

Abstract
One of the current limitations of gene transfer protocols involving mammalian genomes is the lack of spatial and temporal control over the desired gene manipulation. Starting from a human keratin gene showing a complex regulation as a template, we identified regulatory sequences that confer inducible gene expression in a subpopulation of keratinocytes in stratified epithelia of adult transgenic mice. We used this cassette to produce transgenic mice with an inducible skin blistering phenotype mimicking a form of epidermolytic hyperkeratosis, a keratin gene disorder. Upon induction by topical application of a phorbol ester, the mutant keratin transgene product accumulates in the differentiating layers of epidermis, leading to keratinocyte lysis after application of mechanical trauma. This mouse model will allow for a better understanding of the complex relationship between keratin mutation, keratinocyte cytoarchitecture, and hypersensitivity to trauma. The development of an inducible expression vector showing an exquisite cellular specificity has important implications for manipulating genes in a spatially and temporally controlled fashion in transgenic mice, and for the design of gene therapy strategies using skin as a tissue source for the controlled delivery of foreign substances.
The development of gene transfer technologies has led to an understanding of the regulation and function of a great variety of genes in their natural environment. They have been also exploited to produce animal models mimicking the key manifestations of specific human diseases, providing critical data relative to their molecular basis and biology, and an opportunity to assess new therapeutic approaches (1, 2). One of the standing limitations of gene transfer is the lack of control over the timing of the desired gene manipulation (3, 4). In mouse, transgenesis usually involves the introduction of DNA in a one-cell embryo or embryonic stem cells (5), and thus the consequences of the modification introduced depend upon the regulatory sequences of the transgene or targeted locus. Since a substantial fraction of genes in mammalian genomes is activated during development, so are the consequences of the gene manipulation. When this latter aims at gain-of-function or loss-of-function phenotypes, it ensues that the viability of the transgenic animals may be compromised, or that the consequences of the primary alteration may be blurred by secondary homeostatic changes. The recent adaptation of the Cre-loxP recombination system of bacteriophage P1 for manipulating the mouse genome in a regulated fashion represents a major breakthrough (4). While strategies akin to the Cre-loxP will undoubtedly be further refined for manipulating genomes, a major remaining limitation is the development of inducible expression vectors that would confer the desired expression pattern for the Cre recombinase or its functional equivalent.
Starting from a human keratin-encoding gene that features a complex pattern of expression, we surmised that it may be possible to develop a vector that confers inducible expression in epidermis and other stratified epithelia of transgenic mice. The starting template was the human keratin 6a (hK6a) gene, one among several K6 isoform genes clustered on 12q11-13 (ref. 6 and references therein). The K6 isoforms are normally expressed in a variety of stratified epithelia, including the hair follicle outer root sheath in skin, many glandular tissues, oral epithelia, esophagus, and forestomach (6, 7). They are better known, however, for their greatly enhanced expression under conditions associated with hyperproliferation or abnormal differentiation in complex epithelia (8). Thus, K6 is induced in wound-edge keratinocytes as early as 4–6 hr after injury to human skin and disappears after wound closure (ref. 9 and references therein). K6 is also induced in several diseases affecting complex epithelia, including viral infections, squamous metaplasia, carcinoma, and chronic hyperproliferative disorders such as psoriasis (8, 10). In these conditions, K6 protein expression can be abundant and is restricted to the postmitotic, suprabasal compartment of the stratified epithelia involved. In mouse skin, K6 can be induced by topical application of a variety of chemicals—e.g., phorbol esters and retinoic acid (11). K6 induction also occurs in primary cultures of mitotically active stratified epithelial cells (8). The inducible aspect of K6 gene regulation is thus of obvious interest for conditional gene manipulation in mice. Here we report on the identification of a segment in the hK6a gene that enables the inducible expression of heterologous ORFs in the differentiating keratinocytes of epidermis and other stratified epithelia of adult transgenic mice. To illustrate its potential, we exploited this expression vector to produce a transgenic mouse model featuring an inducible skin phenotype mimicking a known inherited skin blistering disease, epidermolytic hyperkeratosis.
MATERIALS AND METHODS
Production of Transgenic Mice.
