

Abstract
Background
Bodo saltans is a free-living kinetoplastid and among the closest relatives of the trypanosomatid parasites, which cause such human diseases as African sleeping sickness, leishmaniasis and Chagas disease. A B. saltans genome sequence will provide a free-living comparison with parasitic genomes necessary for comparative analyses of existing and future trypanosomatid genomic resources. Various coding regions were sequenced to provide a preliminary insight into the bodonid genome sequence, relative to trypanosomatid sequences.
Results
0.4 Mbp of B. saltans genome was sequenced from 12 distinct regions and contained 178 coding sequences. As in trypanosomatids, introns were absent and %GC was elevated in coding regions, greatly assisting in gene finding. In the regions studied, roughly 60% of all genes had homologs in trypanosomatids, while 28% were Bodo-specific. Intergenic sequences were typically short, resulting in higher gene density than in trypanosomatids. Although synteny was typically conserved for those genes with trypanosomatid homologs, strict colinearity was rarely observed because gene order was regularly disrupted by Bodo-specific genes.
Conclusion
The B. saltans genome contains both sequences homologous to trypanosomatids and sequences never seen before. Structural similarities suggest that its assembly should be solvable, and, although de novo assembly will be necessary, existing trypanosomatid projects will provide some guide to annotation. A complete genome sequence will provide an effective ancestral model for understanding the shared and derived features of known trypanosomatid genomes, but it will also identify those kinetoplastid genome features lost during the evolution of parasitism.
Background
The Kinetoplastida (Euglenozoa) are unicellular flagellates that include the trypanosomatid parasites, most notably Trypanosoma brucei, T. cruzi and Leishmania spp. These organisms cause substantial mortality and morbidity in humans and their livestock worldwide as the causative agents of African sleeping sickness, Chagas disease and leishmaniasis respectively. Bodo saltans is a free-living heterotroph found worldwide in freshwater and marine habitats. It possesses the diagnostic kinetoplastid features, such as flagella sited within a specialised flagellar pocket, glycolytic processes confined to a dedicated organelle (the 'glycosome'), and the characteristic concentration of mitochondrial DNA at the base of the flagellum (the 'kinetoplast') [1,2]. When comparing trypanosomatid parasites with each other, or collectively with other eukaryotes, the value of B. saltans is as a non-parasitic near relative, (i.e., an 'outgroup'), that can illuminate their key evolutionary transitions. Five draft genome sequences exist for Trypanosoma spp. and four for Leishmania spp. [3-7]; these will be augmented with further strains and other non-human parasites in the coming years [8]. With such excellent comparative resources in place or in development, there is a critical need for a non-trypanosomatid outgroup. In effect, it will provide a model of the ancestral trypanosomatid to distinguish those derived parts of the parasite genomes (i.e., unique trypanosomatid adaptations) from those which are a legacy of the free-living ancestor. For instance, such a model will help to resolve whether trypanosomatids previously possessed an algal plastid from which 'plant-like' genes in trypanosomatid genomes are derived [9-11]. As a prelude to a complete B. saltans genome sequencing effort, this study sought to establish an initial understanding of the bodonid genome, its structure and content relative to the trypanosomatids.
The most recent kinetoplastid phylogeny has shown that trypanosomatid parasites are just one of many independent acquisitions of parasitism, indeed, a relatively minor component of total diversity [12-15]. Nonetheless, they are, naturally, the most important aspect of kinetoplastid diversity. Many features of their completed genome sequences emphasised the common ancestry of T. brucei, T. cruzi and Leishmania spp., especially with respect to gene repertoire and order [16], but their critical pathological differences were also evident at the genomic level. The three human parasites cause distinct diseases; their genomes contain enigmatic adaptations related to pathogenesis and immune evasion, for instance the bloodstream expression site in T. brucei from which its variant surface glycoproteins (VSG) are expressed [17,18], and surface antigen families in general [16]. Without an historical dimension, these features cannot be compared, nor understood in an evolutionary context. As it is among the closest bodonid relatives of the trypanosomatids [19], Bodo saltans is a suitable outgroup to address three principal comparative issues: i) understanding how human trypanosomatid parasites acquired their distinct pathological strategies; ii) understanding how the ancestral trypanosomatid became parasitic in terms of derived innovations (e.g., cell surfaces) and loss of genomic repertoire; iii) understanding how typical kinetoplastid features (e.g., glycosomes) evolved and how these might have been modified for parasitism.
Quite what to expect from a bodonid genome sequence is an open question. Beyond the basic kinetoplastid features named above, the biological differences between bodonids and trypanosomatids are striking. While B. saltans is a bacteriovore, especially prevalent in polluted waters or other environments with high bacterial densities [1], trypanosomatids are obligate parasites inhabiting a nutrient-rich, but ultimately hostile, host environment, and adept at exploiting their eutrophic environment to maximise proliferation and transmission. By contrast, B. saltans preys on bacterial cells [1,2] and is probably adapted for resource acquisition within its relatively oligotrophic environment. Although bodonids and trypanosomatids are all flagellates, trypanosomatids attach their single flagellum to the cell surface to generate motile force, whereas the anterior flagellum in B. saltans is modified with hair-like mastigonemes, which may assist prey location during feeding [2,20-22]. There are wider cytoskeletal differences also; the subpellicular microtubular cortex is instrumental in maintaining the numerous cell forms adopted by trypanosomatids [23], but is reduced in bodonids, (which lack complex developmental stages), to the region around the cytostome [2,24]. Perhaps most importantly for understanding the evolution of parasitism, we can expect substantial differences between trypanosomatid cell surfaces that function primarily to manipulate and frustrate the host immune response and bodonid membranes that are perhaps largely concerned with cellular homeostasis.
Rather than providing definitive answers to these questions, the preliminary sequence data presented here provides an initial insight into a few comprehensively resolved locations in the B. saltans genome, indicating what to expect from gene content and arrangement, and testing the feasibility of a complete sequence project. The sequence contigs were compared with corresponding regions in trypanosomatids (based on conserved gene order, where this existed), to examine gene content and the conservation of gene order (i.e., colinearity) and, therefore, the potential for using trypanosomatid genome sequences as scaffolds to assist assembly and annotation of the B. saltans sequence.
