

Abstract
Historically, duplicate genes have been regarded as a major source of novel genetic material. However, recent work suggests that chimeric genes formed through the fusion of pieces of different genes may also contribute to the evolution of novel functions. To compare the contribution of chimeric and duplicate genes to genome evolution, we measured their prevalence and persistence within Drosophila melanogaster. We find that ∼80.4 duplicates form per million years, but most are rapidly eliminated from the genome, leaving only 4.1% to be preserved by natural selection. Chimeras form at a comparatively modest rate of ∼11.4 per million years but follow a similar pattern of decay, with ultimately only 1.4% of chimeras preserved. We propose two mechanisms of chimeric gene formation, which rely entirely on local, DNA-based mutations to explain the structure and placement of the youngest chimeric genes observed. One involves imprecise excision of an unpaired duplication during large-loop mismatch repair, while the other invokes a process akin to replication slippage to form a chimeric gene in a single event. Our results paint a dynamic picture of both chimeras and duplicate genes within the genome and suggest that chimeric genes contribute substantially to genomic novelty.
IDENTIFYING the genetic origins of novel traits is a problem that lies at the heart of evolutionary theory. All biological diversity must ultimately have a source. However, the relative importance of different mutational sources is far from certain. Different types of mutations may have very different phenotypic consequences and may act on different timescales. It is possible that simple sequence change may be the predominant form of mutation, while simultaneously providing little in the way of biological novelty. More complex mutations may actually provide a richer substrate upon which selection can act. One such complex mutation is the rare event that fuses pieces of gene sequences to create a chimeric gene. Such chimeric genes may be more likely than other mutations to serve as an important source of novel genetic material.
Duplicate genes have long been regarded as a fundamental source of genetic novelty (Ohno 1970; Lynch and Conery 2000). This view implicitly assumes that gene functions are to some extent mutually exclusive in that the same gene cannot perform multiple functions simultaneously. Through duplication, one copy can maintain the ancestral function, leaving the other copy free to develop a new function. This process of duplication and preservation by natural selection is called “neofunctionalization” (Lynch et al. 2001). However, duplicates can also be preserved through subfunctionalization, acquiring tissue-specific or stage-specific activity without the evolution of novel function (Force et al. 1999; Lynch and Force 2000). The relative probabilities of neofunctionalization and subfunctionalization remain unclear. However, growing evidence suggests subfunctionalization is common (Van Hoof 2005).
The development of novel functions may often require the formation of novel protein conformations. However, vast mutational distances often separate alternative protein structures from one another (Bogarad and Deem 1999; Cui et al. 2002). In such cases, duplicate gene evolution via point mutations may have difficulty acquiring novel structures, because a gene that has accumulated only a portion of the necessary mutations to reach a novel folding pattern is likely to misfold completely (Bogarad and Deem 1999; Cui et al. 2002). Without the constraint of selection, a misfolded duplicate is likely to decay into a pseudogene before acquiring a novel functional conformation. When a chimeric gene is formed, pieces of functional genes contribute to the formation of a new protein that is immediately different from either of its parental genes. These gene pieces may be more likely than random genetic material to fold correctly into appropriate three-dimensional structures. The resultant chimeric genes may contain novel combinations of folding domains that point mutations have difficulty reaching. Hence, chimera formation may create new genes that have reasonably stable structures while at the same time effecting large jumps through the mutational landscape. Indeed, results from theoretical simulations and in vitro mutagenesis experiments confirm that shuffling sequence fragments, especially unrelated fragments, is frequently more successful at attaining new structures than evolution by point mutation alone (Giver and Arnold 1998; Cui et al. 2002). Furthermore, results from in vitro gene splicing indicate that rearrangement of even highly divergent sequences often results in stable chimeric forms (Voigt et al. 2002).
Early results indicate that chimeric genes may be a promising source of novel proteins. The well-characterized chimeric gene jingwei is a copy of the Adh gene with new 5′ exons that confer preference for novel substrates (Long and Langley 1993; Long et al. 1999; Wang et al. 2000; Zhang et al. 2004). Analysis of evolutionary rates in three Adh-derived chimeric genes in various Drosophila species reveals elevated rates of replacement substitutions after chimera formation that are consistent with positive selection driving amino acid replacements in young chimeric genes (Jones and Begun 2005).
Still, even with these encouraging results, very little is known about the general behavior of chimeric genes. Previous work on duplicate genes has revealed that duplicates form and decay rapidly in the genomes of several taxa (Lynch and Conery 2000, 2003; Hahn et al. 2005, 2007; Demuth et al. 2006). However, none of these studies has estimated the likelihood of preservation through the forces of subfunctionalization or neofunctionalization. In light of these deficiencies, we undertook a genomewide investigation of chimeric genes and duplicate genes in D. melanogaster. We estimate and compare independent rates of formation, decay, and preservation in recently arisen chimeric and duplicate genes. We find that chimeric genes are formed in appreciable numbers and often persist long enough to provide a potential source of novelty in Drosophila melanogaster. We also propose two possible molecular mechanisms of chimeric gene formation that are entirely dependent on local mutational events.
