

Abstract
To study retinal immunity in a defined system, a CD4+ T cell receptor (TCR) transgenic (Tg) mouse line (βgalTCR) specific for β-galactosidase (βgal) was created and used with Tg mice that expressed βgal in retinal photoreceptor cells (arrβgal mice). Adoptive transfer of resting βgalTCR T cells, whether naive or antigen-experienced, into arrβgal mice did not induce retinal autoimmune disease (experimental autoimmune uveoretinitis, EAU), and gave no evidence of antigen recognition. Generation of βgalTCR T cells in arrβgal mice by use of bone marrow grafts, or double Tg mice, also gave no retinal disease or signs of antigen recognition. Arrβgal mice were also resistant to EAU induction by adoptive transfer of in vitro activated βgalTCR T cells, even though the T cells were pathogenic if the βgal was expressed elsewhere. In vitro manipulations to increase T cell pathogenicity prior to transfer did not result in EAU. The only strategy that induced a high frequency of severe EAU was transfer of naive, CD25-depleted, βgalTCR T cells into lymphopenic arrβgal recipients, implicating regulatory T cells (Tregs) in the T cell inoculum, and in the recipients, in the resistance to EAU. Surprisingly, activation of the CD25-depleted βgalTCR T cells prior to transfer into the lymphopenic recipients reduced EAU. Together, the results suggest that endogenous regulatory mechanisms, as well as peripheral induction of Tregs, play a role in the protection from EAU.
Keywords: Autoimmunity, Tolerance/Suppression, Neuroimmunology
Introduction
T cell recognition of central nervous system (CNS)3 antigens (Ag) induces both tolerance that reinforces CNS immune privilege, and immunopathogenesis, when immune privilege fails to protect from autoimmune disease. The mechanisms that control the outcome of T cell recognition of CNS Ags form the basis for maintenance of peripheral tolerance in CNS tissues (1). The retina is a distinct subset of CNS tissue with an extraordinary concentration of tissue-specific proteins for visual transduction, the absence of meninges, and a lack of lymphatic drainage, which distinguish it from brain and spinal cord (2, 3).
Retinal immune privilege is maintained by several activities. Aire-related mechanisms are important, as aire-driven expression of retinal interphotoreceptor retinoid-binding protein (IRBP) affected pathogenesis in murine experimental autoimmune uveoretinitis (EAU) (4, 5). A positive correlation between mRNA transcripts of photoreceptor cell Ags in the thymus and resistance to EAU has been reported for rodents (6, 7). Negative thymic selection of T cells specific for retinal Ag has been observed (8-10), as has positive selection of retinal-protective regulatory T cells (Tregs) (11). Induction of EAU was most potent when heterologous Ags or non-immunodominant epitopes of retinal Ags were used for immunizations (12), suggesting that endogenous photoreceptor cell Ags induced partial self-tolerance. Immunological ignorance of retinal Ags has been suggested from several studies (13, 14), and it has been proposed that the lack of retinal self-Ag in draining lymph nodes (LN) can compromise the generation of peripheral tolerance against autoreactive T cells, making the retina more vulnerable to autoimmune disease (10). This concept is supported by studies testing the consequences of peripheral expression of retinal Ag via retroviral transduction of bone marrow grafts (15, 16), by extraocular transgenic (Tg) expression using a class II MHC promoter or collagen promoter (13, 17), and by extraocular DNA vaccination (18). These studies showed that even exceptionally low levels of extra-retinal Ag expression yielded animals whose susceptibility to EAU was lost, while other measures of the immune response were little changed.
Given the breadth of activities working to maintain immune privilege, T cells from T cell receptor transgenic (TCR-Tg) mice that recognize either endogenous or neo-self Ags will be crucial in advancing our understanding of the multifaceted processes of self-tolerance and autoimmunity. Of particular relevance are TCR-Tg mice specific to endogenous or Tg Ags expressed within the retina associated with EAU (9, 10). Using mice that express hen egg lysozyme (HEL) on retina-specific promoters, in conjunction with HEL-specific 3A9 TCR-Tg T cells, a high incidence of severe, spontaneous EAU was found in the double Tg mice, despite extensive thymic deletion (9, 10). To further understand the mechanisms and conditions of T cell recognition of retinal Ags leading to either tolerance or immunopathology, we created TCR-Tg mice (βgalTCR mice) using the TCR from our CD4+, βgal-specific, 3E9 T cell clone (19) for use in experiments with Tg mice that express Escherichia coli β-galactosidase (βgal) as a neo-self Ag specifically in photoreceptor cells (arrβgal mice). Unlike the HEL/3A9 model, we found the retina to be highly resistant to disease mediated by the βgalTCR T cells even in arrβgal × βgalTCR double Tg mice, but susceptible if the βgal was expressed in brain astrocytes. The only potent enhancer of retinal photoreceptor autoimmune disease was lymphopenia-induced proliferation (LIP) induced by adoptive transfer of CD25-depleted, naive βgalTCR T cells into arrβgal mice on the Rag(-/-) background. Tregs present in, or generated from, the T cell inoculum contributed to the resistance to EAU in arrβgal mice.
Materials and Methods
Generation and analysis of βgalTCR transgenic mice
Generation of the CD4+ 3E9 T cell clone specific for the βgal peptide (YVVDEANIETHGMV) has been described (19). Founder mice carrying transgenes for 3E9 TCR α and β chains were generated (unpublished data) and crossed to normal B10.A mice. The transgene positive F1 mice were then crossed to generate stable 3E9-α or 3E9-β Tg mouse lines. Functional 3E9-TCR Tg mice (βgalTCR mice) were generated by crossing the 3E9-α and 3E9-β Tg mice. F1 mice were analyzed by PCR for both the α and β transgenes. Mice positive for both transgenes were assayed for proliferative responses in peripheral blood mononuclear cells (PBMC) to either βgal peptide or whole βgal protein. Mice whose PBMCs responded to βgal Ag were crossed to propagate the βgalTCR-Tg mouse line. All subsequent βgalTCR-Tg mice were assayed for the ability of PBMCs to respond in vitro to βgal Ag prior to any other analysis.