Results
Gene structure
Clones were selected from the B. saltans fosmid library according to random end-sequences and positive results for specific PCR probes. Inserts from 12 fosmid clones were shotgun sequenced, comprising 0.403 Mbp in total and an average size of 33.6 Kbp. Table 1 describes the composition of the 12 contigs in terms of the affinity shown by each putative coding sequence to sequence databases. 178 putative coding sequences are specified; genes could be predicted by eye because of a definite elevation in GC content in coding regions. Subsequent matches to sequence databases showed these features to be correct. The boundaries between coding and flanking regions are marked by a transition from GC-rich to AT-rich signatures; the sequences shown in Figure 1 clearly demonstrate the GC troughs that appear between coding sequences. This pattern is repeated in other contigs, as shown in subsequent figures. Gene density is high relative to corresponding regions in the L. major and T. brucei genome sequences, reflected by the consistently short intercoding sequences across all contigs (average = 377.2 bp). Figure 2 compares the gene order of one region (average interceding sequence length = 439.7 bp) with positionally orthologous regions in L. major (average = 1480.6 bp) and T. brucei (average = 1129.4 bp); this, like most fosmid inserts, contains more genes in Bodo than in trypanosomatids.
Table 1.
Description of genomic fragments sequenced in this study.
| Clone | Probe† | Length (bp) | Genes by taxonomic affinity* | GenBank accession number | ||||
| K | E | P | U | TOTAL | ||||
| 5 e 15 | Rab1 | 35803 | 4 | 2 | 0 | 7 | 13 | FJ168547 |
| 14 l 17 | α-tubulin | 38184 | 8 | 1 | 0 | 5 | 14 | FJ168548 |
| 16 k 02 | HSP70 | 32908 | 8 | 2 | 0 | 2 | 12 | FJ168549 |
| 23 g 24 | GPDH | 37591 | 11 | 1 | 0 | 7 | 19 | FJ168550 |
| 45 a 09 | 38203 | 7 | 1 | 1 | 5 | 14 | FJ168551 | |
| 45 a 12 | 41499 | 16 | 3 | 0 | 5 | 24 | FJ168552 | |
| 46 a 11 | 30067 | 8 | 3 | 1 | 3 | 15 | FJ168553 | |
| 5 m 18 | β-tubulin | 28046 | 9 | 1 | 0 | 3 | 13 | FJ168554 |
| 93 d 02 | 18199 | 6 | 2 | 0 | 2 | 10 | FJ168555 | |
| 93 e 01 | 40818 | 12 | 2 | 0 | 3 | 17 | FJ168556 | |
| 96 f 03 | 38993 | 12 | 0 | 0 | 2 | 14 | FJ168557 | |
| 96 g 09 | 31028 | 5 | 2 | 0 | 6 | 13 | FJ168558 | |
| TOTAL | 411339 | 106 | 20 | 2 | 50 | 178 | ||
Note: † Clones for which no probe is stated were selected after end-sequencing.* Genes were classified according to their affinity: kinetoplastid (K), other eukaryotic (E), prokaryotic (P) or unknown (U).
Figure 1.
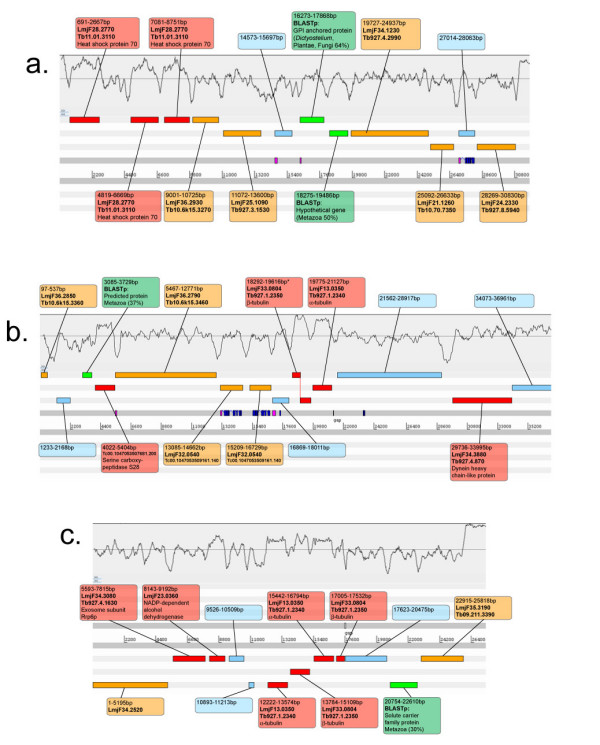
Schematic representation of three regions of the B. saltans genome sequence, as shown in the Artemis genome browser. Six reading frames are shown as parallel grey bars; scale in base-pairs. Base composition is plotted above. Putative coding sequences are shown as coloured boxes: red (homolog of trypanosomatid gene with known function), orange (homolog of hypothetical trypanosomatid gene), green (hypothetical gene with no trypanosomatid homolog but a positive functional match to a sequence database), blue (hypothetical gene with no matches to sequence databases). Labels attending these coding sequences contain the GeneDB identification numbers of homologous trypanosomatid genes where possible, or the description of homologous genes detected by BLAST comparisons (with % identity). Predicted transmembrane helices (blue) and signal peptides (purple) are shown on the DNA strands below the coding sequence. a. Clone '16k02' containing a tandem gene array of heat-shock protein 70. b. Clone '14l17' containing a tandem gene array of α- and β-tubulin. An asterisk * denotes a β-tubulin gene disrupted by a single base deletion at position 589. c. Clone '5m18' containing a second tandem gene array of α- and β-tubulin.
Figure 2.

Screenshot from the Artemis Comparison Tool (ACT), showing a 41.5 Kb fragment of B. saltans genome sequence (clone '45a12') and corresponding regions from chromosome 18 of L. major (top) and chromosome 6 of T. brucei (bottom). Key to B. saltans coding sequence annotation: 1. RNA-binding protein (homolog of Tb927.7.5380); 2. Hypothetical, no matches; 3. Serine-threonine protein kinase (Metazoa 46%, Plantae 43%); 4. Hypothetical, no matches; 5. Homolog of Tb10.61.3155; 6. Hypothetical lipase; 7. Serine-threonine protein kinase (Tb10.61.3140); 8. Homolog of Tb10.61.3130; 9. Possible ornithine decarboxylase (Bacteria 27%); 10. Dephospho-CoA kinase (Tc00.1047053511277.500); 11. Homolog of Tb10.61.3120; 12. Homolog of Tb10.61.3115; 13. Homolog of Tb10.61.3110; 14. DNAJ chaperone (Tb10.61.3100); 15. Homolog of Tb10.61.3080; 16. Homolog of Tb10.61.3070; 17. GPI-anchor transamidase (Tb10.61.3060); 18. Tubulin tyrosine ligase (Tb10.61.3050); 19. Ubiquitin-conjugating enzyme (Tb927.5.1000); 20. Homolog of Tb10.61.3040.