METHODS
Methods for chimera identification:
We performed an all-by-all BLASTn comparison (Altschul et al. 1990), considering only nonself matches with E < 10−10 for the D. melanogaster r.5.2-all-CDS data set obtained from FlyBase (accessed August 2007; ftp://ftp.flybase.net/releases/) (Adams et al. 2000). Chimeric genes were identified using the following criteria. The two most significant matches identify putative “parental genes.” One parental gene provides the exons that contribute to the 5′ end of the candidate chimera and the second parental gene contributes to the remainder of the candidate chimera. The two parental genes must hit regions of the chimera that do not overlap by >15 bp, and the chimeric gene must be the best hit for each parent. We removed genes that physically overlapped with their two parental genes, and we excluded heterochromatic sequences where assembly and annotations are still in their initial phase. Prior to further analysis, we confirmed the existence of each chimeric gene with PCR amplification from D. melanogaster reference strain y1 cn1 bw1 sp1 genomic DNA. These qualifications produced a final list of 14 putative chimeric genes (Table 1). The genomic sequence for each chimeric and parental gene was obtained from FlyBase and aligned using a blast2seq (Tatusova and Madden 1999) to determine the breakpoints of chimera formation (Figure 1, detailed alignments available as supplemental information). Genomic sequences of parental genes were also aligned to one another, although no significant similarity was found.
TABLE 1.
Identity and divergence of chimeric genes
| Chimeric gene | Parental gene A | Parental gene B | dN | dS | t | Location | Intron gain/loss |
|---|---|---|---|---|---|---|---|
| CG31904-PD | CG13796-PA | CG7216-PA (acp1) | 0.000 | 0.000 | 0.003 | T | 0 |
| CG18853-PA | CG12822-PA | CG11205-PA (phr) | 0.000 | 0.000 | 0.004 | NT | 0 |
| CG32318-PA | CG9191-PA (Klp61F) | CG9187-PA (psf1) | 0.000 | 0.000 | 0.008 | T | 0 |
| CG31864-PA | CG12264-PA | CG5202-PA (escl) | 0.000 | 0.000 | 0.010 | NT | 0 |
| CG12592-PA | CG18545-PA | CG12819-PA (sle) | 0.007 | 0.017 | 0.017 | T | −1 |
| CG31687-PA | CG2508-PA (cdc23) | CG31688-PA | 0.015 | 0.023 | 0.025 | NT | 0 |
| CG18217-PA | CG17286-PA (spd2) | CG4098-PA | 0.011 | 0.025 | 0.027 | NT | 1 |
| CG30457-PA | CG10953-PA | CG13705-PA | 0.000 | 0.093 | 0.125 | 0 | |
| CG17196-PA | CG17197-PA | CG17195-PA | 0.113 | 0.412 | 0.414 | T | 0 |
| CG11961-PA | CG9416-PA | CG30049-PA | 0.052 | 0.501 | 0.501 | 0 | |
| CG3978-PA (pnr) | CG9656-PA (grn) | CG10278-PA (GATAe) | 0.053 | 0.579 | 0.576 | −1 | |
| CG6844-PA (nAcRα-96Ab) | CG5610-PA (nAcRα-96Aa) | CG11348-PA (nAcRβ-64B) | 0.107 | 0.733 | 0.727 | 0 | |
| CG6653-PA (Ugt86De) | CG31002-PA (Gga) | CG17200-PA (Ugt86Dg) | 0.129 | 0.745 | 0.743 | 0 | |
| CG31668-PB | CG33124-PB | CG8451-PA | 0.054 | 0.513 | 0.513 | 0 |
T, parental genes located in tandem with chimera; NT, parental genes located within two genes of chimera.
Figure 1.—

Placement and structure of chimeric genes. (A) The seven youngest chimeric genes were all found in close proximity with their parental genes. The parental gene that contributed the 5′ end is found downstream, and the parental gene that contributed to the 3′ end is upstream. (B) Genomic sequences were aligned to find the breakpoints of chimera formation. Breakpoints fall in exons for six of the seven youngest chimeric genes. Portions of the chimera that align to parental gene A are highlighted in yellow, and portions aligning to parental gene B are highlighted in blue. Portions of parental genes that do not align to the chimera are shown in gray. Structures are adapted from FlyBase (http://flybase.org). Gene structure was largely conserved with one intron loss in CG12592 and one intron gain in CG18217.
Duplicate gene identification:
We identified duplicate genes using similar methodology. In an all-by-all BLASTn comparison (Altschul et al. 1990) at E < 10−10 with self-hits removed, duplicate genes were taken as reciprocal best-hit pairs. Our list of duplicate genes excludes all known chimeric genes as well as all heterochromatic sequences. A large multigene family that is under diversifying selection is likely to operate under different dynamics from duplicate gene pairs. In the tradition of previous research (e.g., Nadeau and Sankoff 1997; Lynch and Conery 2000; Moore and Purugganan 2003), we removed genes with significant BLAST hits to more than five genes. These qualifications produced a final list of 584 putative pairs of duplicate genes. Additionally, we repeated our analysis on members of large gene families. Associated parameter estimates can be found in supplemental Table 1.
Chimeric gene phylogeny:
To identify orthologous relationships, we performed a reciprocal best-hit BLASTn search at E < 10−10 for each chimeric gene against GLEANR consensus annotations for the D. simulans, D. sechellia, D. yakuba, D. erecta, D. ananassae, and D. pseudoobscura genomes obtained from the AAA wiki website (accessed January 2008; http://rana.lbl.gov/drosophila/wiki/index.php) (Drosophila 12 Genomes Consortium et al. 2007). We further required that chimeric gene ortholog alignments span the boundary of chimera formation to ascertain that each putative ortholog was indeed a chimera and not merely related to a single parental gene.