All DNA constructs involved the proximal 5.2 kb or 2.5 kb of 5′ upstream sequence from the hK6a gene (6). Two coding sequences were used. (i) We used a lacZ coding sequence modified with a nuclear localization sequence at its 5′ coding end and a simian virus 40 poly(A) sequence at its 3′ end (12) (Fig. 1A). (ii) We used a mutated K6a cDNA bearing an in-frame deletion of 156 nucleotides in exon 1, such that residues 125–176 were deleted from the deduced protein sequence (6). Also, the sequences encoding the C-terminal 4 amino acids of K6a (SYKH-stop; 6) were replaced with a sequence (NH2-MEQKLISEEDLN-stop-COOH) encoding an epitope derived from the myc oncoprotein (13). This cDNA was designated ΔK6a-myc (Fig. 1B). A 545-bp segment corresponding to the 3′ noncoding sequence of K6a was placed at the 3′ end of this construct. Transgenic founders were generated by pronuclear injection of DNA in C57B6/BalbC3 embryos, and lines were established by matings in the same background. The transgenic lines produced featured 1–16 transgene copies per mouse genome (data not shown).
Figure 1.
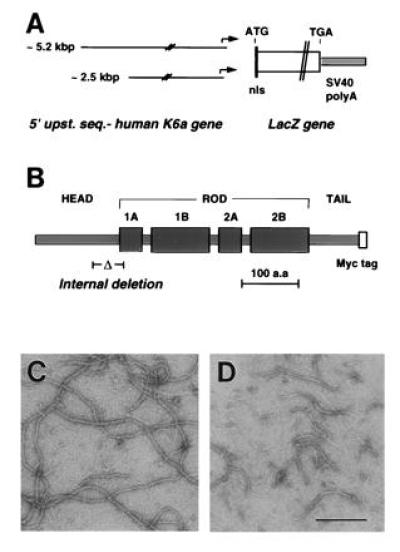
Transgene constructs and mutant K6 properties. (A) Structure of the lacZ transgenes. Both constructs feature the lacZ reporter sequence, modified to contain a nuclear localization signal (nls) at its N terminus, and a simian virus 40 poly(A) sequence at the 3′ end. They differ with respect to the amount of 5′upstream sequence from the hK6a gene. (B) Predicted secondary structure of hK6a as deduced from the cDNA sequence (7). A centrally located rod domain features four subdomains with heptad repeats of apolar residues and α-helical structure (1A, 1B, 2A, and 2B). The rod domain is flanked by non-helical head and tail domains. The internal deletion (Δ) encompasses the last 36 residues of the head domain and the first 16 residues of the rod domain. (C and D) Electron micrographs of negatively stained filaments assembled in vitro from purified recombinant keratins. (C) Filaments assembled from K6a-myc, K17, K5, and K14 (≈1:1:1:1 ratio). (D) Mutant filament assembled from ΔK6a-myc, K17, K5, and K14. Assemblies were subjected to a pelleting assay (9) to assess polymerization efficiency. The control keratin sample polymerized with >80% efficiency, while the mutant ΔK6a-myc containing sample polymerized with 40% efficiency. (Bar = 200 nm.)
Protocols for Manipulating Skin and Other Epithelia.
Studies involving animals were reviewed by the Johns Hopkins University Animal Use and Care Committee. Mice were anesthetized with avertin and their backs epilated with Nair cream. To produce skin injury, the surgical area was disinfected and full thickness wounds were produced using a 4-mm punch (Acuderm, Ft. Lauderdale, FL). For injury to foot pad epidermis, cornea, oral mucosa, and tongue, short superficial incisions were made using a scalpel under similar conditions. Tissues were harvested after ≈24 hr and processed for LacZ histochemistry. Chemical induction of transgene expression was done by topical application of phorbol-12-myristate-13-acetate (PMA; 150 μl of a 50 mM stock in acetone; Sigma) or all-trans-retinoic acid (150 μl of a 100 μg/ml stock in ethanol; Sigma) on Nair-epilated skin three times every third day. Controls consisted in topical application of the vehicle. For tape-stripping studies, the back skin of 8-week-old mice was treated with PMA topically three times every third day. On day 9 mice were anesthetized, and Scotch Guard (3M) tape was firmly applied onto back skin and removed in a sudden motion. Tape stripping was repeated three to five times and skin tissue was processed for histology.
Skin Tissue Analysis.