Gene content
Table 1 shows that 106/178 coding sequences (59.6%) are homologs of known trypanosomatid genes. The percentage nucleotide identity between bodonid and trypanosomatid proteins varies greatly; genes of known conservatism display high identity (α-tubulin, 98%; β-tubulin, 99%; HSP70, 95%; GAPDH, 81%), but on average coding sequences are 44.38% identical and the most abundant identity class is 30–39%. Hence, most orthologs in these two classes have diverged by two-thirds or more. Of those coding sequences without trypanosomatid homologs, 20 show homology with other eukaryotes, 2 are of bacterial affinity, and the remainder (28.1%) are without matches to any database, i.e., Bodo-specific. Despite the bacterial contamination inevitable in DNA preparations (see methods), we can be certain that these bacterial-type coding sequences are not artefacts because they are present in fosmid inserts otherwise composed of eukaryotic sequences, and individual sequence clones span both the bacterial-type gene and surrounding eukaryotic-type sequence. Although present in B. saltans, some of the familiar genes intensively studied in trypanosomatids are found in novel contexts. Figure 1 describes tandem gene arrays of HSP70 and tubulin, which are found in locations unlike those in trypanosomatids. An alternating tandem array containing α and β-tubulin is found in two distinct inserts (Figure 1b and 1c); α and β-tubulin isoforms contain no amino acid differences but had dissimilar (unalignable) 3' untranscribed regions.
Coding sequences without trypanosomatid homologs were compared to sequence and structural databases (see Table 2). Many of the gene products are homologous to proteins beyond the Kinetoplastida, suggesting that they are core eukaryotic proteins subsequently lost from trypanosomatids; for example, contiguous genes in Figure 1a homologous to a GPI-anchored protein in plants and fungi (16273–17686 bp) and a hypothetical gene in Metazoa (18275–19486 bp). Gene products in other regions (not shown) contain protein domains known elsewhere, for example an ABC transporter protein (clone '5 e 15', 21065 bp) and a nucleotide-sugar transporter protein (clone '45 a 12', 41227 bp), strongly indicating that these are Bodo-specific members of ubiquitous gene families. Some otherwise uncharacterised hypothetical proteins are predicted to expressed on the cell surface. The region shown in Figure 3a is notable not only for the base composition of coding regions and admixture of trypanosomatid and Bodo-specific genes mentioned above, but also for a hypothetical protein (16737–17978 bp) with 7 predicted transmembrane helices and a signal peptide. Table 2 contains other examples of Bodo-specific hypothetical genes predicted to be surface expressed, including those shown in Figure 1a (27014–28063 bp) and b (16869–18011 bp).
Table 2.
Bodo-specific hypothetical genes, with evidence of protein domains, transmembrane domains (TMH) signal peptides (SP) and affinities to sequence databases where available.
| Clone | Position | Size (bp) | Interpro match | TMH | SP | Best BLASTp match | Best taxonomic match (% amino acid identity) |
| 14 l 17 | 3085 | 645 | Oxygenase (IPR005123) | Prolyl hydroxylase | Metazoa (53%) | ||
| 16869 | 1143 | Zn-binding protein (IPR001841) | 1 | ||||
| 21613 | 7356 | Leucine-rich repeat (IPR001611) | 2 | ||||
| 16 k 02 | 14573 | 1125 | Yes | ||||
| 16273 | 1596 | Yes | GPI-anchored protein | Plantae/Fungi (64%) | |||
| 18275 | 1212 | Hypothetical protein | Metazoa (50%) | ||||
| 27014 | 1050 | 7 | Yes | ||||
| 23 g 24 | 391 | 1626 | Pleckstrin-like (IPR001849) | ||||
| 7655 | 2781 | Zinc metallopeptidase (IPR006025) | |||||
| 21361 | 23428 | EF-hand protein | Tetrahymena (47%) | ||||
| 45 a 09 | 4161 | 535 | Methyltransferase (IPR013216) | ||||
| 30026 | 1134 | Phosphoribulokinase (IPR006083) | Bacteria (50%) | ||||
| 45 a 12 | 12191 | 1695 | Lipase (IPR008265) | 1 | |||
| 31394 | 455 | Serine-Threonine protein kinase (IPR017442) | Protein kinase | Metazoa (46%) | |||
| 41227 | 264 | Nucleotide-sugar transporter (IPR007271) | 3 | Yes | |||
| 46 a 11 | 211 | 753 | Endonuclease (IPR001604) | 1 | Yes | Endonuclease | Bacteria (53%) |
| 1166 | 1416 | Thioredoxin (IPR000866) | Nucleoredoxin | Plantae (53%) | |||
| 12286 | 1092 | Fucosyltransferase | Plantae (36%) | ||||
| 14365 | 765 | Hypothetical protein | Metazoa (55%) | ||||
| 5 e 15 | 477 | 10617 | Phosphatidylinositol-4-phosphate kinase (IPR003409) | ||||
| 14913 | 1296 | Lyzosomal lipase (IPR006693) | 1 | Lipase | Metazoa (59%) | ||
| 16510 | 2199 | Serine endopeptidase (IPR001254) | Serine protease | Plantae (46%) | |||
| 21065 | 546 | ABC transporter (IPR013525) | 4 | Yes | |||
| 32153 | 2613 | 1 | |||||
| 35286 | 546 | Zinc-finger protein (IPR001841) | |||||
| 5 m 18 | 5577 | 1857 | Sodium/hydrogen exchanger (IPR006153) | 14 | Yes | Sodium/hydrogen exchanger | Metazoa (50%) |
| 17069 | 321 | Ankyrin repeat protein (IPR002110) | |||||
| 93 d 02 | 5528 | 1758 | Yes | ||||
| 7529 | 2589 | Chloride channel (IPR0014743) | 9 | Chloride channel | Metazoa (56%) | ||
| 14924 | 1788 | WD40 repeat (IPR001680) | Hypothetical protein | Plantae (56%) | |||
| 93 e 01 | 56 | 5505 | 14 | ||||
| 16439 | 1995 | Forkhead associated protein (IPR000253) | |||||
| 18802 | 4260 | 8 | Yes | ||||
| 24535 | 855 | Protein tyrosine phosphatase (IPR000340) | Protein phosphatase | Plantae (56%) | |||
| 96 g 09 | 10627 | 2559 | PDZ protein binding motif (IPR001478) | Yes | |||
| 16737 | 1242 | 7 | Yes | ||||
| 26182 | 948 | Leucine-rich repeat kinase (IPR001611) | Receptor-type protein kinase | Plantae (54%) | |||
| 27597 | 483 | Leucine-rich repeat kinase (IPR001611) | Receptor-type protein kinase | Plantae (34%) |
Figure 3.