Estimating time t since formation:
The age of a duplicate or chimeric gene is not directly observable. However, the time since formation t should be largely reflected in the mutational distance dS. We used BLAST coordinates to match regions of each chimera to parental gene sequences. For genes with more than one chimeric transcript, we selected the one that had the most extensive BLAST coverage. We aligned amino acid sequences for each chimera with each parental gene using ClustalW v1.8 (Thompson et al. 1994) and then back translated to produce nucleotide alignments that preserved the reading frame. Frameshift mutations in CG31864 and CG31904 were removed for the purposes of the alignment. We then concatenated the aligned segments and used the CODEML package of PAML v3.15 (Yang 1997) to estimate dN and dS for each chimera. We assumed no across-site rate variation (α-parameter set = ∞), estimated transition–transversion bias from each gene (estimated κ), and calculated equilibrium codon frequencies on the basis of overall nucleotide frequencies (F1 × 4). We generated in-frame alignments for duplicate genes and estimated dN and dS as described above. Because of the difficulties in estimating dN and dS when divergence is large, we restricted our analysis to those chimeras and duplicates with dS < 1, leaving 14 chimeras and 213 duplicate genes.
Estimates of dN and dS from PAML represent maximum-likelihood (ML) point estimates. The accuracy of these estimates is affected by the number of sites examined such that the variance of dS is greater in shorter sequences. We used a Bayesian framework to correct maximum-likelihood estimates of dS for the effects of sequence length. We estimate time t as the mean of the posterior distribution of t. The probability of observing dS with time t and S synonymous sites is binomially distributed according to
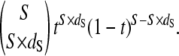 |
If t has a noninformative prior uniformly distributed from 0 to 1, then the Bayesian posterior density of t is
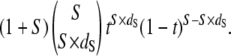 |
This gives the mean posterior estimate of t as
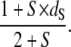 |
As expected, the limit of this estimate as S grows large is equal to dS.
Maximum-likelihood estimation of duplicate and chimera dynamics:
We model the distribution of duplicate and chimera ages according to a birth–death–preservation process in which new genes form at a constant rate λ and after formation are subject to one of two mutually exclusive fates, either loss at rate μ or preservation at rate ν. After formation, a gene will be lost with probability μ/(μ+ν) and preserved with probability ν/(μ+ν). This process gives the following function for describing the number of genes expected with a particular age t:
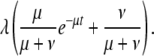 |
Assuming μ ≫ 1, the total number of genes expected with t between 0 and 1 is
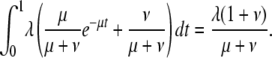 |
Combining these equations gives the probability density function of t:
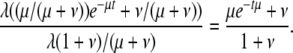 |
Here we see that the age distribution of genes depends only on μ and ν, while the total count of genes depends on λ, μ, and ν. We used numerical optimization to find the values of μ and ν that maximize the likelihood of observing the t distribution of duplicate genes and chimeric genes. We then used the estimated values of μ and ν to find the ML estimate of λ on the basis of the total number of genes present with t between 0 and 1. Additionally, we estimated the 95% confidence intervals of our ML point estimates, using bootstrap replicates obtained by sampling with replacement from the observed distribution of t values. This approach assumes that formation events occur independently of one another. The mean estimates are robust to this assumption. However, if duplicates form in clusters due to segmental duplication events, then the process will have greater variance and our confidence intervals will underestimate the underlying extent of variation.
Repetitive elements:
Each chimeric and duplicate gene used to fit this model was checked for potential similarity to transposable elements, using Repeat Masker 3.2.6 (http://www.repeatmasker.org) against the RepBase Update database (accessed Oct 2008; http://www.girinst.org) (Jurka 2000; Jurka et al. 2005; Kapitonov and Jurka 2008). None of our 213 duplicates or 14 chimeric genes with t < 1.0 had any similarity to transposons.
RESULTS
We identified 14 putative chimeric genes in D. melanogaster whose origin is recent enough that we can be reasonably certain of their evolutionary history; i.e., t < 1.0 (Table 1). In contrast, we found 213 putative duplicate genes that show t < 1.0. Here, we measure time since formation t in terms of the evolutionary distance separating duplicate pairs and chimeric genes from their progenitor genes. In this case, t is measured in the same units as dS, substitutions per synonymous site, but reflects a more comprehensive Bayesian estimate (see methods). Our definitions of chimeric genes are exceptionally stringent, requiring that coding sequences of two parental genes contribute to the coding sequence of chimeric genes. There are almost certainly other types of chimeric genes in the D. melanogaster genome. However, we restrict our analysis to a subset of genes whose chimeric origins are most clearly supported.
Placement and structure of chimeric genes:
Seven chimeric genes in D. melanogaster are nearly identical to their parental genes (t < 0.05), suggesting a very recent evolutionary origin. All are found with no more than two annotated genes between the chimera and each parental gene, and the intervening genes appear to be duplicates of one another (Table 1). All seven of these chimeras lie in the center of the construct and the parental gene contributing to the 5′ end is found downstream of the chimera (Figure 1A). Gene structure was largely conserved with one intron loss and one intron gain occurring in separate genes (Figure 1B, Table 1). Six of the seven breakpoints occurred within exons, suggesting that breakpoints of chimera formation are not biased toward introns. Coding sequences and genomic sequences of parental gene pairs do not hit one another in a BLASTn search at E < 10−10, suggesting that chimeric genes form from structurally distinct proteins.