Other mice
The βgal-expressing arrβgal (previously named hi-arr-βgal), GFAPβgal, and ROSA26 Tg mice on the B10.A background have been described in detail elsewhere (13, 19-21). βgal expression in rod photoreceptor cells of arrβgal mice mimics expression of endogenous arrestin (22, 23), producing 150 ng βgal/retina and <0.5 ng/pineal gland. Rare, unidentified βgal-positive brain cells were seen. No other sites of expression were found. GFAPβgal mice express βgal in astrocytes of brain (175 ng/brain), retina, and optic nerve, based on activity of the GFAP promoter (24). Expression was not found in thymus of arrβgal or GFAPβgal mice. Expression of βgal in adult ROSA26 mice was low, but present in many tissues, including the thymus. Mice were handled in accordance with the ARVO Statement for the Use of Animals in Ophthalmic and Vision Research, and University of Minnesota animal use and care guidelines. Mice were housed under specific pathogen-free (SPF) conditions on lactose-free chow.
In-vitro T cell, PBMC, and cytokine analysis
Spleen or LN cell suspensions were filtered through a 70 μm cell strainer and the red blood cells (RBC) were lysed in 0.17 M NH4Cl. The cells were then washed 2X in phosphate-buffered saline (PBS), and resuspended in RPMI-1640 supplemented with 10 % fetal calf serum (FCS). PBMCs were prepared from heparinized blood. Routine in vitro Ag stimulation was performed with 1μM βgal peptide using βgalTCR splenocytes or purified T cells plus irradiated (3000 R) B10.A splenocytes, as Ag presenting cells (APC), at an APC/T cell ratio of 10:1. Proliferation and cytokine assays were performed in 96-well plates using 5 × 105 βgalTCR splenocytes, or 5 × 105 APC with either 5 × 104 purified βgalTCR T cells or 5-25 × 104 PBMCs, and Ag as indicated in a final volume of 200 μL. For proliferation assays the cells were pulsed with [3H]thymidine at 48 hr and counted at 72 hr post-stimulation. Cytokines were assayed at 48 hr post-stimulation by ELISA using antibodies and reagents from R&D Systems (Minneapolis, MN) or by cytometric bead array (CBA, BD Bioscience, San Jose, CA) per manufacturer’s instructions. When indicated, mice were immunized by subcutaneous injection of 100 μg βgal peptide emulsified in complete Freund’s adjuvant (CFA; Sigma, St. Louis, MO).
T cell and bone marrow transfers
Purified βgalTCR T cells were prepared from spleen and LN using anti-CD90.2 (Thy1.2) and/or anti-CD25 microbeads (Miltenyi, Auburn, CA) per manufacturer’s protocol. The cells were washed and resuspended to a concentration of 5 × 107 cells/mL in PBS. Where indicated, the cells were labeled with 4 μM 5(6)-carboxyfluorescein diacetate N-succinimidyl ester (CFSE; Molecular Probes, Eugene, OR) for 10 min at 37° C with mixing. Labeling was terminated by addition of 10 mL RPMI-1640 media supplemented with 10 % FCS. The cells were washed and resuspended to 2 × 107 cells/mL in saline. Recipient mice received the indicated number (10 × 106) of βgalTCR T cells via ip injection. βgalTCR bone marrow (BM) was extruded by femoral and tibial lavage with PBS. BM cells were PBS washed, RBC lysed, washed again, and resuspended in saline. BM recipient mice were irradiated (2 × 600 R, 4 hr interval) and received 4-5 × 107 cells in 100 μL ip.
In vivo treatment of βgalTCR T cell recipients
Groups of arrβgal and B10.A control mice were given an iv inoculation of 5 × 106 in vitro Ag-experienced, CFSE-labeled, resting βgalTCR T cells on day 0. Recipient mice were treated with 0.5 μg/mouse of pertussis toxin (PTx; Sigma, St. Louis, MO) on days 12 and 19, or given 20,000 U/mouse of IFNγ (R & D Systems, Minneapolis, MN) on days 12, 13, 14, 17, and 18. Control mice received saline only. Spleens were harvested for flow cytometry on day 28.
Flow cytometry
Spleen, thymus, or LN cell suspensions were prepared as described above except that the final resuspension was made in FACS buffer (PBS with 2 % FCS and 0.02 % sodium azide). 0.25-2.0 μL/106 cells of the appropriate fluorescent-labeled antibodies (BD Biosciences, San Diego, CA or eBioscience, San Diego, CA) were added and the suspensions incubated on ice for 30 min. The cells were then washed and resuspended in FACS buffer and analyzed on a FACSCalibur flow cytometer using CellQuest (BD Biosciences, San Jose, CA) or FlowJo (Tree Star, Ashland, OR) software.
Induction of autoimmune disease by adoptive transfer of T cells
Adoptive transfer for induction of EAU or brain inflammation was done with pooled, unfractionated βgalTCR splenocytes and LN cells stimulated with βgal peptide (0.5 μM), with or without IL-12 (5.0 ng/mL) or the combination of IL-1 (5 ng/mL), IL-6 (10 ng/mL), and TGFβ1 (2 ng/mL) to induce IL-17 production. At 48 hr post-stimulation, IL-2 was added (10 U/ml) and the βgalTCR T cells were cultured for an additional 6 hr. The cultures were washed, resuspended in PBS to 2 × 107 cells/mL, and inoculated at 5-20 × 106 βgalTCR T cells or splenocyte/lymphocyte cells per mouse, ip. Where indicated, T cells were isolated by Thy1.2 selection and activated with βgal peptide and irradiated B10.A × Rag(-/-) splenocytes at a splenocyte/T cell ratio of 10:1 for 48 hr. At the indicated times post-transfer, tissues were harvested, fixed in 10 % buffered formalin, paraffin embedded, sectioned (5 micron), and stained with hematoxylin and eosin. The slides were examined in a masked fashion with EAU scores of 0 (no disease) to 4 (complete loss of photoreceptor cells) based on histopathological changes to the retina (25). Brain inflammation was scored from 0 (none) to 4 (severe) based on histopathology (26).
Induction of autoimmune disease in double transgenic mice
Arrβgal or arrβgal × βgalTCR double Tg mice were given a single subcutaneous inoculation of 100 μg βgal protein emulsified in CFA with or without 0.5 μg PTx per mouse via ip injection. Eyes were harvested at 21 days post-immunization and analyzed for histopathology. The ability of soluble Ag and Ptx to induce disease was assayed by injecting mice ip with 200 μg βgal protein mixed with 0.5 μg Ptx. Eyes (from arrβgal × βgalTCR mice) and brains (from GFAPβgal × βgalTCR mice used as controls) were harvested at 35 and 21 days post-immunization, respectively, and analyzed for histopathology.
