightforward way using nonhomogeneous Poisson processes, in which the rate of the Poisson process at a given location will reflect the local GC content, for example. We also note that since isochores, regions of relatively constant GC content, are ∼300 kb long (Constantini et al. 2006), then if we are considering regions of the order of 100 kb (say), as in the examples in this article, an assumption of homogeneity is more reasonable. It is also likely that the locations of reads are correlated across fragments in the sequencing library. We have shown in other work (Jiang et al. 2006) that procedures such as those used here continue to work in the context of single-feature polymorphism (SFP) data, in which the positions of SFPs are also highly correlated across fragmentations of a given chromosome (theoretical results will change, of course). One might also consider Poisson processes of different rates for different regions or allow the probability that a read is sequenced to depend on the local GC content.
Our method for estimating recombination rate is admittedly somewhat ad hoc. This approach represents an attempt to finesse the principal difficulty in using resequencing data to estimate recombination rates: the fact that while, under reasonable assumptions, reads can be mapped to a genomic location within an individual, it is, at least under current incarnations of the technologies, not known which copy of the chromosome has been read. Our method relies upon use of a threshold NT, and we show performance under a range of values for NT. It is worth noting that in other scenarios, such as varying recombination rates, say, performance as a function of NT may vary. However, we note that, while we demonstrated results for estimating recombination rate using the composite-likelihood estimator of Hudson in this article, other methods, for example LDHat (McVean et al. 2004), might also be usefully adapted to the present context by using a similar threshold scheme and might improve robustness to variation in recombination rates.
In summary, resequencing data provide an exciting, economically efficient way of generating large quantities of sequence data. Such data will typically result in a level of coverage that varies from locus to locus. In this article we have shown that this complication can be dealt with in a reasonably simple way, allowing for successful estimation of evolutionary parameters from such data.
Acknowledgments
We gratefully acknowledge the criticism of the reviewers, which resulted in a considerably improved article. We are also grateful to Ines Hellmann and Rasmus Nielsen for help in providing a preformatted version of the data of Venter et al. (2001) and with running the analysis program used to generate the results in Hellmann et al. (2008). This work was funded in part by National Institutes of Health grants HG002790, GM67243, HL084706, GM069890, and MH084678. S.T. is a Royal Society–Wolfson Research Merit Award holder.
APPENDIX: DERIVATIONS
We now derive the results of Equations 1–4. First consider the probability that a random point in the genome is covered by reads or the expected fraction of the genome is covered by reads. We assume the reads occur on the real line (−∞, ∞) according to a Poisson process with rate λ. The region of interest corresponds to the interval (0, G) in base pairs. The probability that a point x is not covered by reads of length l is the probability that no events occur in the interval (x − l, x), which is e−λl. The expected number of reads in (0, G) is Gλ, covering an average of Gλl bp. The coverage per base is therefore X = Gλl/G = λl. To achieve a coverage of X we choose λ = X/l. The probability that a base is not covered is therefore
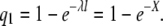 |
With imperfect coverage of the region, we do not have full sequence information for the entire chromosome. However, we can still detect segregating sites with sequenced reads, unless either the site is entirely unsequenced or we see only ancestral (or mutant) alleles at that segregating site. Suppose there are b copies of the mutant allele at a segregating site of interest. The probability that this segregating site is detected is the product of the probability of reading at least one of the b mutant and at least one of the (n − b) ancestral alleles. This gives formula (2). To find the probability that a segregating site is detected by reads without conditioning on b, we average over the probability qnb of having b mutants in a sample of n chromosomes given in Griffiths and Tavaré (1998) for coalescent samples with constant population size.
To derive the expected number of segregating sites detected by reads, we use indicator functions I such that Ii = 1 if the ith segregating site is detected by reads; otherwise Ii = 0. Note that 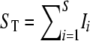 , where S is the total number of segregating sites in the sample. Conditioning on S = s, we have
, where S is the total number of segregating sites in the sample. Conditioning on S = s, we have
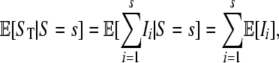 |
since Ii is independent of S. The distribution of the Bernoulli random variable, Ii, is determined by 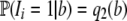 , where b is the number of mutants at the ith segregating site. Hence
, where b is the number of mutants at the ith segregating site. Hence
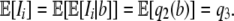 |
Thus we have
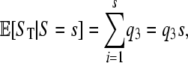 |
so that 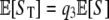 .