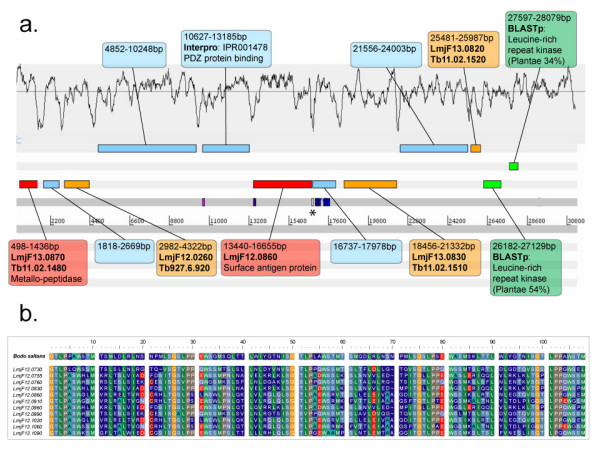
a. Schematic representation of a 32.9 Kb fragment of B. saltans genome sequence (clone 96g09) in the Artemis genome browser. The asterisk * denotes a physical sequence gap of uncertain length. b. Protein sequence alignment showing the repetitive region of 11 'surface antigen' proteins from L. major and their putative homolog from B. saltans, shown in panel a. (13440–16665 bp).
Colinearity
The extent of conserved gene order, or colinearity, between bodonid and trypanosomatid genome sequences was assessed using the Artemis Comparison Tool (ACT, see methods). One region of excellent colinearity is shown in Figure 2 and, despite disruption by some eukaryotic genes not seen in trypanosomatids, this contig corresponds unmistakably with chromosome 18 in L. major and chromosome 10 in T. brucei. Conversely, the presence of so many non-trypanosomatid genes meant that colinearity disappears entirely in some locations, as shown in Figure 1. Across the 12 genomic regions however, both patterns were atypical; most regions shows brief patches of colinearity, perhaps 2 or 3 genes with conserved synteny, set among larger regions of Bodo-specific genes or homologs to trypanosomatid genes from elsewhere in the genome. In this sense, the sequence presented in Figure 3 is representative because several coding sequences are homologs of trypanosomatid genes on chromosomes 13 (L. major) and 11 (T. brucei); these are roughly colinear but the order is disrupted by genes present on other chromosomes or by Bodo-specific genes.
Discussion
In this study, various locations in B. saltans genome, amounting to ~0.4 Mbp, were sequenced. Assuming that the bodonid genome is approximately the same size as a trypanosomatid haploid genome, i.e., 35–55 Mbp [16], these sequences comprise ~1% of the complete genome sequence, which will therefore contain roughly 14,000 genes. The success and utility of a B. saltans genome project will depend on its relationship with existing trypanosomatid genome sequences. This study shows that coding regions of the B. saltans genome share several structural features with trypanosomatids, indicating that the project is both feasible and likely to provide a useful comparative resource. Putative B. saltans genes lack introns, as in most trypanosomatid genes [3-5]. They display a conspicuous elevation in GC content, which will greatly assist gene finding. No evidence of strand-switching was observed in B. saltans, corroborating the view that it operates polycistronic transcription [25], i.e., transcription of many contiguous loci within a single nascent transcript [26-28], which is subsequently trans-spliced and polyadenylated to produce mature mRNA, as in trypanosomatids [29-33].
Although the arrangement of coding regions along the bodonid chromosome may be conserved with trypanosomatids, it is clear that gene order was not. The extent of conserved synteny, or rather colinear gene order, between bodonid and trypanosomatid genomes is of particular importance to the assembly of any B. saltans genome sequence. The coding regions presented here indicate that trypanosomatid genome sequences will be of limited value in the global assembly of a B. saltans genome sequence. Strict colinearity was not normally observed, if only because of the large number of Bodo-specific genes interposed between trypanosomatid homologs. Colinearity tended to persist over a distance of 3–5 genes, although some regions displayed conspicuous conservation (e.g., Figure 2), while others showed none at all (e.g., Figure 1). Therefore, this initial exploration of the B. saltans genome demonstrates that it should be possible to resolve a complete genome sequence, but, while the existing trypanosomatid resources will provide some useful guides for annotation, they could not be used as scaffolds for assembly, which should proceed de novo.
The purpose of a completed B. saltans genome sequence would be for understanding the evolution of trypanosomatid genome sequences. The mixture of familiar and novel features in the regions sequenced here indicates the value of a bodonid genome sequence in distinguishing trypanosomatid characters inherited from free-living ancestors (and still shared with them) from characters evolved since the origin of trypanosomatids. Hence, the first application would be in determining which parts of the trypanosomatid genome reflect the genomic legacy inherited from free-living ancestors, and show how they have been co-opted and modified for parasitism. Bodonid and trypanosomatid cells share various structural features, principally those that characterise kinetoplastid cells. Bodonids arrange their mitochondrial DNA in kinetoplasts, although their position within the cell differs from trypanosomatids [1], and conduct their glycolytic pathways within a dedicated organelle (the glycosome) [2]. Bodonids construct their flagella in a similar manner to trypanosomatids, but deploy them very differently [1]. While B. saltans uses one flagellum for movement and another for feeding, trypanosomatids flagella perform their motility function within the context of their sophisticated cell forms.
One might expect these structural similarities to be reflected at the genomic level. α- and β-tubulin, the proteins that facilitate the development of flagella in trypanosomatids, are known to be arranged in tandem gene arrays, with an alternating, heterotypic α-β array in Trypanosoma spp. and distinct, monotypic α and β arrays in Leishmania spp. [34-37]. Bodonids were shown to share the alternating conformation, suggesting that Leishmania spp. and their relatives had abolished the ancestral locus and evolved novel genomic repertoires [38]. However, two B. saltans regions containing tubulin in this study show that modification of tubulin repertoire has also occurred in Trypanosoma, since neither of the α-β arrays in B. saltans was found at the genomic position occupied in trypanosomes. This demonstrates the utility of the B. saltans genome in resolving the evolutionary causes of structural or compositional differences between trypanosomatid genomes.