Induction and analysis of autoimmune disease in lymphopenic mice
CD25+ and CD25- fractions were isolated from βgalTCR spleen and LN cell suspensions using a CD25+ selection kit and then enriched for CD4+ cells by negative selection (Miltenyi). The cells were washed and resuspended in PBS to 5 × 106/ml and 3 × 105 cells were injected iv into Rag(-/-) or arrβgal × Rag(-/-) mice. The eyes were harvested at the indicated time points and analyzed as described above. Analysis of the delayed-type hypersensitivity (DTH) response to βgal was done by injection of βgal (50 μg in 10 μL) into the ear pinna as described previously (20).
Results
Analysis of βgalTCR T cells
Construction and initial identification of βgalTCR mice was made by PCR showing the presence of both the α and β TCR genes in genomic DNA (unpublished data). Unprimed βgalTCR mice exhibited a positive DTH response while single chain 3E9 α and β Tg mice did not (unpublished data). Analysis of βgalTCR splenocytes by flow cytometry showed 55-60 % of CD3+ cells were CD4+Vβ10.1+, and more than 85 % of CD4+ cells expressed Vβ10.1 (Fig. 1A). Further, comparison of naïve B10.A and βgalTCR CD4+VB10.1+ T cells showed similar expression of the activation markers CD25, CD44, CD45RB, and CD69, at levels consistent with the cells being phenotypically naïve (Fig. 1B). The number of CD62Llo cells from spleen was modestly increased in βgalTCR mice (Fig. 1B). In vitro proliferation assays demonstrated that βgalTCR splenocytes and/or lymphocytes responded to both βgal peptide and native βgal protein in a dose-dependent manner (Fig. 1C). Ag stimulation of unfractionated βgalTCR splenocytes produced significant amounts of IL-2 and IFNγ consistent with Th1 cells (27, 28) (Table I), but little IL-4, IL-5, IL-10, and TNFα. IL-17 was also produced, consistent with the Th17 phenotype of autoimmune disease-inducing T cells (29, 30). The Ag specificity of βgalTCR T cells was confirmed by the lack of cytokine production when stimulated with ovalbumin (OVA).
Figure 1.
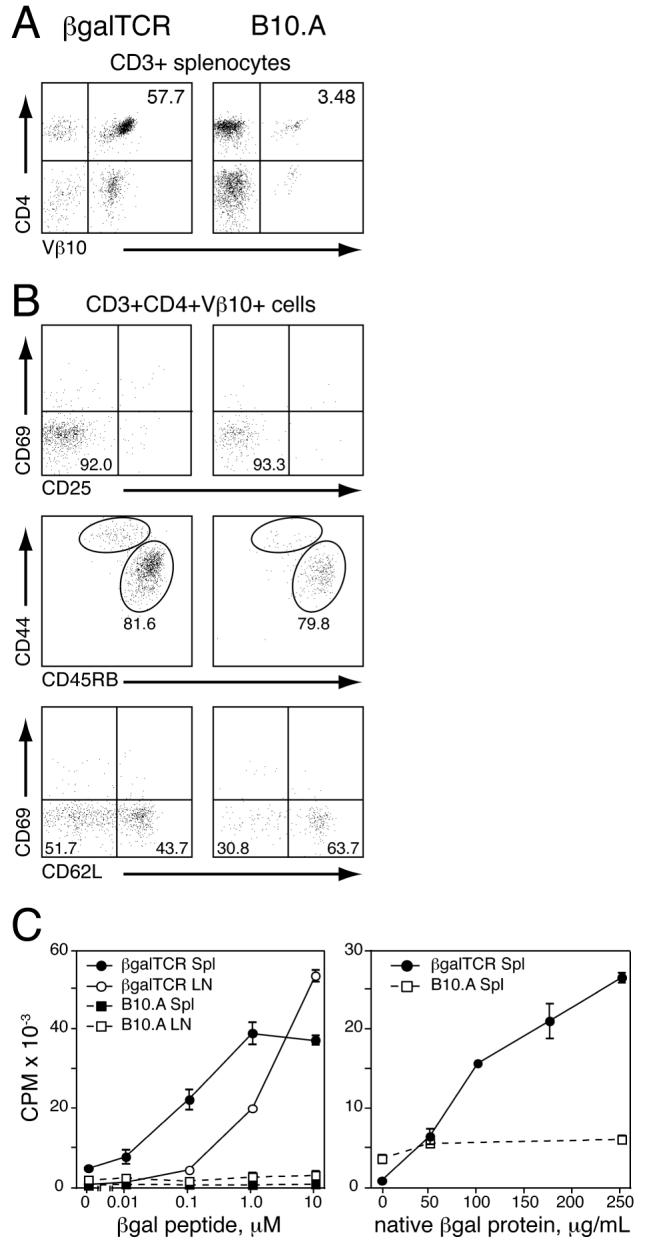
Analysis of phenotype and Ag response of T cells from βgalTCR Tg mice. (A) FACS analysis of CD3+ splenocytes from B10.A control (right) and βgalTCR Tg (left) mice. Percentage of cells with a specific phenotype is indicated. (B) FACS analysis of CD3+CD4+Vβ10+ splenocytes from naïve B10.A control (right) and βgalTCR Tg (left) mice for cell surface activation markers. (C) Proliferative response of control B10.A and TCRβgal Tg mouse lymph node cells and/or splenocytes to βgal peptide (left) or native βgal protein (right). Values shown are the mean of three animals, each done in triplicate, with error bars indicating standard error of mean (SEM).
Table I. Cytokine production by Ag stimulated βgalTCR splenocytes.
| Cytokine, pg/mLa | |||||||
|---|---|---|---|---|---|---|---|
| Agb | IL-2 | IL-4 | IL-5 | IL-10 | IL-17 | INFγ | TNFα |
| none | 5.6 ± 2.9 | 0.7 ± 1.0 | 2.5 ± 1.6 | BDc | 4.1 ± 3.6 | 1.7 ± 0.8 | 15.2 ± 8.5 |
| OVA | 6.8 ± 1.0 | BDc | 2.1 ± 0.5 | NDc | 21.1 ± 23.0 | 14.5 ± 1.4 | 178 ± 21 |
| βgal | 3470 ± 2275 | 5.0 ± 2.4 | 31 ± 15 | 460 ± 70 | 6605 ± 2195 | 8071 ± 3835 | 1434 ± 38 |
± standard deviation.
100 ug/ml ovalbumin, 10 μM βgal peptide.
BD = below detection, ND = not determined.