For β-galactosidase (β-gal) histochemistry, tissues were prefixed in 1% formaldehyde/0.2% glutaraldehyde (60 min) and incubated overnight in X-Gal solution (5-bromo-4-chloro-3-indolyl β-d-galactoside; ref. 14), postfixed in Bouins’ solution, paraffin-embedded, and sections stained with eosin. For immunohistochemistry, Bouins’-fixed, paraffin-embedded tissue sections were reacted with antisera directed against mouse K6 (15), β-gal (Promega) or myc (MBL International, Watertown, MA) followed by a peroxidase conjugate (Kirkegaard & Perry Laboratories). To assay β-gal activity in soluble tissue extracts, control and transgenic skin was homogenized and supernatants were assayed for activity following the manufacturer’s instructions (Promega). Keratin protein extraction from skin samples was performed as described (16), and final pellets were solubilized in urea/2-mercaptoethanol buffer. Known amounts of protein were electrophoresed (SDS/PAGE), and the gels were Coomassie blue stained or blotted onto nitrocellulose. Membranes were incubated with the antisera to K6 or myc and bound antibodies were detected by alkaline phosphatase (Bio-Rad). The amount of ΔK6a-myc protein relative to total skin proteins was determined as described (see ref. 16).
Characterization of Keratin Mutant Properties in Vitro. Human keratins K5, K14, K17, K6a-myc, and ΔK6a-myc were produced in a recombinant form in Escherichia coli and purified as described (17).
Heterotypic complexes of K5/K14, K6a-myc/K17, and ΔK6a-myc/K17 were isolated in 6.5 M urea buffer as described (9, 17). Equimolar amounts of K5/K14 and either K6a-myc/K17 or ΔK6a-myc/K17 complexes were mixed in urea, and 10 nm filament formation was induced by dialysis against low-ionic strength buffer (17). Assemblies were visualized by negative staining and electron microscopy, and subjected to a pelleting assay (100,000 × g for 30 min) to assess polymerization efficiency (9).
RESULTS AND DISCUSSION
Analysis of the Activity of the hK6a Gene 5′ Upstream Sequence in Transgenic Mice.
We characterized six transgenic lines with [5.2-kb hK6a 5′]-LacZ and two transgenic lines with [2.5-kb hK6a 5′]-LacZ to analyze the activity of this potential regulatory segment of the hK6a gene. When organs and tissues, including skin, are systematically surveyed, no β-gal activity can be detected in most transgenic lines made with either of these constructs (Fig. 2D). [Note: In many transgenic lines, sporadic LacZ expression is detected in occasional outer root sheath keratinocytes in a small subset of hair follicles in trunk skin. In addition, some lines show sporadic and weak expression of the transgene in other tissues (e.g., tongue, retina, spleen, muscle, liver). The activity of these K6a gene promoter segments has been detailed and will be reported elsewhere (unpublished data).] In striking contrast, strong β-gal activity is detected at the proximal edge of the wound site within 4 hr after experimental injury to the skin, and extends further away from the wound site at later time points in a manner analogous to mouse endogenous K6 (Fig. 2 C, E, and F). Injury to other stratified epithelia, including tongue, oral mucosa, cornea, and footpad epidermis, also cause transactivation of the lacZ transgene (data not shown). Finally, topical application of the phorbol ester PMA (Fig. 2 G and H) or retinoic acid (data not shown) on skin also causes a strong activation of lacZ transgene expression in epidermis and to a lesser extent, in the outer root sheath of hair follicles. These two chemicals are known to be potent inducers of K6 expression in these mouse skin epithelia (Fig. 2B; refs. 7 and 11, and references therein). Antibody staining shows that in transgenic mouse epidermis subjected to injury (Fig. 2I) or PMA application (not shown), the β-gal protein is restricted to the nuclei of differentiating suprabasal keratinocytes. Induced K6 protein localizes to the same compartment in wounded or psoriatic human epidermis (9, 10). The histochemistry data were confirmed by β-gal enzymatic assays performed on soluble extracts prepared from skin (control mouse skin, 0.06 ± 0.01 milliunits/mm2 tissue, n = 3; intact transgenic mouse skin, 0.08 ± 0.01 milliunits/mm2 tissue, n = 7; PMA-induced transgenic mouse skin, 1.70 ± 0.50 milliunits/mm2 tissue, n = 5). Differences were seen in the extent of skin LacZ expression among transgenic lines, and the transgene featuring 5.2 kb of hK6a 5′ upstream sequence generally led to a greater expression after skin injury or chemical treatment (data not shown). Injury or appropriate chemical treatment induces transgene expression in skin for seven out of the eight transgenic mouse lines produced with the [5.2-kb hK6a 5′]-LacZ and [2.5-kb hK6a 5′]-LacZ constructs. These results thus demonstrate that the proximal 2.5 kb of 5′ upstream sequence can mediate inducible expression in a hK6a-like fashion in several stratified epithelia of adult transgenic mice, whereas as much as 5.2 kb of 5′ upstream sequence fails to direct sustained expression in a detectable fashion in the tissues surveyed. Studies are in progress to further characterize the regulation of the hK6a gene in transgenic mice. The results obtained so far indicate that we identified a regulatory region in a human keratin gene that can direct the expression, in an inducible and cell type-directed manner, of an heterologous reporter sequence to the epidermis and other stratified epithelia of adult transgenic mice.