The second application of a B. saltans genome sequence would be to identify which components of the free-living legacy have been lost from trypanosomatids, and therefore, how reductive genome evolution has contributed to the parasite genomes. Table 2 describes many predicted proteins identified in B. saltans that have no trypanosomatid homologs. Among these, mostly Bodo-specific, genes are membrane transporters, various protein kinases, and other proteins containing domains commonly associated with cell surfaces. These and other Bodo-specific proteins must include those metabolism pathways, intracellular transport, cellular signalling and subcellular structures that exist in free-living kinetoplastids, but which have been deleted during the evolution of parasitism. Many of these proteins will be widespread among eukaryotic lineages, as is evident in Table 2; yet we should also expect to encounter a considerable genetic repertoire unique to the Kinetoplastida and so entirely new.
Having identified those features of trypanosomatid genomes that reflect their free-living ancestry, a B. saltans genome sequence would also reveal the additions to each parasite genome; structures derived from existing genes and co-opted for novel uses, and genuinely novel genes involved in parasite-specific adaptations. These enigmatic genes include the numerous and diverse families of surface glycoprotein that form the protective coats around trypanosomatid parasites. T. brucei, T. cruzi and L. major each display highly derived and complex surface coats to frustrate host immunity, yet they differ in structure and substance and it is not known how each acquired its distinct solution to their common problem. Understanding the origins of these surface architectures will only be achieved with an historical perspective; one principal objective of a B. saltans genome project would be to identify the precursors of proteins such as VSG in T. brucei, mucins and trans-sialidase in T. cruzi, and proteophosphoglycans in Leishmania spp. (amongst others). A glimpse of this potential is seen in Figure 3, which includes a predicted protein with a complex 24 amino acid repeat (13440–16655 bp). The protein had a high affinity (42% amino acids identical) with a gene family on chromosome 12 in Leishmania spp., (currently annotated as 'surface antigens'), and a more distant affinity with proteophosphoglycans. Figure 3b shows a sequence alignment of the repeat domain from the B. saltans protein and its leishmanial homologs, where the level of amino acid identity rises to 50%.
Conclusion
Thorough sequencing of a few locations in the B. saltans genome has revealed clear similarities with trypanosomatids, but has also shown that trypanosomatid genome sequences will not be effective guides for any complete bodonid project, due to significant differences in content and gene order. This mixture of familiar and novel features suggests that B. saltans will indeed provide an effective outgroup for comparisons of trypanosomatid parasites, and, as with the evolution of tubulin repertoire, the historical perspective to understand which aspects of trypanosomatid biology have been retained from their common ancestry, which have been lost, and what has been uniquely derived since.
Methods
Fosmid library preparation
A freshwater strain of Bodo saltans ('Lake Konstanz'; courtesy of Dr Julius Lukes, University of South Bohemia, Czech Republic), was cultured in tap water in the presence of environmental bacteria. Bodonid cells were concentrated through a gentle centrifugation step (3,000 g for 2 minutes). Genomic DNA was prepared after resuspension of the pellet using phenol-chloroform extraction. This preparation contained a residuum of bacterial DNA. Genomic DNA was sheared and blunt-end repaired before being electrophoresed on a CHEF gel, from which the 25–40 kb band region was excised. The DNA was electroluted from the gel slice and ligated into a pCC1 fosmid vector (CopyControl Fosmid Kit; Epicentre Biotechnologies). Fosmid ligations were packaged into lambda bacteriophage (Gigapack XL2 Packaging Extract; Stratagene) and used to transform XL2-Blue MRF ultracompetent cells. Positive transformants were picked from chloramphenicol plates and cultured under drug selection. The B. saltans genome library contained 9600 individual clones (approximately 300 Mb).
Clone selection and sequencing
As B. saltans cannot be grown axenically, 96 fosmid inserts were end-sequenced to examine the relative contributions of bodonid and bacterial DNA to the library. 16% of clones had end-sequences with affinity for eukaryotic coding sequences when compared to databases. Another 19% of clones had matches to bacterial sequences. Hence, although a larger proportion of end-sequences may have been genuine bodonid non-coding sequences (without representation in sequence databases), the library included a considerable, perhaps equal, component of bacterial DNA. Seven clones with positive end-sequence matches to trypanosomatid genes were sequenced in full (see below). Filters were prepared for the library by spotting bacterial culture on to a charged nylon membrane (Nytran Supercharge membrane: Schleicher and Schuell Bioscience) and lysing the cells; denaturation and fixation of the fosmid DNA produced a filter representing 8,832 genome fragments of 25–40 Kb. The filter was probed for five known B. saltans genes using radiolabelled PCR products, generated with the following primers: α-tubulin (F: AACGCSTGCTGGGAGYTGT; R: GTTGATRCCGCACTTGAAGCC; 1 kb), β-tubulin (F: AACCAGATCGGCTCNAAGTT; R: GATGTTGTTSGGGATCCACTC; 1 kb), Glyceraldehyde-6-phosphate dehydrogenase (F: CGGTCAAGGTAGGCATCAAC; R: TTGGGAAGGTTGTTCTGGAG; 800 bp), Heat shock protein 70 (F: TTCAAGAACGACCAGGTTGA; R: ACCAAGTCCGGCAACAATAG; 1.4 kb) and Rab1 (F: TTTGACAACCGBTACAAGGC; R: CCTTTGCGGACGTCTCAAAGTA; 500 bp). Clones corresponding to positive spots on the filter were cultured, and purified fosmid DNA was fully sequenced using a shotgun sequencing method to approximately 8× coverage.
Assembly and analysis
Fosmid inserts were assembled using Phrap [39] and arranged within Gap4 [40]. PCR products were generated to close residual gaps between finished contigs. Finished sequence was annotated within Artemis [41,42] and coding sequences were initially defined by eye. Whole sequences were compared to EMBL sequence databases using both BLASTn and BLASTp algorithms. Coding sequences were scrutinised for possible transmembrane helices and signal peptides using TMHMM [43] and SignalP [44] respectively. Each coding sequence was checked for known protein domains using all options within the Interproscan suite [45]. Conserved synteny was assessed by aligning B. saltans contigs with T. brucei and L. major chromosomal regions using ACT [46] and existing trypanosomatid sequences downloaded from the GeneDB website [7].