In vivo antigen recognition by naive βgalTCR T cells
Interactions in vivo between naïve βgal-specific T cells and retinal βgal were tested using purified, resting βgalTCR T cells from pooled spleen and LN. T cells were labeled with CFSE and transferred into βgal Tg and control mice. Labeled βgalTCR T cells were recovered from recipient LN and spleens ten days post-transfer and analyzed for evidence of Ag recognition. βgalTCR T cells transferred into ROSA26 mice, or control B10.A mice immunized with βgal peptide after transfer, showed clear evidence of Ag recognition compared to βgalTCR T cells transferred into normal B10.A recipients. Proliferation of βgalTCR T cells was indicated by dilution of CFSE in Vβ10+ lymphocytes from ROSA26 and B10.A/peptide-inoculated mice (Fig. 2A) and in B10.A mice inoculated with whole βgal (Fig. 2B). Their expression of cell surface molecules associated with Ag recognition by T cells was also skewed toward the activated phenotype (increased CD44, CD69 and decreased CD45RB, CD62L; Fig. 2A). Conversely, βgalTCR T cells recovered from normal B10.A recipients, or from arrβgal mice, did not dilute their CFSE and maintained the naïve phenotype (Fig. 2A). Ag-specificity was further shown by inoculation with OVA, which did not induce CFSE dilution (Fig. 2B). βgalTCR T cells recovered from submandibular and cervical LN of recipient arrβgal mice were not phenotypically different than those recovered from inguinal and popliteal LN (unpublished data).
Figure 2.
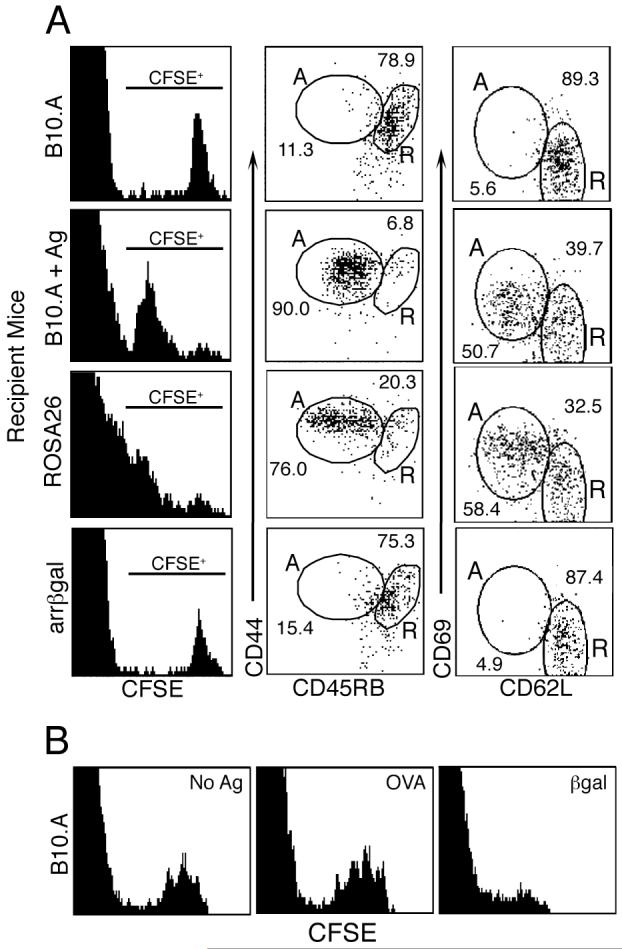
Analysis of Ag recognition in vivo by adoptively transferred, CFSE-labeled naïve βgalTCR T cells. (A) Representative FACS analysis of Vβ10+ lymphocytes recovered from recipient mice 10 days post-transfer for CFSE dilution and the indicated cell surface molecules. The percentage of lymphocytes having a resting/naïve (R) or an activated (A) phenotype is indicated. B10.A + Ag = 100 μg βgal peptide given ip to B10.A recipient mice three days post-T cell transfer. (B) Analysis of Ag specificity. FACS analysis of Vβ10+ lymphocytes from spleens of B10.A mice that received 100 rads irradiation prior to transfer of naive, CFSE-labeled βgalTCR T cells. Cells were harvested 14 days post-transfer. Recipients were given ip injections of saline (No Ag); 1 mg OVA; or 300 μg native βgal three days post-T cell transfer.
Development and function of βgalTCR T cells maturing in βgal Tg mice
Since short term residence of naïve βgalTCR T cells in arrβgal Tg mice offered little evidence of recognition of endogenous βgal, we asked if βgalTCR T cells maturing in arrβgal mice were altered in their development or ability to respond to βgal as measures of Ag recognition. Introduction of a more physiologically relevant number of developing βgal-specific T cells was achieved using BM grafting. βgalTCR mouse-BM was engrafted into irradiated B10.A or arrβgal mice, and T cell development and function were analyzed at 40 days post-transfer. FACS analysis of PBMC showed similar levels of βgalTCR T cell engraftment in both arrβgal and B10.A recipients (Fig. 3A). CD4+Vβ10+ T cells represented an average of 26.6 % and 29.9 % of all CD3+ lymphocytes in the recipient B10.A and arrβgal mice respectively. A vast majority of these T cells are likely to be bona fide βgalTCR T cells since the endogenous Vβ10 TCR gene is expressed on only about 5 % of all T cells in control mice. βgalTCR T cells did not repopulate well in ROSA26 mice transferred with βgalTCR-BM as shown by the reduced percentage of CD4+Vβ10+ in CD3+ lymphocytes (5.4 %). Unlike arrβgal mice, ROSA26 mice express βgal in the thymus, therefore this result is consistent with central tolerance and negative selection of maturing βgalTCR T cells. Analysis of CD3+Vβ10+ thymocytes from βgalTCR-BM engrafted arrβgal vs. B10.A mice showed nearly identical distributions of T cells into either CD4 or CD8 single positive, or CD4/CD8 double positive or double negative subsets (Fig. 3B), suggesting little or no central tolerance to βgal in arrβgal mice. Arrβgal mice receiving βgalTCR-BM did not develop EAU. Under these conditions, the results indicate that expression of βgal from the rod photoreceptor cell arrestin promoter had little effect on βgalTCR T cell development.
Figure 3.

Analysis of βgalTCR T cells derived from βgalTCR-BM engrafted mice. (A) Representative FACS analysis of CD3+ lymphocytes from peripheral blood for percentage of CD4+Vβ10+ cells 40 days post-BM transfer. The normal B10.A mice were not irradiated or grafted; all other groups were irradiated and given βgalTCR-BM where indicated. (B) FACS analysis of CD3+Vβ10+ thymocytes from control (B10.A) and βgalTCR-BM engrafted mice for T cell subset distribution. Also shown is the percentage of CD3+ thymocytes expressing Vβ10. Assays performed 40 days post BM transfer. (C) FACS analysis of CD4+Vβ10+ splenocytes for the indicated cell surface activation marker from B10.A, βgalTCR, and βgalTCR-BM engrafted mice 60 days post BM transfer. (D) Proliferative response of splenocytes from βgalTCR-BM engrafted mice to βgal peptide. Values represent the mean of three animals, each done in triplicate, with error bars indicating SEM. (E) FACS analysis of CD4+Vβ10+ splenocytes for cell surface markers of memory phenotype in immunized and non-immunized βgalTCR-BM engrafted recipient mice. For B, C, and E, bars indicate the mean for all animals in each group (n ≥ 3) with error bars indicating SEM.