Figure 2.
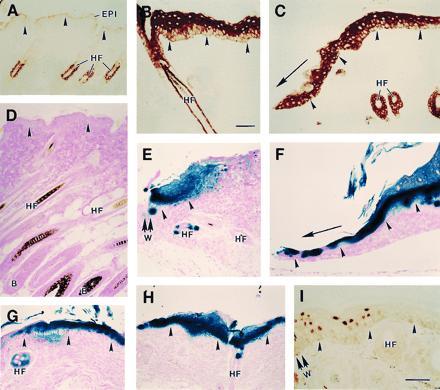
Inducible expression of lacZ transgene in transgenic mouse skin. (A–C) Cross-sections of control mouse skin reacted with an anti-mouse K6 antiserum followed by a peroxidase conjugate. Shown are intact skin (A), PMA-treated skin (B), and wound edge tissue (C) at 48 hr after skin injury. (D–H) Cross-sections of skin tissue from LacZ transgenic mice processed for β-gal histochemistry. (D) Intact skin from a [5.2-kb hK6a]-LacZ transgenic mouse. (E and F) Wound edge tissue at 4 hr (E) and 3 days (F) after skin injury in a [5.2-kb hK6a]-LacZ transgenic mouse (W, wound site). (G and H) PMA-treated skin tissue from a [5.2-kb hK6a]-LacZ transgenic mouse (G) and a [2.5 kb hK6a]-LacZ transgenic mouse (H). (I) The wound edge (W) of a [2.5-kb hK6a]-LacZ transgenic mouse at 24 hr after full-thickness skin injury reacted with an anti-β-gal antibody followed by a peroxidase conjugate. In all frames the dermo-epidermal interface is highlighted with arrowheads. In C and F, the direction of migration of the epithelium is depicted with an arrow. EPI, epidermis; HF, hair follicle; B, HF bulb region. (Bars = 100 μm.)
Creating a Mouse Model with an Inducible Skin Blistering Phenotype.
Mutations in epidermal keratin genes underlie several inherited skin bullous diseases in which the epidermis lyses in response to trivial trauma, producing painful blisters and compromising barrier function. Since epidermal keratin expression is tightly regulated in a differentiation-specific fashion, the identity of the keratin gene bearing the mutation dictates the subepidermal location of the plane of cleavage after trauma and the clinical manifestations of the disease (18, 19). These bullous diseases are usually dominantly inherited (18, 19), consistent with the dominant negative behavior of mutations affecting selective portions of the α-helical domain of keratin proteins (20). This domain is critical to their polymerization in 10 nm intermediate filaments and bears the majority of mutations discovered in patients suffering from these diseases (18–20). Along with genetic linkage analyses, the production of mouse models involving the tissue-specific expression of dominant negative keratin mutants played a key role in the discovery of the molecular basis underlying two bullous diseases, epidermolysis bullosa simplex and epidermolytic hyperkeratosis (16, 21–25). As is typical of patients, the skin blistering phenotype is present at birth in these mice, owing to the fact that the transcriptional activation of these keratin genes coincides with epidermal morphogenesis during development (26). While these mouse models led to a better understanding of the etiology and clinical features of inherited bullous diseases, the early onset of blisters and the associated secondary complications often cause death shortly after birth. Even in transgenic lines with a milder phenotype and better prognosis, the lack of control over mutant keratin synthesis and blister formation has precluded a thorough analysis of the relationship between mutant keratin expression, keratinocyte cytoarchitecture, and hypersensitivity to mechanical trauma. Yet, a better understanding of this complex relationship is likely to provide useful clues for the development of therapeutic strategies that are effective for the management of the skin condition in these patients.