There are two possible molecular mechanisms that could produce chimeric genes with the observed structure. The first involves short, segmental duplications with a subsequent deletion during large loop mismatch repair, resulting in the removal of the stop codon of one gene and the initial exons of the other (Figure 2). An alternative mechanism involves a process akin to replication slippage in which replication stalls and then the two strands misalign before synthesis resumes to produce the observed structure in a single event (Figure 3). In agreement with the data, both of these mechanisms would produce chimeric genes through local, DNA-based events with few substitutions separating chimeras from their parental genes. Thus, we suggest that most chimeras form close to their parental genes through these two mechanisms and only later are relocated to different parts of the genome.
Figure 2.—

Large-loop mismatch repair mechanism of chimera formation. A short segmental duplication occurs on a given chromosome, copying genes PB and PA. During meiosis or mitosis, the duplicated region pairs with an unduplicated chromatid. The outer genes PB and PA′ pair with the unduplicated genes on the opposite chromatid, producing a loop of unpaired DNA, initiating large-loop mismatch repair. Imprecise excision of the loop creates a chimeric gene with the observed structure.
Figure 3.—

Replication slippage-based mechanism of chimera formation. During the synthesis of the parental gene PA, replication stalls. While searching for the proper template to resume replication, genes misalign as shown so that PA pairs with PB. New DNA is synthesized on the misaligned strand, producing a chimeric gene with the structure and placement observed in the seven youngest chimeras.
The two genes CG31668 and CG6844 display a more complicated structure where one parental gene contributes different segments to both the 5′ and the 3′ portion of the chimera while material from the second parental gene lies in the middle. As both of these genes have t > 0.5, it is conceivable that secondary events may have added exons to the gene after initial formation. Only 1 of the 14 chimeric genes showed evidence of nonequal ages of the segments (supplemental Figure 1), although this disparity could easily be attributed to gene conversion.
Age distribution of chimeric and duplicate genes:
Duplicate and chimeric gene birth and death is a highly dynamic process, involving forces of formation, decay, and preservation. These conflicting forces do well to explain the observed distribution of gene ages, which is characterized by large numbers of very young genes and much smaller numbers of older genes (Figure 4). After formation, a given duplicate or chimera is subject to one of two fates: decay through nonfunctionalizing mutations or preservation through neofunctionalization or subfunctionalization. We can construct a model for duplicate and chimeric genes that accounts for the relative rates of birth, death, and preservation. Such a model predicts the following age distribution of duplicate or chimeric genes,
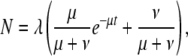 |
where N represents the number of genes present after a given time t, λ represents the rate of formation, μ represents the rate of decay, and ν represents the rate of preservation. We used maximum likelihood to fit λ, μ, and ν from this model to our data for chimeric and duplicate genes (Figure 4, Table 2). Additionally, we used bootstrap resampling to estimate the 95% confidence intervals of these parameters (Table 2).
Figure 4.—

The age distribution for (A) duplicate and (B) chimeric genes fitted with our model of birth (formation), death (decay), and preservation. Histogram bins show the observed distribution of t values, while dashed lines show the maximum-likelihood (ML) model fit. The rate of formation λ scales the overall number of duplicates/chimeras observed, but does not affect the shape of the distribution. The rate of decay μ determines how quickly the distribution drops to a constant threshold, while both μ and the rate of preservation ν together determine the height of the constant tail. ML estimates of rate parameters are shown in Table 2.
TABLE 2.
Maximum-likelihood rate estimates with corresponding 95% bootstrap intervals (numbers in parentheses) of birth, decay, and preservation in duplicate and chimeric genes
| Birth λ | Decay μ | Preservation ν | Preservation probability | |
|---|---|---|---|---|
| Duplicates | ||||
| Per 1.0 t | 3470 (2300–4800) | 47.4 (30.4–67.1) | 2.03 (1.42–2.86) | 4.1% (2.8–6.4%) |
| Per 106 yr | 76.4 (50.9–105.9) | 1.04 (0.67–1.48) | 0.045 (0.031–0.063) | – |
| Chimeras | ||||
| Per 1.0 t | 518 (146–1090) | 76.5 (22.5–157.5) | 1.10 (0.29–3.84) | 1.4% (0.4–6.0%) |
| Per 106 yr | 11.4 (3.22–23.97) | 1.68 (0.50–3.47) | 0.024 (0.006–0.085) | – |
| P (dup > chim)a | 1.000 | 0.112 | 0.8437 | 0.9322 |
One-sided P-value determined from proportion of 10,000 bootstrap replicates that show greater estimates for duplicates than for chimeras.
Our parameter estimates suggest that significantly more duplicates are formed than chimeras with 3470 duplicates and 518 chimeric genes arising in the time it takes to accumulate one synonymous substitution per synonymous site (t = 1.0). Both duplicate and chimeric genes are eliminated quickly; duplicates show an average half-life of 0.0146 t and chimeras show an average half-life of 0.0091 t. Under the assumption that synonymous sites in Drosophila evolve at a rate of 1.1 × 108 substitutions per site per year (Tamura et al. 2004), duplicates and chimeras should accumulate 1.0 t over a period of 45.5 million years. This conversion yields a half-life of ∼0.66 MY for duplicate genes and 0.44 MY for chimeras, with no significant difference between the two. Using the same conversion factor, we find that 76.4 duplicate genes and 11.4 chimeras form per million years. On the basis of our calculated rates of formation and preservation, we find that after formation 4.1% of duplicate genes and 1.4% of chimeric genes are eventually preserved, with no significant difference. Thus, we find that the number of chimeric genes within the D. melanogaster genome is primarily limited by a moderate rate of formation relative to duplicate genes and not by an exceptionally high rate of decay or low rate of preservation.