To determine if βgalTCR T cells that developed after βgalTCR-BM engrafting of βgal Tg recipients were altered by βgal in retinal photoreceptor cells, the phenotype, response, and effector functions of βgal specific T cells recovered from TCRβgal-BM engrafted recipient mice were compared at sixty days post-transfer. Cell surface activation makers were similar between naïve CD4+Vβ10+ T cells recovered from spleens of βgalTCR-BM engrafted arrβgal and B10.A mice as well as to control βgalTCR and B10.A mice (Fig. 3C). Similar results were observed in CD4+Vβ10+ cells recovered from LN (unpublished data). Splenocytes from βgalTCR-BM engrafted arrβgal and B10.A mice exhibited similar dose-dependent proliferative responses to βgal peptide (Fig. 3D).
To compare the development of memory βgalTCR T cells in mice whose naïve T cells matured with or without βgal expressed in photoreceptor cells, βgalTCR-BM recipient mice or control mice were immunized with βgal peptide, and splenocytes were analyzed twenty-one days post-immunization. Immunized βgalTCR-BM recipient arrβgal mice and B10.A mice showed similar increases in CD44 levels and decreases in CD62L levels (Fig. 3E), indicating that the development of the effector memory T cell phenotype was not affected by the presence of βgal in photoreceptor cells. In summary, these results suggested that the presence of βgal in photoreceptor cells had little effect on the phenotype, response, or effector functions of naïve or memory βgalTCR T cells. Further, no spontaneous EAU, or EAU induction by immunization with βgal/CFA, was observed in any of the βgal Tg mice grafted with βgalTCR-BM.
Manipulation of antigenic ignorance
It has been proposed that a lack of Ag presentation of retina-derived Ags in peripheral LN limits the generation of peripheral tolerance, leading to increased susceptibility to EAU (10). Since Ag-experienced T cells are widely thought to have increased ability to detect and respond to Ag, adoptive transfer of Ag-experienced, CFSE-labeled, resting βgalTCR T cells, and treatments to promote Ag presentation of retinal βgal, were used to probe for evidence of peripheral retinal Ag recognition (Fig. 4). Although none of the mice receiving the βgalTCR T cells along with IFNγ or PTx treatment developed EAU, there were small, but significant, Ag-dependent changes in: 1) recovery of CFSE-labeled βgalTCR T cells from spleen, 2) their percentage of all CD4+Vβ10+ cells and, 3) the level of CFSE fluorescence in CFSE+ cells recovered from IFNγ-treated, or PTx-treated arrβgal vs. B10.A recipients. These parameters were unchanged in control comparisons of saline-treated arrβgal vs. B10.A mice and IFNγ vs. saline-treated B10.A mice. These results suggest that manipulations designed to increase Ag presentation produced a limited, non-pathogenic, T cell recognition of retinal βgal.
Figure 4.
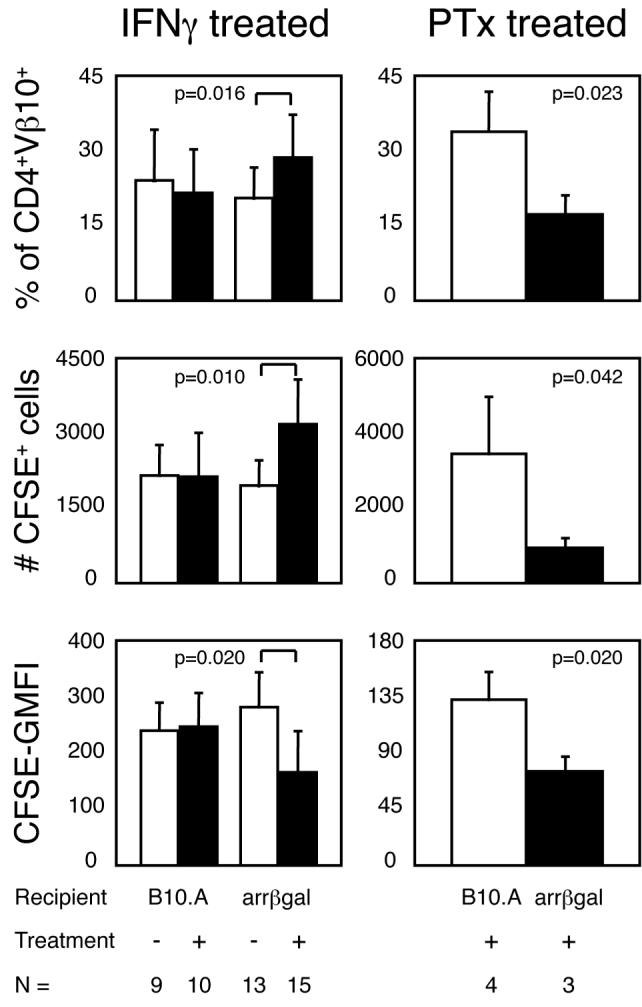
In vivo administration of IFNγ or PTx leads to Ag-dependent changes in βgalTCR T cells. Resting, CFSE-labeled βgalTCR T cells were transferred into mice that were treated with IFNγ (left) or PTx (right) as described in materials and methods. Bars indicate mean value for all animals in each group with error bars indicating SEM. p values determined by student’s t test.
Susceptibility of single and double Tg mice to EAU induction
Several TCR Tg mouse models of autoimmune disease have used double Tg mice in which both the target Ag and Ag-specific T cells are present (9, 10, 31, 32). Accordingly, we generated arrβgal × βgalTCR double Tg mice and compared the ability to induce EAU in these double Tg mice versus single Tg arrβgal mice. Arrβgal mice were moderately susceptible to induction of EAU by immunization with βgal in CFA, followed by PTx given iv (Table II). Conversely, double Tg arrβgal × βgalTCR mice were highly resistant to the same immunization protocol, and did not develop spontaneous EAU when aged up to 14 months in SPF conditions.