Abbreviations
ACT: Artemis comparison tool; PCR: Polymerase chain reaction; BLAST: Basic local alignment search tool; CDS: Coding sequence; IGS: Intergenic sequence; CHEF: Clamped homogeneous electric field.
Authors' contributions
APJ prepared and probed the genomic library, assembled and annotated sequence contigs, analysed the data and prepared the manuscript. MAQ oversaw DNA preparation and sequencing. MB provided funds for sequencing and editorial guidance in producing the manuscript.
Acknowledgments
Acknowledgements
B. saltans culture was kindly collected and donated by Dr Julius Lukes. APJ is a Wellcome Trust Sanger Institute Postdoctoral Research Fellow. All sequencing was carried out by the Pathogen Genomics Group at the Wellcome Trust Sanger Institute. The manuscript benefited from editorial comments and the criticism of two anonymous referees.
Contributor Information
Andrew P Jackson, Email: aj4@sanger.ac.uk.
Michael A Quail, Email: mq1@sanger.ac.uk.
Matthew Berriman, Email: mb4@sanger.ac.uk.
References
- Vickerman K, Preston TM. Comparative cell biology of the kinetoplastid flagellates. In: Lumsden WHR, Evans DA, editor. Biology of the Kinetoplastida. London: Academic Press; 1976. pp. 35–130. [Google Scholar]
- Vickerman K. Organization of the Bodonid Flagellates. In: Patterson DJ, Larsen J, editor. The Biology of Free-living Heterotrophic Flagellates. Oxford: Clarendon Press, Oxford; 1991. pp. 159–176. [Google Scholar]
- Berriman M, Ghedin E, Hertz-Fowler C, Blandin G, Renauld H, Bartholomeu DC, Lennard NJ, Caler E, Hamlin NE, Haas B, Bohme U, Hannick L, Aslett MA, Shallom J, Marcello L, Hou L, Wickstead B, Alsmark UC, Arrowsmith C, Atkin RJ, Barron AJ, Bringaud F, Brooks K, Carrington M, Cherevach I, Chillingworth TJ, Churcher C, Clark LN, Corton CH, Cronin A, Davies RM, Doggett J, Djikeng A, Feldblyum T, Field MC, Fraser A, Goodhead I, Hance Z, Harper D, Harris BR, Hauser H, Hostetler J, Ivens A, Jagels K, Johnson D, Johnson J, Jones K, Kerhornou AX, Koo H, Larke N, Landfear S, Larkin C, Leech V, Line A, Lord A, Macleod A, Mooney PJ, Moule S, Martin DM, Morgan GW, Mungall K, Norbertczak H, Ormond D, Pai G, Peacock CS, Peterson J, Quail MA, Rabbinowitsch E, Rajandream MA, Reitter C, Salzberg SL, Sanders M, Schobel S, Sharp S, Simmonds M, Simpson AJ, Tallon L, Turner CM, Tait A, Tivey AR, Van Aken S, Walker D, Wanless D, Wang S, White B, White O, Whitehead S, Woodward J, Wortman J, Adams MD, Embley TM, Gull K, Ullu E, Barry JD, Fairlamb AH, Opperdoes F, Barrell BG, Donelson JE, Hall N, Fraser CM, Melville SE, El-Sayed NM. The genome of the African trypanosome Trypanosoma brucei. Science. 2005;309:416–422. doi: 10.1126/science.1112642. [DOI] [PubMed] [Google Scholar]
- Ivens AC, Peacock CS, Worthey EA, Murphy L, Aggarwal G, Berriman M, Sisk E, Rajandream MA, Adlem E, Aert R, Anupama A, Apostolou Z, Attipoe P, Bason N, Bauser C, Beck A, Beverley SM, Bianchettin G, Borzym K, Bothe G, Bruschi CV, Collins M, Cadag E, Ciarloni L, Clayton C, Coulson RMR, Cronin A, Cruz AK, Davies RM, De Gaudenzi J, Dobson DE, Duesterhoeft A, Fazelina G, Fosker N, Frasch AC, Fraser A, Fuchs M, Gabel C, Goble A, Goffeau A, Harris D, Hertz-Fowler C, Hilbert H, Horn D, Huang YT, Klages S, Knights A, Kube M, Larke N, Litvin L, Lord A, Louie T, Marra M, Masuy D, Matthews K, Michaeli S, Mottram JC, Muller-Auer S, Munden H, Norbertczak H, Oliver K, O'Neil S, Pentony M, Pohl TM, Price C, Purnelle B, Quail MA, Rabbinowitsch E, Reinhardt R, Rieger M, Rinta J, Robben J, Robertson L, Ruiz JC, Rutter S, Saunders D, Schafer M, Schein J, Schwartz DC, Seeger K, Seyler A, Sharp S, Shin H, Sivam D, Squares R, Squares S, Tosato V, Vogt C, Volckaert G, Wambutt R, Warren T, Wedler H, Woodward J, Zhou SG, Zimmermann W, Smith DF, Blackwell JM, Stuart KD, Barrell B, Myler PJ. The genome of the kinetoplastid parasite, Leishmania major. Science. 2005;309:436–442. doi: 10.1126/science.1112680. [DOI] [PMC free article] [PubMed] [Google Scholar]
- El-Sayed NM, Myler PJ, Bartholomeu DC, Nilsson D, Aggarwal G, Tran AN, Ghedin E, Worthey EA, Delcher AL, Blandin G, Westenberger SJ, Caler E, Cerqueira GC, Branche C, Haas B, Anupama A, Arner E, Aslund L, Attipoe P, Bontempi E, Bringaud F, Burton P, Cadag E, Campbell DA, Carrington M, Crabtree J, Darban H, da Silveira JF, de Jong P, Edwards K, Englund PT, Fazelina G, Feldblyum T, Ferella M, Frasch AC, Gull K, Horn D, Hou L, Huang Y, Kindlund E, Klingbeil M, Kluge S, Koo H, Lacerda D, Levin MJ, Lorenzi H, Louie T, Machado CR, McCulloch R, McKenna A, Mizuno Y, Mottram JC, Nelson S, Ochaya S, Osoegawa K, Pai G, Parsons M, Pentony M, Pettersson U, Pop M, Ramirez JL, Rinta J, Robertson L, Salzberg SL, Sanchez DO, Seyler A, Sharma R, Shetty J, Simpson AJ, Sisk E, Tammi MT, Tarleton R, Teixeira S, Van Aken S, Vogt C, Ward PN, Wickstead B, Wortman J, White O, Fraser CM, Stuart KD, Andersson B. The genome sequence of Trypanosoma cruzi, etiologic agent of Chagas disease. Science. 2005;309:409–415. doi: 10.1126/science.1112631. [DOI] [PubMed] [Google Scholar]