We took advantage of the hK6a gene-based expression cassette to produce a transgenic mouse model in which the skin blistering phenotype is strictly inducible. We engineered an internal deletion of 52 amino acids at the junction between the head and α-helical domains of hK6a (Fig. 1B). This region of the protein is the target of the majority of mutations discovered so far in keratin genes of patients suffering from dominantly inherited skin blistering diseases (18–20). Consistent with a dominant negative behavior (20), the ΔK6a-myc mutant protein is not impaired in its ability to form stable heterotetramers in a chemical crosslinking assay (9) involving either K14 or K17 as type I keratin pairing partners (data not shown). However, this mutant interferes with the formation of 10 nm filaments when induced to polymerize in vitro in the presence of wild-type keratins (Fig. 1 C and D), documenting a dominant negative effect on intermediate filament assembly. Given the promiscuous nature of the pairing and assembly of type I and type II keratins (27), the mutated hK6a protein was expected, once expressed in transgenic mice, to interact with the type I keratins normally present in epidermis (for related mouse studies involving interactions between type I and type II keratins of different tissues and species of origin; see refs. 28 and 29). We analyzed two transgenic lines with [5.2-kb hK6a 5′]-ΔK6a-myc and seven transgenic lines with [2.5-kb hK6a 5′]-ΔK6a-myc. The distribution of the transgene product in mouse tissues was reproducible within and between these transgenic lines (data not shown).
As expected from our studies involving lacZ as a reporter gene, the intact skin of transgenic mice harboring these constructs shows no transgene expression and a normal histological appearance (Fig. 3A). Repeated topical application of the inducer PMA on the epilated skin of ΔK6a-myc transgenic mice resulted, in some lines (see below), in macroscopically detectable changes at the skin surface, with erythematous papules that progressed to an overt scaly hyperkeratotic appearance upon continued treatment. Morphological studies of induced skin confirmed the accumulation of the myc-tagged keratin in differentiating keratinocytes of transgenic epidermis, along with characteristic alterations in their cytoarchitecture (Fig. 3 B–E). Depending on the levels of transgenic mutant keratin in skin epithelia, which vary between transgenic lines (Fig. 4) and according to the treatment regimen, these alterations can progress to cellular vacuolization and lysis (Fig. 3E). No significant alteration was detected in the hair follicle outer root sheath (data not shown), consistent with the lower levels of transgene products induced in this deeper tissue (Fig. 3B). Such epidermal lesions are not seen in the PMA-treated skin of control nontransgenic mice (Fig. 3F), in [hK6a 5′]-LacZ transgenic mice (Fig. 2), or in transgenic lines with less abundant expression of the ΔK6a-myc transgene (data not shown). Intraepidermal blisters can be produced by tape-stripping in PMA-treated skin of ΔK6a-myc transgenic mice that express the transgene at higher level, but not in similarly treated control skin (Fig. 3 H and I). Biochemical analyses of protein extracts prepared from intact and PMA-treated skin shows that the skin phenotype is directly related to the extent of mutant keratin expression relative to mouse K6 (Fig. 4).
Figure 3.
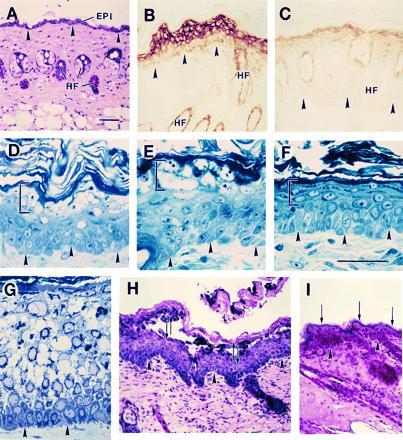
Induction of mutant keratin expression results in trauma-induced epidermolysis in mouse skin. (A) Hematoxylin/eosin stained section of intact skin in a [5.2-kb hK6a]-ΔK6a-myc mouse (21–1p line). (B) Myc antibody-stained section of paraffin-embedded, PMA-treated skin of a transgenic mouse that expresses the mutant transgene at high level (Fig. 4, lane 6; line 21–1p), whereas C shows myc staining in a mouse that expresses the transgene at low level (Fig. 4, lane 7; line 21–1 m). (D–F) Toluidine blue-stained sections of epoxy-embedded mouse skin. (D and E) PMA-treated skin of 21–1p transgenic mice; (F) PMA-treated control skin. Bracketed areas depict histological aberrations in transgenic epidermis but not in control. (G) Toluidine blue-stained section of epoxy-embedded skin from a individual suffering from severe epidermolytic hyperkeratosis. (H and I) Application of mechanical trauma by repeated tape-stripping in PMA-treated skin of a 21–1p transgenic mouse (H) and a control mouse (I). In H the opposing arrows denote the rupture of epidermis in the uppermost spinous layers, while the arrows in I denote the maintenance of the integrity of the granular layer. Arrowheads highlight the location of the dermo-epidermal interface. EPI, epidermis; HF, hair follicle. (Bars = 100 μm.)