Table II. Inhibition of EAU induction in arrβgal × βgalTCR double Tg mice.
| arrβgal | arrβgal × βgalTCR | ||||
|---|---|---|---|---|---|
| Immunization | incidence | severity | incidence | severity | P valuea |
| βgal + CFA | 0/8 | 0 | 0/15 | 0 | |
| βgal + CFA + PTx | 12/36 | 0.5, 0.5, 0.5, 1, 1, 1, 2, 2, 2.5, 3, 3.5, 4 | 0/12 | 0 | 0.023 |
Fisher’s exact test: 2-tailed; arrβgal vs. arrβgal × βgalTCR
Adoptive transfer of βgalTCR T cells
Using βgal peptide to activate pooled spleen and LN cells from βgalTCR mice in vitro led to potent activation as shown by cytokine production (Table I). After two days, 5-20 × 106 viable cells were transferred into the βgal Tg mice. Preliminary studies showed that GFAPβgal mice, which express βgal in CNS astrocytes, developed CNS infiltrates and lesions following adoptive transfer of βgalTCR T cells (unpublished data). Therefore, they were used as positive controls for the activity of the various preparations of βgalTCR T cells that follow. None of 18 arrβgal mice developed EAU, but 4 of 8 GFAPβgal mice developed inflammatory lesions in the brain (Table III). The large difference in susceptibility of the retina as compared to brain was unexpected, as the concentration of Ag in retina is much higher than in brain; i.e. the 150 ng of βgal/retina is concentrated in 3 μL of retinal tissue whereas the 175 ng in brain is contained in approximately 500 μL of tissue.
Table III. arrβgal mice resisted induction of EAU by adoptive transfer of pathogenic T cells.
| Recipient Mice | |||||
|---|---|---|---|---|---|
| arrβgala | GFAPβgalb | ||||
| Cells Transferredc | incidence | severity | incidence | severity | P value |
| untreated | 0/18 | 0 | 4/8 | 0.5, 1.0, 1.5, 2.0 | 0.0047d |
| IL-12 (exogenous) | 0/17 | 0 | ND | ||
| IL-17 (induced) | 1/23 | 1.0 | 5/13 | 0.5, 1.0, 1.0, 2.0, 3.5 | 0.0161d |
EAU 21 days post-transfer.
brain inflammation 5-7 days post-transfer.
Unfractionated spleen and LN cells plus Ag and indicated cytokine treatment.
Fisher’s exact test; 2-tailed; arrβgal vs. GFAPβgal.
To determine if more potent activation of the T cells would enhance disease induction, cultures were activated in the presence of agents known to promote development of pathogenic CD4+ T cells in EAU, including IL-12 (33), which failed to promote EAU induction (Table III). Inclusion of IL-1, LPS, CpG or PTx in the cultures during activation also did not promote disease in arrβgal recipients (unpublished data). Since Th17 cells have been identified as important mediators of EAU (34), βgalTCR T cells were cultured with the combination of IL-1, IL-6, and TGFβ1 to induce differentiation of IL-17 secreting Th17 cells (35). A single Ag activation cycle in the presence of these three cytokines gave a moderate level of IL-17 production, while a second cycle induced very high levels of IL-17 production (Fig. 5A). Subsequent activation cycles in the presence of the IL-17 inducing cytokines did not further increase IL-17 levels, but the inducing cytokines were necessary to maintain IL-17 production (unpublished data). The combination of cytokine supplements had little effect on IL-2 production compared to cultures stimulated without cytokines (Fig. 5B). TNFα and IFNγ levels were also assayed since these cytokines are associated with EAU pathogenesis (36, 37), but they were unaffected (unpublished data). While the IL-17-secreting phenotype was induced in the cytokine-treated βgalTCR T cells, there was no increase in the frequency of EAU in arrβgal mice compared to mice that received βgalTCR T cells stimulated without cytokines. Only one mouse developed mild disease in the peripheral retina (Fig. 6B and Table III). These same IL-17-induced βgalTCR T cells were significantly pathogenic in the GFAPβgal mice.
Figure 5.
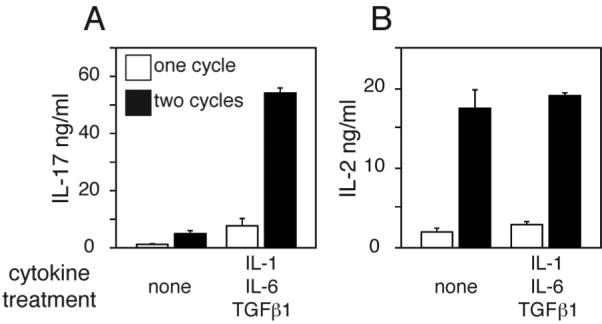
Induction of IL-17 production by βgalTCR T cells. Production of IL-17 (A) and IL-2 (B) following one or two rounds of Ag/APC stimulation with indicated cytokine treatments as describe in Material and Methods. Error bars indicate SEM.
Figure 6.

Representative photomicrographs of autoimmune pathology in arrβgal retina induced by βgalTCR T cells. (A) Normal retina. (B) Retina 21 days post-transfer of 5 × 106 βgalTCR T cells after 4 stimulation cycles under IL-17-inducing culture conditions. Modest loss of peripheral photoreceptor cells was evident after active inflammation subsided (arrow). (C-D) Retinas from an arrβgal × βgalTCR double Tg mouse 35 days after IV inoculation of 0.5 μg PTx and 200 μg βgal showing modest infiltration of the optic nerve head (C, arrow) and substantial active infiltration of the inner layers of the retina plus disruption of the photoreceptor cell layer (D, arrows). (E-G) arrβgal × Rag(-/-) retinas at 49 (E), 62 (F), and 75 (G) days post-transfer of 3 × 105 naïve CD25- βgalTCR T cells showing progressive loss of photoreceptor cells. (A-G), 100 X magnification.
Differential induction of autoimmune disease in βgalTCR × βgal double transgenic mice
Since a recent study reported that soluble Ag along with PTx, in conjunction with transferred, Ag-specific CD4+ T cells, was a potent inducer of autoimmune disease in mice that expressed the Ag in the eye (38), the ability of PTx and soluble βgal to induce EAU in the arrβgal × βgalTCR double Tg mice was analyzed. Only 1 of 15 double Tg mice developed disease using this protocol (Fig. 6C-D and Table IV). To confirm the function of the βgalTCR Tg T cells in double Tg mice, GFAPβgal × βgalTCR mice were tested concurrently and found to develop significant inflammatory infiltrates (Table IV). PTx alone did not induce autoimmune disease in the susceptible GFAPβgal × βgalTCR mice suggesting that Ag availability was a limiting factor.