Phylogenetic distribution of chimeric genes:
We constructed the phylogenetic distribution of D. melanogaster chimeric genes among six other sequenced Drosophila species, on the basis of the latest GLEANR consensus gene models (Figure 5). A substantial number of chimeric genes appear to be specific to D. melanogaster with fewer chimeras preserved across multiple Drosophila species. Six of the 8 D. melanogaster-specific chimeras were found in a recent search for new genes in the D. melanogaster subgroup (Zhou et al. 2008). Some putative gene losses occurred, with CG30457 and CG6844 absent in D. yakuba, CG6844 not found in D. ananassae, and CG6653 missing in D. sechellia. Whether these absences are due to true gene losses or are merely a product of annotation gaps is uncertain. We have repeated our search for chimeric genes in D. erecta and mapped them onto the phylogeny using identical methods (data not shown). We find that 7 of 30 chimeras in D. erecta are novel, suggesting the presence of lineage-specific chimeric genes is not unique to the D. melanogaster genome.
Figure 5.—

Phylogenetic distribution of chimeric genes found in Drosophila melanogaster. Phylogenetic locations were determined through parsimony. Gene names are placed on the branch in which each chimera was formed. Phylogeny and t were consistent for almost all genes considered. Eight chimeric genes (††: CG18853, CG32318, CG31864, CG12592, CG31904, CG31687, CG18217, and CG31668) are specific to D. melanogaster. Putative losses (see results) are not shown.
Gene conversion, if present, will act to deflate t relative to the phylogenetic age of a gene and thereby cause discordance between comparative phylogenetic estimates of chimera formation and single-genome estimates. We compared our results against phylogenetic estimates of time since formation, using the expected number of mutations per site for each Drosophila lineage (Drosophila 12 Genomes Consortium et al. 2007). Regression statistics show a significant relationship between t and phylogenetic-based measurements (coefficient = 0.969, intercept = 0.066, P = 0.028, R2 = 0.340), suggesting that gene conversion is not widely acting on chimeric genes to deflate t. The chimeric gene CG31668 could not be found in any other sequenced Drosophila species despite its high value of t = 0.513. However, it is possible that the observed absences are due to incomplete annotations or gaps in genome assembly, and hence we favor the use of t to measure its age.
DISCUSSION
Through a genomewide study of D. melanogaster, we are able to identify 14 chimeric genes where portions of different coding sequences have contributed to the formation of new proteins (Table 1). From these chimeras we are able to make several conclusions regarding the evolution of chimeric genes. We find that the formation and preservation of chimeric genes contribute significantly to genome content, with one chimera preserved every ∼6.3 million years. Also, we find that local mutational events are sufficient to describe the formation of these chimeric genes. Because of our strict definitions, this data set will not capture several other types of novel genes. The recruitment of novel UTRs, events where previously untranslated sequence contributes to novel coding sequence, or events where domesticated transposable elements contribute to the formation of a novel gene will not be reported by our methods of chimera discovery. We have therefore reported a conservative list of chimeric genes, which should contain excellent candidates for functional analysis. Experimental gene ontology data are not available on FlyBase (accessed May 2008; http://flybase.org) for many of our chimeric and parental genes and full molecular analysis will be necessary to determine the full significance of these chimeric genes.
Although current experimental data are sparse for these genes, in at least one case we can determine the functional significance of chimera formation. The chimeric gene pannier (pnr or CG3978) contains segments that are similar to GATAe and to grain (grn) (Table 1). The phylogenetic distribution of pnr suggests that it was formed prior to the divergence of D. melanogaster and D. pseudoobscura (Figure 5). The parental gene GATAe is a transcription factor responsible for the specification of the endoderm during development (Okumura et al. 2005) while the parental gene grn is a transcription factor responsible for the formation of spiracles, stigmatophore, and adult legs from ectoderm tissue (Brown and Castelli-Gair Hombria 2000). Molecular characterization shows that the chimera pnr is a transcription factor responsible for structuring the cardiac mesoderm (Alvarez et al. 2003), a function that is phenotypically distinct from that of either parental gene. Whether this functional difference is due to development of entirely new activity or specialization among ancestral functions cannot be determined from available data.
Mechanisms of chimera formation:
We find that young chimeric genes are always located in between their two parental genes and that very few changes have occurred in intron–exon structure (Figure 1). To explain the placement and structure of the chimeras observed, we propose two mechanisms of chimera formation that rely entirely on local events without the need for repetitive element mobilization. The first possible mechanism involves imprecise excision of a segmental duplication during large-loop mismatch repair (Figure 2). Here, a segment of DNA duplicates during replication, and then the two outermost genes in the segment pair with a chromatid that lacks the duplication. Recognition of an unpaired stretch of DNA initiates large-loop mismatch repair, which may be resolved imprecisely, creating a chimeric gene. Unaligned portions of parental genes depicted in gray in Figure 1 correspond to the deleted segment. This large-loop mismatch repair can operate only prior to fixation of the duplication, explaining the lack of mutations separating the chimeric and parental genes.