Figure 4.
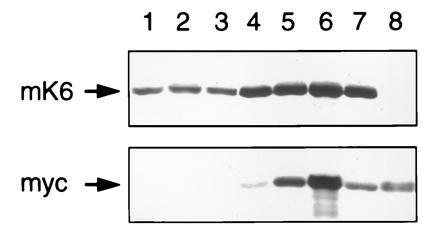
Analysis of transgenic skin protein extracts by SDS/PAGE and immunoblotting. (Upper) Mouse K6 (mK6) immunoblot. (Lower) Myc tag immunoblot. Lanes: 1–3, 15 μg protein loaded; 4–7, 5 μg protein loaded. Samples are as follows: lane 1, control mouse, intact skin; lane 2, 19–1p mouse (2.5-kb promoter), intact skin; lane 3, 21–1p mouse (5.2-kb promoter), intact skin; lane 4, 19–2m mouse (2.5-kb promoter), PMA-treated skin; lane 5, 19–1p mouse (2.5-kb promoter), PMA-treated skin; lane 6, 21–1p mouse (5.2-kb promoter), PMA-treated skin; lane 7, 21–1m transgenic mouse (5.2-kb promoter), PMA-treated skin; lane 8 (myc immunoblot only); 20 ng of purified recombinant ΔK6a-myc protein. Note the strong induction in K6 protein after PMA treatment (lanes 4–7; mK6 blot), and the variable extent of ΔK6a-myc product among various transgenic lines (myc blot).
The phenotype observed in this mouse model is very similar to that previously described for transgenic mice expressing mutated K1 or K10 protein in their skin (22, 23, 25), which had been shown to represent a phenocopy of epidermolytic hyperkeratosis and related bullous diseases (Fig. 3, compare E and G). This group of bullous diseases arises from mutations in keratin genes that are constitutively expressed in the differentiating layers of epidermis of various body sites, such as the type II K1 and K2e, and the type I K9 and K10 (18, 19). Given that the ΔK6a-myc mutant keratin preferentially accumulates in the uppermost layers of epidermis (Fig. 3B), the histopathological attributes of our mouse model appear most related to ichthyosis bullosa of Siemens (30–32). Unlike these previous mouse models, in which the fully developed cytolytic changes in epidermis and their associated secondary manifestations in skin occur shortly after birth, the [hK6a 5′]-ΔK6a-myc transgenic mice offer tight control over mutant keratin biosynthesis and susceptibility of epidermis to trauma-induced lysis. The mouse model introduced here thus offers an unique opportunity to study the initial cytoarchitectural changes that occur in epidermal keratinocytes as they accumulate a mutant keratin protein in situ, whether these changes alone induce a homeostatic response in the cells affected, and how these changes ultimately translate into an hypersensitivity to trivial mechanical trauma.
Starting from a keratin gene showing a complex regulation, we showed that it is possible to dissect out relevant regulatory elements and obtain a pattern of expression featuring inducibility and specificity toward a subpopulation of keratinocytes in epidermis and other stratified epithelia. In adult transgenic mice, the human keratin-based expression vector confers strong activity upon application of a variety of stimuli, including widely available chemicals and injury (>0.2% of total skin proteins, based on analyses of ΔK6a-myc mice). To illustrate the potential usefulness of this expression vector, we produced a mouse model that features an inducible skin blistering phenotype. Future studies on these mice should provide significant insights into the intricate relationship between mutant keratin expression and epidermal blister formation following mechanical trauma to the skin. This expression vector will also allow for an examination of the functional contribution of various types of biological molecules to the re-epithelialization of wounds. Moreover, when exploited in the context of emerging technologies such as Cre-loxP recombination (5), the tetO system (33), or synthetic ligands that can control the activity of artificial promoters (ref. 34 and references therein), this expression vector may well enable the temporally and spatially controlled induction of specific gene rearrangements in a variety of stratified epithelia including epidermis, oral mucosa, esophagus, and cornea. With some modification, this type of vector may also be useful in the design of gene therapy strategies in which local or even systemic delivery of substances can be controlled in a noninvasive fashion, e.g., from the skin (35–37). Potential applications range from the enhancement of wound repair in severely burned patients to the controlled delivery of substances capable of impacting on target tissues, whether locally or systemically after their blood-borne distribution.