- Peacock CS, Seeger K, Harris D, Murphy L, Ruiz JC, Quail MA, Peters N, Adlem E, Tivey A, Aslett M, Kerhornou A, Ivens A, Fraser A, Rajandream MA, Carver T, Norbertczak H, Chillingworth T, Hance Z, Jagels K, Moule S, Ormond D, Rutter S, Squares R, Whitehead S, Rabbinowitsch E, Arrowsmith C, White B, Thurston S, Bringaud F, Baldauf SL, Faulconbridge A, Jeffares D, Depledge DP, Oyola SO, Hilley JD, Brito LO, Tosi LR, Barrell B, Cruz AK, Mottram JC, Smith DF, Berriman M. Comparative genomic analysis of three Leishmania species that cause diverse human disease. Nat Genet. 2007;39:839–47. doi: 10.1038/ng2053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wellcome Trust Sanger Institute, Pathogen Sequencing Unit 'GeneDB' Interface http://www.genedb.org
- Buck G, Berriman M, Donelson J, El-Sayed N, Kissinger J, Simpson L, Tait A, Teixeira M, Beverley S. Pathogenomics of Trypanosomatid Parasites (White Paper) 2007. http://www.genome.gov/26525388
- Hannaert V, Saavedra E, Duffieux F, Szikora JP, Rigden DJ, Michels PA, Opperdoes FR. Plant-like traits associated with metabolism of Trypanosoma parasites. Proc Natl Acad Sci USA. 2003;100:1067–71. doi: 10.1073/pnas.0335769100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bodył A, Mackiewicz P. Were class C iron-containing superoxide dismutases of trypanosomatid parasites initially imported into a complex plastid? A hypothesis based on analyses of their N-terminal targeting signals. Parasitology. 2008;135:1101–10. doi: 10.1017/S0031182008004642. [DOI] [PubMed] [Google Scholar]
- Leander BS. Did trypanosomatid parasites have photosynthetic ancestors? Trends Microbiol. 2004;12:251–8. doi: 10.1016/j.tim.2004.04.001. [DOI] [PubMed] [Google Scholar]
- Dolezel D, Jirků M, Maslov DA, Lukes J. Phylogeny of the bodonid flagellates (Kinetoplastida) based on small-subunit rRNA gene sequences. Int J Syst Evol Microbiol. 2000;50:1943–51. doi: 10.1099/00207713-50-5-1943. [DOI] [PubMed] [Google Scholar]
- Simpson AG, Lukes J, Roger AJ. The evolutionary history of kinetoplastids and their kinetoplasts. Mol Biol Evol. 2002;19:2071–83. doi: 10.1093/oxfordjournals.molbev.a004032. [DOI] [PubMed] [Google Scholar]
- Simpson AG, Gill EE, Callahan HA, Litaker RW, Roger AJ. Early evolution within kinetoplastids (Euglenozoa), and the late emergence of trypanosomatids. Protist. 2004;155:407–22. doi: 10.1078/1434461042650389. [DOI] [PubMed] [Google Scholar]
- Simpson AG, Stevens JR, Lukes J. The evolution and diversity of kinetoplastid flagellates. Trends Parasitol. 2006;22:168–74. doi: 10.1016/j.pt.2006.02.006. [DOI] [PubMed] [Google Scholar]
- El-Sayed NM, Myler PJ, Blandin G, Berriman M, Crabtree J, Aggarwal G, Caler E, Renauld H, Worthey EA, Hertz-Fowler C, Ghedin E, Peacock C, Bartholomeu DC, Haas BJ, Tran AN, Wortman JR, Alsmark UC, Angiuoli S, Anupama A, Badger J, Bringaud F, Cadag E, Carlton JM, Cerqueira GC, Creasy T, Delcher AL, Djikeng A, Embley TM, Hauser C, Ivens AC, Kummerfeld SK, Pereira-Leal JB, Nilsson D, Peterson J, Salzberg SL, Shallom J, Silva JC, Sundaram J, Westenberger S, White O, Melville SE, Donelson JE, Andersson B, Stuart KD, Hall N. Comparative genomics of trypanosomatid parasitic protozoa. Science. 2005;309:404–9. doi: 10.1126/science.1112181. [DOI] [PubMed] [Google Scholar]
- Pays E. The variant surface glycoprotein as a tool for adaptation in African trypanosomes. Microbes Infect. 2006;8:930–937. doi: 10.1016/j.micinf.2005.10.002. [DOI] [PubMed] [Google Scholar]
- Taylor JE, Rudenko G. Switching trypanosome coats: what's in the wardrobe? Trends Genet. 2006;22:614–620. doi: 10.1016/j.tig.2006.08.003. [DOI] [PubMed] [Google Scholar]
- Heyden S von der, Cavalier-Smith T. Culturing and environmental DNA sequencing uncover hidden kinetoplastid biodiversity and a major marine clade within ancestrally freshwater Neobodo designis. Int J Syst Evol Microbiol. 2005;55:2605–21. doi: 10.1099/ijs.0.63606-0. [DOI] [PubMed] [Google Scholar]
- Karpov SA, Zhukov BF. Prosteishie aktivnogo ila (Protozoa of the Active Silt) Leningrad: Nauka; 1983. Ultrathin Structure of Pleuromonas jaculans Perty (Kinetoplastida, Zoomastigophorea) pp. 53–156. [Google Scholar]
- Mitchell GC, Baker JH, Sleigh MA. Feeding of a freshwater flagellate, Bodo saltans, on diverse bacteria. J Protozool. 1988;35:219–222. [Google Scholar]
- Jezbera J, Horòák K, Šimek K. Food selection by bacterivorous protists: insight from the analysis of the food vacuole content by means of fluorescence in situ hybridization. FEMS Microbiol Ecol. 2005;52:351–363. doi: 10.1016/j.femsec.2004.12.001. [DOI] [PubMed] [Google Scholar]
- Gull K. The cytoskeleton of Trypanosomatid Parasites. Annu Rev Microbiol. 1999;53:629–655. doi: 10.1146/annurev.micro.53.1.629. [DOI] [PubMed] [Google Scholar]