Table IV. ArrβGal × βgalTCR double Tg mice were resistant to EAU.
| Recipient Mice | |||||
|---|---|---|---|---|---|
| arrβgal × βgalTCRa | GFAPβgal × βgalTCRb | ||||
| Immunization | incidence | severity | incidence | severity | P value |
| βgal + PTx | 1/15 | (1.0, 2.5) | 11/18 | 1.0, 1.0, 1.0, 1.5,1.5, 1.5, 1.5, 1.0,1.0, 1.5, 2.0 | 0.0019c |
| PTx only | ND | ND | 0/5 | 0 | 0.007d |
EAU 35 days post-immunization.
brain inflammation 21 days post-immunization.
Fisher’s exact test; 2-tailed; arrβgal × βgalTCR vs. GFAPβgal × βgalTCR.
Fisher’s exact test; 2-tailed; GFAPβgal × βgalTCR mice; βgal + PTx vs. PTx only incidence.
Lymphopenia-induced autoimmune disease
LIP has been associated with acquisition of an activated T cell phenotype, and inflammatory disease in murine models (39-42). A small number (3 × 105) of CD25- T cells isolated from βgalTCR × Rag(-/-) mice were transferred into arrβgal × Rag(-/-) mice and a control group of Rag(-/-) mice. Eyes harvested between 2 and 11 weeks post-transfer showed a slowly progressive, but relentless destruction of the entire photoreceptor layer (Fig. 7) by a process that was minimally inflammatory, but highly destructive (Fig. 6E-G). Transfer into Rag(-/-) mice lacking retinal βgal expression gave no EAU (0/12), nor did transfer of an equivalent number of CD25+ T cells from βgalTCR × Rag(-/-) donors into arrβgal × Rag(-/-) recipients (0/4).
Figure 7.
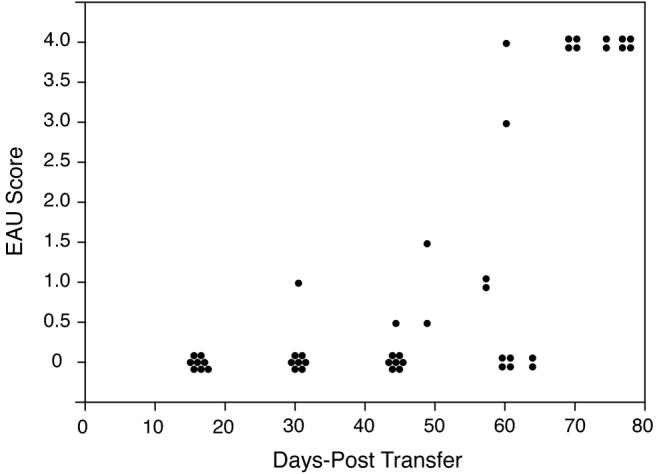
LIP leads to EAU in arrβgal mice. 3 × 105 CD25- βgalTCR T cells were transferred into arrβgal × Rag(-/-) recipients and the eyes were harvest at the indicated time post-transfer and scored for EAU. Each dot represents one eye.
Retinal Ag affected the recovery of βgalTCR T cells
In addition to the retinal destruction associated with LIP by naïve CD25-depleted βgalTCR T cells in the arrβgal × Rag(-/-) recipients, evidence for other Ag-dependent effects on the number, phenotype, and function of the recovered cells was sought. Approximately twice as many βgalTCR T cells were recovered from Rag(-/-) recipients compared to arrβgal × Rag(-/-) recipients (Table V). Each of the populations contained similar portions of CD25+Foxp3+ T cells, approximately 4 %, but the number of cells with a CD25+Foxp3+ Treg phenotype was greater in the Rag(-/-) recipients without βgal. CD25+Foxp3+ T cells could be detected at three weeks post-transfer (unpublished data) and were similarly present in both types of recipients at ten weeks post-transfer (Fig. 8A).
Table V. Analysis of T cells from Rag(-/-) and arrβgal × Rag(-/-) mice transferred with CD4+CD25-βgalTCR T cells.
| T cell Recovery | ||||||
|---|---|---|---|---|---|---|
| Recipient Mice | N | number of CD4+Vβ10+ (x 106)a | number of CD4+Vβ10+ CD25+ (x 104) a | number of CD4+Vβ10+ Foxp3+ (x 104) a | number of CD4+Vβ10+ CD25+ Foxp3+ (x 104) a | CD4+Vβ10+ % Tregs |
| Rag(-/-) | 12 | 1.06 ± 0.69 | 17.60 ± 9.4 | 5.21 ± 3.94 | 4.77 ± 3.86 | 4.29 ± 3.27 |
| arrβgal × Rag(-/-) | 11 | 0.59 ± 0.24 | 7.72 ± 2.28 | 2.03 ± 1.55 | 1.79 ± 1.53 | 3.75 ± 3.08 |
| P valueb | 0.0414 | 0.0024 | 0.0206 | 0.0267 | 0.6870 | |
Mean of total cells per animal ± standard deviation ten weeks post-transfer.
Student’s t test, two-tailed.
Figure 8.