A second mechanism of formation that could explain the structures observed involves an event similar to replication slippage (Figure 3). Here, during the synthesis of gene PA, replication stalls. While searching for the proper template to resume replication, genes misalign as shown so that PA pairs with PB. New DNA is synthesized on the misaligned strand, producing a chimeric gene with the structure and placement observed in the seven youngest chimeras. Such misalignments are likely to be rare. Searches of the genomic regions for each parental gene pair in a blast2seq could find no similar regions that might facilitate alignment in the seven youngest chimeric genes, even with an exceptionally permissive E-cutoff of 100. Manual inspection revealed 9 bp of overlap in the genomic sequences contributing to CG31904. One other gene had 4 bp of overlap, and the remaining five young chimeric genes had ≤3 bp of overlap (see supplemental information). With such minimal nucleotide identity between these two genes, aberrant alignment must occur infrequently, especially when an appropriate substrate lies such a short distance downstream.
Under both these mechanisms, chimeric genes will arise in tandem with their parental genes, and may later relocate to other parts of the genome. Such a model brings with it certain implications concerning gene expression as well as gene formation. Any newly formed chimeric gene will inherit the promoter from its downstream parental gene and will initially be governed by the same chromatin modeling patterns that regulate both of its parental genes. These factors make it likely that a given chimeric gene will display an initial expression pattern that is largely similar to that of its 3′ downstream parental gene. Later relocalization and possible recruitment of alternative regulatory elements may be an important step in optimizing the function of the novel gene. Unfortunately, existing microarray experiments use probes that cannot always distinguish between parental and chimeric genes. Assessing expression patterns in the youngest chimeras will require unique probes that span the boundary of chimera formation.
Our large-loop mismatch repair mechanism depends upon the formation of segmental duplications of two or more genes. Using 35 mapped pairs with t ≤ 0.01, we characterized probable segmental duplications. We found six events leading to double duplications of the tandem form ABA′B′ and one event leading to a triple duplication of the form ABCA′B′C′. Thus, the mean duplication event results in 1.296 duplicates. This result may underestimate the clustering of duplications. If a double duplication forms but one copy is quickly eliminated, it will be recorded as a single gene duplication. Hence, it appears that at least 43% of duplicate genes form in clustered segmental duplication events. We believe that this provides an ample substrate for the formation of chimeric genes through large-loop mismatch repair.
While it may be possible to generate chimeric genes via deletions after fixation, we find little evidence in our data set that this phenomenon occurs (see supplemental Figure 2). The half-life of duplicate genes is exceptionally short, indicating that nonfunctionalizing mutations accumulate very quickly during the lives of duplicate genes. Formation of chimeric genes must then occur quickly to capture available genetic material before decay into pseudogenes. Older duplicate genes that are separated from their parental genes by multiple mutations are preserved by selection and have become immune to elimination. Hence, they may be less likely to contribute to the formation of chimeric genes, as loss of their existing function following partial deletion would be selectively disadvantageous.
Previous observations (Yang et al. 2008) have suggested that transposable elements play an important role in the formation of chimeric genes in Drosophila by facilitating ectopic recombination. The observed disparity between this TE-mediated mechanism and our local mutation models is likely due to a difference in experimental methodology. The in situ hybridization used by Yang et al. (2008) reports only duplicates separated by at least 100 kb. Therefore, it will not identify any chimeric genes found near their parental genes. In contrast, our methods do not discriminate between near and distant genes. We find that young chimeric genes are always found in tandem with their parental genes (Table 1), suggesting that chimeric genes found distant from their parental genes likely represent secondary translocations. Consequently, the chimeric genes found by in situ hybridization may not reflect the patterns observed in the youngest chimeras and thus may be biased toward a TE-mediated mechanism of chimera formation. Still, the observed transposable element association may be important to the process of chimeric gene relocation and hence may affect the ultimate expression patterns of novel chimeric genes.
Some evidence indicates that retrotransposition may be a key player in chimeric gene formation. The neofunctionalized chimeric gene jingwei found in D. yakuba and D. tesseri originated through a retrotransposition event (Long and Langley 1993; Long et al. 1999; Wang et al. 2000; Zhang et al. 2004). Similarly, results from rice (Oryza sativa) have indicated that many retrogenes have chimeric structures (Wang et al. 2006). However, we do not see any evidence that intron loss predominates in D. melanogaster chimeras (Figure 1, Table 1). If chimeric coding sequences do indeed originate through retrotransposition, we would expect some young dispersed chimeric genes. Yet, all seven of the youngest chimeric genes are found in tandem with their parents. Therefore, retrotransposition of coding sequences is probably not a common mechanism that generates coding sequence chimeras in D. melanogaster, consistent with results that show low numbers of new retrogenes in Drosophila (Zhou et al. 2008).
Patterns of preservation:
We find that that large numbers of duplicate genes form and then are rapidly eliminated. This pattern is consistent with recent work on expansion of gene families, which shows large numbers of recent copy number differences between species and fewer shared ancestral differences (Hahn et al. 2007). We estimate that only a small proportion of duplicate genes that form is eventually preserved, either by subfunctionalization or by neofunctionalization. Chimeric genes form in modest numbers relative to their duplicate counterparts but show similar patterns of preservation and decay (Figure 4, Table 2). Hence, chimeric genes seem to be formed often enough and show signs that they may occasionally persist long enough to be an important factor in genome evolution.