Acknowledgments
We thank Ms. S. Brust and A. Chen (Johns Hopkins University Transgenic Core Facility) for the production of mice, Ms. B. Prasad for the analysis of the keratin mutant, and Dr. Q. C. Yu for providing sectioned patient skin samples. We thank Drs. D. Paulin, E. Colucci-Guyon, E. Fuchs, C. Byrne, D. Roop, K. McGowan, and C. Parent for their advice or reagents. P.A.C. is the recipient of a Junior Faculty Research Award from the American Cancer Society. K.T. is a fellow supported by the Dermatology Foundation. This work was supported by National Institutes of Heath Grant AR42047.
Footnotes
Abbreviations: K, keratin; hK6a, human keratin 6a isoform; PMA, phorbol-12-myristate-13-acetate; β-gal, β-galactosidase.
References
- 1.Capecchi M R. Sci Am. 1994;270:52–99. doi: 10.1038/scientificamerican0394-52. [DOI] [PubMed] [Google Scholar]
- 2.Rossant J, Hopkins N. Genes Dev. 1992;6:1–13. doi: 10.1101/gad.6.1.1. [DOI] [PubMed] [Google Scholar]
- 3.Furth P A, St O L, Boger H, Gruss P, Gossen M, Kistner A, Bujard H, Hennighausen L. Proc Natl Acad Sci USA. 1994;91:9302–9306. doi: 10.1073/pnas.91.20.9302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Kühn R, Schwenk F, Aguet M, Rajewsky K. Science. 1995;269:1427–1429. doi: 10.1126/science.7660125. [DOI] [PubMed] [Google Scholar]
- 5.Hogan B, Beddington R, Costantini F, Lacy E. Manipulating the Mouse Embryo: A Laboratory Manual. Plainview, NY: Cold Spring Harbor Lab. Press; 1994. [Google Scholar]
- 6.Takahashi K, Paladini R D, Coulombe P A. J Biol Chem. 1995;270:18581–18592. doi: 10.1074/jbc.270.31.18581. [DOI] [PubMed] [Google Scholar]
- 7.Ramirez A, Vidal M, Bravo A, Larcher F, Jorcano J L. Proc Natl Acad Sci USA. 1995;92:4783–4787. doi: 10.1073/pnas.92.11.4783. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Weiss R A, Eichner R, Sun T T. J Cell Biol. 1984;98:1397–1406. doi: 10.1083/jcb.98.4.1397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Paladini R D, Takahashi K, Bravo N S, Coulombe P A. J Cell Biol. 1996;132:381–397. doi: 10.1083/jcb.132.3.381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Stoler A, Kopan R, Duvic M, Fuchs E. J Cell Biol. 1988;107:427–446. doi: 10.1083/jcb.107.2.427. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Schweizer J. Molecular Biology of the Skin: The Keratinocyte. San Diego: Academic; 1993. pp. 33–78. [Google Scholar]
- 12.Li Z, Marchand P, Humbert J, Babinet C, Paulin D. Development (Cambridge, UK) 1993;117:947–959. doi: 10.1242/dev.117.3.947. [DOI] [PubMed] [Google Scholar]
- 13.Gill S R, Wong P C, Monteiro M J, Cleveland D W. J Cell Biol. 1990;111:2005–2019. doi: 10.1083/jcb.111.5.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Byrne C, Fuchs E. Mol Cell Biol. 1993;13:3176–3190. doi: 10.1128/mcb.13.6.3176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Roop D R, Cheng C K, Titterington L, Meyers C A, Stanley J R, Steinert P M, Yuspa S H. J Biol Chem. 1984;259:8037–8040. [PubMed] [Google Scholar]
- 16.Coulombe P A, Hutton M E, Vassar R, Fuchs E. J Cell Biol. 1991;115:1661–1674. doi: 10.1083/jcb.115.6.1661. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Coulombe P A, Fuchs E. J Cell Biol. 1990;111:153–169. doi: 10.1083/jcb.111.1.153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Fuchs E, Coulombe P A, Cheng J, Chan Y-M, Hutton E, Syder A, Degenstein L, Yu Q C, Letai A, Vassar R. J Invest Dermatol. 1994;103:25S–30S. doi: 10.1111/1523-1747.ep12398924. [DOI] [PubMed] [Google Scholar]