- Brugerolle G, Lom J, Nohynkova E, Joyon L. Comparison et evolution des structures cellulaire chez plusiers espèces de Bodonidés et Crytobiidés appartenant aux genres Bodo, Cryptobia et Trypanoplasma (Kinetoplastida, Mastigophora) Protistologica. 1979;15:197–221. [Google Scholar]
- Santana DM, Lukes J, Sturm NR, Campbell DA, et al. Two sequence classes of kinetoplastid 5S ribosomal RNA gene revealed among bodonid spliced leader RNA gene arrays. FEMS Microbiol Lett. 2001;204:233–237. doi: 10.1111/j.1574-6968.2001.tb10890.x. [DOI] [PubMed] [Google Scholar]
- Imboden MA, Laird PW, Affolter M, Seebeck T. Transcription of the intergenic regions of the tubulin gene cluster of Trypanosoma brucei: evidence for a polycistronic transcription unit in a eukaryote. Nucleic Acids Res. 1987;15:7357–68. doi: 10.1093/nar/15.18.7357. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Flinn HM, Smith DF. Genomic organisation and expression of a differentially-regulated gene family from Leishmania major. Nucleic Acids Res. 1992;20:755–62. doi: 10.1093/nar/20.4.755. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wong S, Morales TH, Neigel JE, Campbell DA. Genomic and transcriptional linkage of the genes for calmodulin, EF-hand 5-protein, and ubiquitin extension protein-52 in Trypanosoma brucei. Mol Cell Biol. 1993;13:207–216. doi: 10.1128/mcb.13.1.207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Van Hamme L, Pays E. Control of gene expression in trypanosomes. Microbiol Rev. 1995;59:223–240. doi: 10.1128/mr.59.2.223-240.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stiles JK, Hicock PI, Shah PH, Meade JC. Genomic organization, transcription, splicing and gene regulation in Leishmania. Ann Trop Med Parasitol. 1999;93:781–807. doi: 10.1080/00034989957781. [DOI] [PubMed] [Google Scholar]
- Campbell DA, Thomas S, Sturm NR. Transcription in kinetoplastid protozoa: why be normal? Microbes Infect. 2003;5:1231–1240. doi: 10.1016/j.micinf.2003.09.005. [DOI] [PubMed] [Google Scholar]
- Myler PJ, Beverley SM, Cruz AK, Dobson DE, Ivens AC, McDonagh PD, Madhubala R, Martinez-Calvillo S, Ruiz JC, Saxena A, Sisk E, Sunkin SM, Worthey E, Yan S, Stuart KD. The Leishmania genome project: new insights into gene organization and function. Med Microbiol Immunol. 2001;190:9–12. doi: 10.1007/s004300100070. [DOI] [PubMed] [Google Scholar]
- Monnerat S, Martinez-Calvillo S, Worthey E, Myler PJ, Stuart KD, Fasel N. Genomic organization and gene expression in a chromosomal region of Leishmania major. Mol Biochem Parasitol. 2004;134:233–43. doi: 10.1016/j.molbiopara.2003.12.004. [DOI] [PubMed] [Google Scholar]
- Thomashow LS, Milhausen M, Rutter WJ, Agabian N. Tubulin genes are tandemly linked and clustered in the genome of Trypanosoma brucei. Cell. 1983;32:35–43. doi: 10.1016/0092-8674(83)90494-4. [DOI] [PubMed] [Google Scholar]
- Seebeck T, Whittaker PA, Imboden MA, Hardman N, Braun R. Tubulin genes of Trypanosoma brucei: A tightly clustered family of alternating genes. Proc Natl Acad Sci USA. 1983;80:4634–4638. doi: 10.1073/pnas.80.15.4634. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maingon R, Gerke R, Rodriguez M, Urbina J, Hoenicka J, Negri S, Aguirre T, Nehlin J, Knapp T, Crampton J. The tubulin genes of Trypanosoma cruzi. Eur J Biochem. 1988;71:285–291. doi: 10.1111/j.1432-1033.1988.tb13788.x. [DOI] [PubMed] [Google Scholar]
- Das S, Adhya S. Organization and chromosomal localization of beta-tubulin genes in Leishmania donovani. J Biosci. 1990;15:239–248. [Google Scholar]
- Jackson AP, Vaughan S, Gull K. Evolution of tubulin gene arrays in Trypanosomatid parasites: genomic restructuring in Leishmania. BMC Genomics. 2006;7:261. doi: 10.1186/1471-2164-7-261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Phrap sequence assembly program http://www.phrap.org/phredphrapconsed.html
- Gap4: genome assembly program http://staden.sourceforge.net/manual/gap4_unix_2.html
- Rutherford K, Parkhill J, Crook J, Horsnell T, Rice P, Rajandream MA, Barrell B. Artemis: sequence visualization and annotation. Bioinformatics. 2000;16:944–5. doi: 10.1093/bioinformatics/16.10.944. [DOI] [PubMed] [Google Scholar]
- Berriman M, Rutherford K. Viewing and annotating sequence data with Artemis. Brief Bioinform. 2003;4:124–32. doi: 10.1093/bib/4.2.124. [DOI] [PubMed] [Google Scholar]
- TMHMM Server v. 2.0: Prediction of transmembrane helices in proteins http://www.cbs.dtu.dk/services/TMHMM/
- Emanuelsson O, Brunak S, von Heijne G, Nielsen H. Locating proteins in the cell using TargetP, SignalP, and related tools. Nature Protocols. 2007;2:953–971. doi: 10.1038/nprot.2007.131. [DOI] [PubMed] [Google Scholar]
- Mulder N, Apweiler R. InterPro and InterProScan: tools for protein sequence classification and comparison. Methods Mol Biol. 2007;396:59–70. doi: 10.1007/978-1-59745-515-2_5. [DOI] [PubMed] [Google Scholar]
- Carver TJ, Rutherford KM, Berriman M, Rajandream MA, Barrell BG, Parkhill J. ACT: the Artemis Comparison Tool. Bioinformatics. 2005;21:3422–3. doi: 10.1093/bioinformatics/bti553. [DOI] [PubMed] [Google Scholar]