We estimate the rate at which duplicate and chimeric gene decay occurs. This rate μ encompasses the effects of drift, mutation, and selective processes. Selective effects will increase or decrease the probability of gene fixation and affect the parameter μ relative to the neutral rate. Any action of selection against the fixation of these genes would be displayed in a higher μ and a shorter half-life. While it may be possible to calculate the relative contributions of selection, population dynamics, and mutational decay to our model, this requires data beyond the scope of this present work. Our estimate of duplicate gene half-life in D. melanogaster based on μ for an updated data set is over five times shorter than that from earlier results (Lynch and Conery 2000, 2003), indicating even more rapid elimination than was previously thought. The improved assembly of highly similar tandem duplicates in more recent genome releases and the addition of a parameter of preservation largely explain such a difference. Because preservation explains a portion of older genes, we fit a much sharper slope to our duplicate genes and find a much more rapid rate of decay.
A slightly lower proportion of chimeric genes than duplicate genes seems to be preserved in D. melanogaster, although the difference is not significant (Table 2). Unlike duplicate genes, a chimeric gene is immediately different from either parental gene. As it does not contain the complete gene sequence of either parental gene, it may not carry the same full functional redundancy as a typical duplicate. Thus, preservation by subfunctionalization may be less likely than with duplicate genes. Hence, we suspect that the set of chimeric genes that is retained over long periods of time could contain a greater proportion of neofunctionalized proteins relative to subfunctionalized proteins than the set of duplicate genes, even if the actual number retained is lower.
Chimeric genes found in our search must have both parental genes present in the D. melanogaster genome. Loss of parental genes after formation would yield chimeric genes that are absent from this data set. Hence, our results may be biased toward lower estimates of preservation and higher estimates of loss compared to what actually occurs. However, chimeric genes whose parental genes have been lost may be more likely to have fully redundant functions than chimeras preserved with parental genes still present. Thus, the preserved chimeric genes in our data set may be enriched for chimeras that have either neofunctionalized or subfunctionalized.
Conclusions:
Previous estimates of duplicate gene birth and death utilized a phylogenetic approach to detect formation and loss (Hahn et al. 2005, 2007; Demuth et al. 2006). Such approaches are highly sensitive to differences in sequence coverage, genomic assembly, and gene annotation among species. Our method dates chimeric and duplicate genes solely on the basis of comparisons within a single genome and hence should be robust to inconsistencies in genome annotation across taxa. Additionally, our estimates of duplicate and chimeric gene formation should be broadly applicable to any annotated genome, without the need for sequenced sister groups. Finally, current phylogenetic approaches do not explicitly account for preservation of duplicates by selection. Our model, on the other hand, allows characterization of genes that have become immune to stochastic decay processes and are preserved by selective constraint.
We found significant turnover of chimeric and duplicate genes within the D. melanogaster genome. Chimeric genes appear in sufficient numbers and seem to survive long enough after formation to contribute significantly to genomic content. Still, it will require rigorous molecular work to ascertain their ultimate importance and propensity to develop novel functions. Careful functional characterization will be needed to determine what cellular roles chimeric genes play and to what extent these roles represent true evolutionary novelty.
Acknowledgments
We thank Marna S. Costanzo, Pierre Fontanillas, Rob J. Kulathinal, Scott W. Roy, Jeffrey P. Townsend, and Qi Zhou for moral support and helpful discussions. We also thank David Rand and two anonymous reviewers for their comments, which improved this manuscript. This work was supported by a National Science Foundation Predoctoral Fellowship to T.B. and by National Institutes of Health grants GM068465 and GM065169.
References
- Adams, M. D., S. E. Celniker, R. A. Holt, C. A. Evans, J. D. Gocayne et al., 2000. The genome sequence of Drosophila melanogaster. Science 287 2185–2195. [DOI] [PubMed] [Google Scholar]
- Altschul, S. F., W. Gish, W. Miller, E. W. Myers and D. J. Lipman, 1990. Basic local alignment search tool. J. Mol. Biol. 215 403–410. [DOI] [PubMed] [Google Scholar]
- Alvarez, A. D., W. Shi, B. A. Wilson and J. B. Skeath, 2003. Pannier and pointedP2 act sequentially to regulate Drosophila heart development. Development 130 3015–3026. [DOI] [PubMed] [Google Scholar]
- Bogarad, L. D., and M. W. Deem, 1999. A hierarchical approach to protein molecular evolution. Proc. Natl. Acad. Sci. USA 96 2591–2595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brown, S., and J. Castelli-Gair Hombria, 2000. Drosophila grain encodes a GATA transcription factor required for cell rearrangement during morphogenesis. Development 127 4867–4876. [DOI] [PubMed] [Google Scholar]
- Cui, Y., W. H. Wong, E. Bornberg-Bauer and H. S. Chan, 2002. Recombinatoric exploration of novel folded structures: a heteropolymer-based model of protein evolutionary landscapes. Proc. Natl. Acad. Sci. USA 99 809–814. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Demuth, J. P., T. De Bie, J. E. Stajich, N. Cristianini and M. W. Hahn, 2006. The evolution of mammalian gene families. PLoS ONE 1 e85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Drosophila 12 Genomes Consortium, A. G. Clark, M. B. Eisen, D. R. Smith, C. M. Bergman et al., 2007. Evolution of genes and genomes on the Drosophila phylogeny. Nature 450 203–218. [DOI] [PubMed] [Google Scholar]