.
References
- Bentley, D. R., 2006. Whole-genome re-sequencing. Curr. Opin. Genet. Dev. 16 545–552. [DOI] [PubMed] [Google Scholar]
- Brockman, W., P. Alvarez, S. Young, M. Garber, G. Giannoukos et al., 2008. Quality scores and SNP detection in sequencing-by-synthesis systems. Genome Res. 18 763–770. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chaisson, M., P. Pevzner and H. Tang, 2004. Fragment assembly with short reads. Bioinformatics 20 2067–2074. [DOI] [PubMed] [Google Scholar]
- Constantini, M., O. Clay, F. Auletta and G. Bernadi, 2006. An isochore map of human chromosomes. Genome Res. 16 536–541. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ewing, B., and P. Green, 1998. Base-calling of automated sequencer traces using Phred. II. Error probabilities. Genome Res. 8 186–194. [PubMed] [Google Scholar]
- Griffiths, R. C., and S. Tavaré, 1998. The age of a mutation in a general coalescent tree. Stoch. Models 14 273–295. [Google Scholar]
- Halushka, M. K., J. B. Fan, K. Bentley, L. Hsie, N. Shen et al., 1999. Patterns of single-nucleotide polymorphisms in candidate genes for blood-pressure homeostasis. Nat. Genet. 22 239–247. [DOI] [PubMed] [Google Scholar]
- Hellmann, I., Y. Mang, Z. Gu, P. Li, F. M. De La Vega et al., 2008. Population genetic analysis of shotgun assemblies of genomic sequence from multiple individuals. Genome Res. 18 1020–1029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hillier, L., G. T. Marth, A. R. Quinlan, D. Dooling, G. Fewell et al., 2008. Whole-genome sequencing and variant discovery in C. elegans. Nat. Methods 5 183–188. [DOI] [PubMed] [Google Scholar]
- Hudson, R. R., 2001. Two-locus sampling distributions and their application. Genetics 159 1805–1817. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hudson, R. R., and N. L. Kaplan, 1985. Statistical properties of the number of recombination events in the history of a sample of DNA sequences. Genetics 111 147–164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jarvie, T., 2005. Next generation sequencing technologies. Drug discovery today. Technologies 2 255–260. [DOI] [PubMed] [Google Scholar]
- Jiang, R., P. Marjoram, J. O. Borevitz and S. Tavaré, 2006. Inferring population parameters from single feature polymorphism data. Genetics 173 2257–2267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johnson, P. L. F., and M. Slatkin, 2006. Inference of population genetic parameters in metagenomics: a clean look at messy data. Genome Res. 16 1320–1327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Korbel, J. O., A. E. Urban, J. P. Affourtit, B. Godwin, F. Grubert et al., 2007. Paired-end mapping reveals extensive structural variation in the human genome. Science 318 420–426. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Margulies, M., M. Egholm, W. E. Altman, S. Attiya, J. S. Bader et al. 2005. Genome sequencing in microfabricated high-density picolitre reactors. Nature 437 376–380. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McVean, G. A., S. R. Myers, S. Hunt, P. Deloukas, D. R. Bentley et al., 2004. The fine-scale structure of recomination rate variation in the human genome. Science 304 581–584. [DOI] [PubMed] [Google Scholar]
- Shendure, J., R. D. Mitra, C. Varma and G. M. Church, 2005. a Advanced sequencing technologies: methods and goals. Nat. Rev. Genet. 5 335–344. [DOI] [PubMed] [Google Scholar]
- Shendure, J., G. J. Porreca, N. B. Reppas, X. Lin, J. P. McCutcheon et al., 2005. b Accurate multiplex polony sequencing of an evolved bacterial genome. Science 309 1728–1732. [DOI] [PubMed] [Google Scholar]
- Terwilliger, J. D., and T. Hiekkalinna, 2006. An utter refutation of the ‘Fundamental Theorem of the HapMap’. Eur. J. Hum. Genet. 14 426–437. [DOI] [PubMed] [Google Scholar]
- Venter, J. C., M. D. Adams, E. W. Myers, P. W. Li, R. J. Mural et al., 2001. The sequence of the human genome. Science 291 1304–1351. [DOI] [PubMed] [Google Scholar]
- Watterson, G. A., 1975. On the number of segregating sites in genetical models without recombination. Theor. Popul. Biol. 7 256–276. [DOI] [PubMed] [Google Scholar]