- 19.McLean W H I, Lane E B. Curr Opin Cell Biol. 1995;7:118–125. doi: 10.1016/0955-0674(95)80053-0. [DOI] [PubMed] [Google Scholar]
- 20.Coulombe P A. Curr Opin Cell Biol. 1993;5:17–29. doi: 10.1016/s0955-0674(05)80004-3. [DOI] [PubMed] [Google Scholar]
- 21.Vassar R, Coulombe P A, Degenstein L, Albers K, Fuchs E. Cell. 1991;64:365–380. doi: 10.1016/0092-8674(91)90645-f. [DOI] [PubMed] [Google Scholar]
- 22.Fuchs E, Esteves R A, Coulombe P A. Proc Natl Acad Sci USA. 1992;89:6906–6910. doi: 10.1073/pnas.89.15.6906. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Rothnagel J A, Greenhalgh D A, Wang X J, Sellheyer K, Bickenbach J R, Dominey A M, Roop D R. Arch Dermatol. 1993;129:1430–1436. [PubMed] [Google Scholar]
- 24.Lloyd C, Yu Q C, Cheng J, Turksen K, Degenstein L, Hutton E, Fuchs E. J Cell Biol. 1995;129:1329–1344. doi: 10.1083/jcb.129.5.1329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Porter R M, Leitgeb S, Melton D M, Swensson O, Eady R A J, Magin T M. J Cell Biol. 1996;132:925–936. doi: 10.1083/jcb.132.5.925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Fuchs E, Byrne C. Curr Opin Genet Dev. 1994;4:725–736. doi: 10.1016/0959-437x(94)90140-x. [DOI] [PubMed] [Google Scholar]
- 27.Hatzfeld M, Franke W W. J Cell Biol. 1985;101:1826–1841. doi: 10.1083/jcb.101.5.1826. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Blessing M, Ruther U, Franke W W. J Cell Biol. 1993;120:743–755. doi: 10.1083/jcb.120.3.743. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Albers K M, Davis F E, Perrone T N, Lee E Y, Liu Y, Vore M. J Cell Biol. 1995;128:157–169. doi: 10.1083/jcb.128.1.157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Rothnagel J A, Traupe H, Wojcik S, Huber M, Hohl D, Pittelkow M, Saeki H, Ishibashi Y, Roop D R. Nat Genet. 1994;7:485–489. doi: 10.1038/ng0894-485. [DOI] [PubMed] [Google Scholar]
- 31.McLean I, Morley S, Labe E B, Eady R, Griffiths W A, Paige D, Harper J, Higgins C, Leigh I. J Invest Dermatol. 1994;103:277–281. doi: 10.1111/1523-1747.ep12394307. [DOI] [PubMed] [Google Scholar]
- 32.Syder A J, Yu Q C, Paller A S, Giudice G, Pearson R, Fuchs E. J Clin Invest. 1994;93:1533–1542. doi: 10.1172/JCI117132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Gossen M, Freundlieb S, Bender G, Muller G, Hillen W, Bujard H. Science. 1995;268:1766–1769. doi: 10.1126/science.7792603. [DOI] [PubMed] [Google Scholar]
- 34.Ho S N, Biggar S, Spencer D, Schreiber S L, Crabtree G R. Nature (London) 1996;362:822–824. doi: 10.1038/382822a0. [DOI] [PubMed] [Google Scholar]
- 35.Greenhalgh D A, Rothnagel J A, Roop D R. J Invest Dermatol. 1994;103:63S–69S. doi: 10.1111/1523-1747.ep12399070. [DOI] [PubMed] [Google Scholar]
- 36.Fenjves E S, Smith J, Zaradic S, Taichman L B. Hum Gene Ther. 1994;5:1241–1248. doi: 10.1089/hum.1994.5.10-1241. [DOI] [PubMed] [Google Scholar]
- 37.Alexander M Y, Bidichandani S I, Cousins F M, Robinson C J, Duffie E, Akhurst R J. Hum Mol Genet. 1995;4:993–999. doi: 10.1093/hmg/4.6.993. [DOI] [PubMed] [Google Scholar]