- Force, A., M. Lynch, F. B. Pickett, A. Amores, Y. L. Yan et al., 1999. Preservation of duplicate genes by complementary, degenerative mutations. Genetics 151 1531–1545. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Giver, L., and F. H. Arnold, 1998. Combinatorial protein design by in vitro recombination. Curr. Opin. Chem. Biol. 2 335–338. [DOI] [PubMed] [Google Scholar]
- Hahn, M. W., T. De Bie, J. E. Stajich, C. Nguyen and N. Cristianini, 2005. Estimating the tempo and mode of gene family evolution from comparative genomic data. Genome Res. 15 1153–1160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hahn, M. W., M. V. Han and S. G. Han, 2007. Gene family evolution across 12 Drosophila genomes. PLoS Genet. 3 e197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jones, C. D., and D. J. Begun, 2005. Parallel evolution of chimeric fusion genes. Proc. Natl. Acad. Sci. USA 102 11373–11378. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jurka, J., 2000. Repbase update: a database and an electronic journal of repetitive elements. Trends Genet. 16 418–420. [DOI] [PubMed] [Google Scholar]
- Jurka, J., V. V. Kapitonov, A. Pavlicek, P. Klonowski, O. Kohany et al., 2005. Repbase update, a database of eukaryotic repetitive elements. Cytogenet. Genome Res. 110 462–467. [DOI] [PubMed] [Google Scholar]
- Kapitonov, V. V., and J. Jurka, 2008. A universal classification of eukaryotic transposable elements implemented in repbase. Nat. Rev. Genet. 9 411–412. [DOI] [PubMed] [Google Scholar]
- Long, M., and C. H. Langley, 1993. Natural selection and the origin of jingwei, a chimeric processed functional gene in Drosophila. Science 260 91–95. [DOI] [PubMed] [Google Scholar]
- Long, M., W. Wang and J. Zhang, 1999. Origin of new genes and source for N-terminal domain of the chimerical gene, jingwei, in Drosophila. Gene 238 135–141. [DOI] [PubMed] [Google Scholar]
- Lynch, M., and J. S. Conery, 2000. The evolutionary fate and consequences of duplicate genes. Science 290 1151–1155. [DOI] [PubMed] [Google Scholar]
- Lynch, M., and J. S. Conery, 2003. The evolutionary demography of duplicate genes. J. Struct. Funct. Genomics 3 35–44. [PubMed] [Google Scholar]
- Lynch, M., and A. Force, 2000. The probability of duplicate gene preservation by subfunctionalization. Genetics 154 459–473. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lynch, M., M. O'Hely, B. Walsh and A. Force, 2001. The probability of preservation of a newly arisen gene duplicate. Genetics 159 1789–1804. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moore, R. C., and M. D. Purugganan, 2003. The early stages of duplicate gene evolution. Proc. Natl. Acad. Sci. USA 100 15682–15687. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nadeau, J. H., and D. Sankoff, 1997. Comparable rates of gene loss and functional divergence after genome duplications early in vertebrate evolution. Genetics 147 1259–1266. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ohno, S., 1970. Evolution by Gene Duplication. Springer-Verlag, Berlin/New York.
- Okumura, T., A. Matsumoto, T. Tanimura and R. Murakami, 2005. An endoderm-specific GATA factor gene, dGATAe, is required for the terminal differentiation of the Drosophila endoderm. Dev. Biol. 278 576–586. [DOI] [PubMed] [Google Scholar]
- Tamura, K., S. Subramanian and S. Kumar, 2004. Temporal patterns of fruit fly (Drosophila) evolution revealed by mutation clocks. Mol. Biol. Evol. 21 36–44. [DOI] [PubMed] [Google Scholar]
- Tatusova, T. A., and T. L. Madden, 1999. BLAST 2 sequences, a new tool for comparing protein and nucleotide sequences. FEMS Microbiol. Lett. 174 247–250. [DOI] [PubMed] [Google Scholar]
- Thompson, J. D., D. G. Higgins and T. J. Gibson, 1994. CLUSTAL W: improving the sensitivity of progressive multiple sequence alignment through sequence weighting, position-specific gap penalties and weight matrix choice. Nucleic Acids Res. 22 4673–4680. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van Hoof, A., 2005. Conserved functions of yeast genes support the duplication, degeneration and complementation model for gene duplication. Genetics 171 1455–1461. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Voigt, C. A., C. Martinez, Z. G. Wang, S. L. Mayo and F. H. Arnold, 2002. Protein building blocks preserved by recombination. Nat. Struct. Biol. 9 553–558. [DOI] [PubMed] [Google Scholar]
- Wang, W., J. Zhang, C. Alvarez, A. Llopart and M. Long, 2000. The origin of the jingwei gene and the complex modular structure of its parental gene, yellow emperor, in Drosophila melanogaster. Mol. Biol. Evol. 17 1294–1301. [DOI] [PubMed] [Google Scholar]
- Wang, W., H. Zheng, C. Fan, J. Li, J. Shi et al., 2006. High rate of chimeric gene origination by retroposition in plant genomes. Plant Cell 18 1791–1802. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang, S., J. R. Arguello, X. Li, Y. Ding, Q. Zhou et al., 2008. Repetitive element-mediated recombination as a mechanism for new gene origination in Drosophila. PLoS Genet. 4 e3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang, Z., 1997. PAML: a program package for phylogenetic analysis by maximum likelihood. Comput. Appl. Biosci. 13 555–556. [DOI] [PubMed] [Google Scholar]
- Zhang, J., A. M. Dean, F. Brunet and M. Long, 2004. Evolving protein functional diversity in new genes of Drosophila. Proc. Natl. Acad. Sci. USA 101 16246–16250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou, Q., G. Zhang, Y. Zhang, S. Xu, R. Zhao et al., 2008. On the origin of new genes in Drosophila. Genome Res. 18 1446–1455. [DOI] [PMC free article] [PubMed] [Google Scholar]