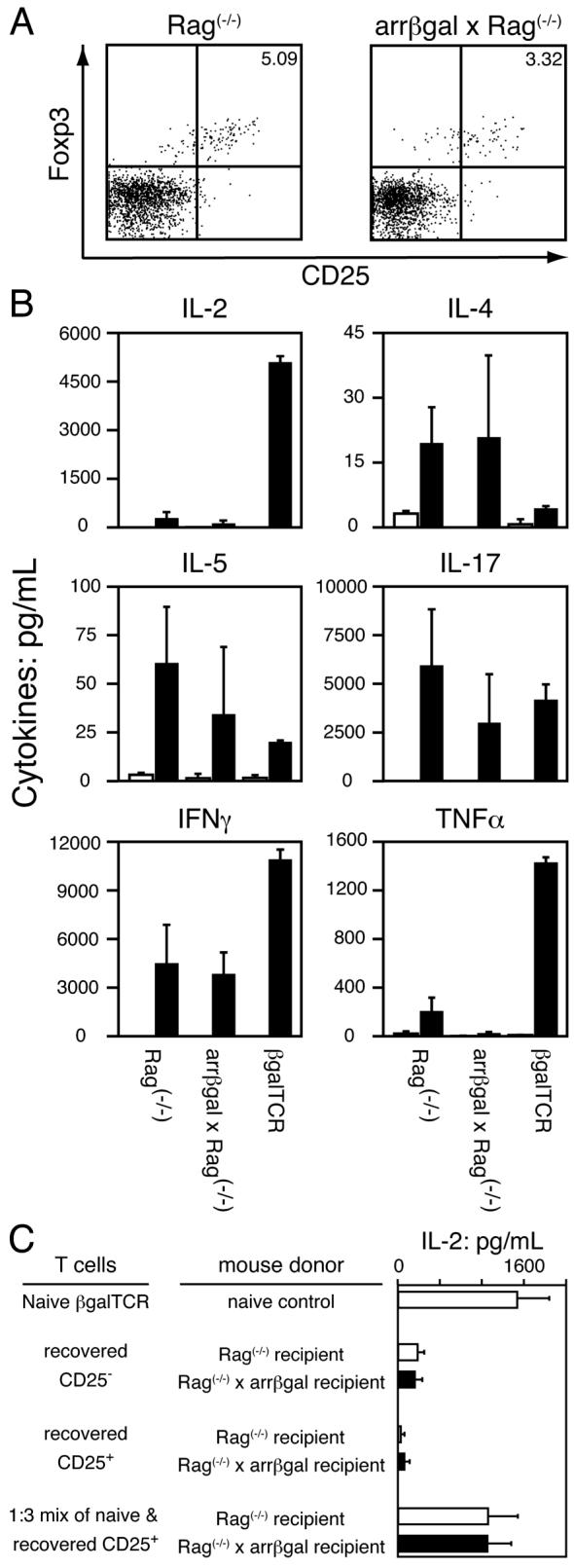
Analysis of T cells recovered from lymphopenic recipient mice reflects the contributions of Th1, Th2, and regulatory T cells. Spleen and LN cells were harvested 9-11 weeks post-transfer of CD25- βgalTCR T cells into Rag(-/-) and arrβgal × Rag(-/-) mice. (A) Representative FACS plots of CD4+Vβ10+ T cells recovered from transferred Rag(-/-) and arrβgal × Rag(-/-) mice showing similar proportions of cells that are CD25+Foxp3+. (B) Analysis of cytokine production by 5 × 105 pooled, Ag-stimulated spleen and LN from normal βgalTCR Tg mice and lymphopenic mice transferred with CD25- βgalTCR T cells. Solid bar = with Ag, open bar = no Ag. (C) IL-2 production by 1 × 105 CD25+ T cells and CD25- T cells recovered from Rag(-/-) and arrβgal × Rag(-/-) recipient mice, and 1 × 105 recovered CD25+ T cells mixed with 3.5 × 104 naïve βgalTCR T cells upon stimulation with Ag and APC.
Cytokine production by recovered βgalTCR T cells was substantially altered by their residence in both types of lymphopenic recipients relative to normal βgalTCR T cells, but the effect of the presence of βgal was minimal (Fig. 8B). The level of cytokine production was numerically lower, in most cases, in cells recovered from arrβgal × Rag(-/-) recipients, but no single significant difference from the Rag(-/-) recipients was found. In another experiment, the CD25+ cells from the lymphopenic recipients of CD25-depleted βgalTCR T cells were isolated and tested for their ability to inhibit Ag-stimulated IL-2 production by naïve βgalTCR T cells. The CD25+ cells had little ability to inhibit the IL-2 production of fresh βgalTCR T cells, and also produced little IL-2 upon stimulation with Ag/APC (Fig. 8C).
Tregs derived from the T cell inoculum
We previously demonstrated the presence of an uncharacterized, transferable regulatory activity in the arrβgal mice (20), and showed above that the arrβgal mice were resistant to adoptive transfer of EAU by the βgalTCR T cells. Since Rag(-/-) mice do not have conventional, endogenous CD4+25+Foxp3+ T cells, we tested the possibility that arrβgal mice on the Rag(-/-) background might be more susceptible to EAU induced by adoptive transfer of in vitro activated βgalTCR T cells. Thy1.2+ cells were isolated from pooled βgalTCR spleen and LN, activated for 48 h with Ag presented by irradiated splenic APC from Rag(-/-) mice, and transferred to arrβgal and arrβgal × Rag(-/-) mice. The modified activation protocol generated T cells that induced a low, but significant incidence of EAU, with minimal severity. However, there was no difference in the incidence and severity of EAU between the two groups of recipients (Fig. 9, left) suggesting that endogenous Tregs had a limited role in preventing pathogenesis.
Figure 9.
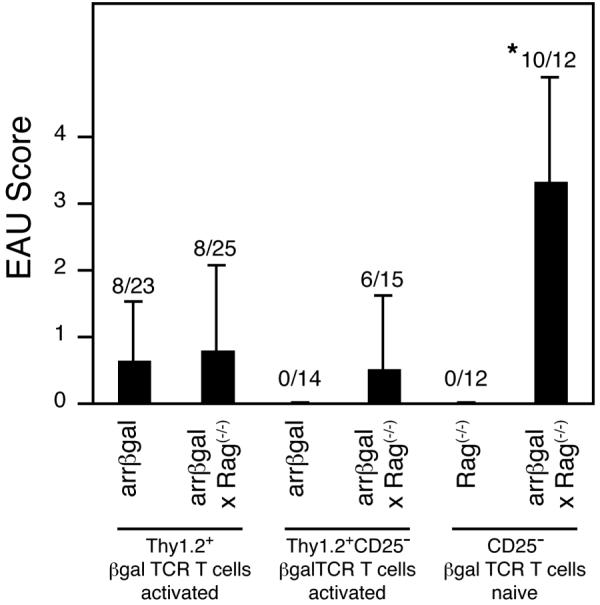
Induction of EAU by adoptive transfer of βgalTCR T cells. The T cells were activated and/or selected as indicated and transferred into indicated recipient mice. * = p < 0.005 compared to all other groups for both incidence (Fisher’s exact test) and severity (Mann-Whitney test).
Since the EAU induced by naïve CD25- T cells in the arrβgal × Rag(-/-) recipients was slow to develop, we asked if activated βgalTCR T cells that had been depleted of CD25+ cells prior to activation would induce EAU more rapidly and be more pathogenic in arrβgal × Rag(-/-) recipients. CD25-depleted, pre-activated βgalTCR T cells were much less pathogenic in lymphopenic recipients than were the naïve CD25-depleted cells. Further, the normal arrβgal recipients remained resistant to EAU induction using either naïve or activated, CD25-depleted βgalTCR T cells (Fig. 9). These results suggested the possibility that Tregs were present in the T cell inoculum. In preliminary studies, we noted that in vitro activation of T cells from βgalTCR mice yielded an increase in the population of Foxp3+ T cells, which could be Tregs. As shown in Fig. 10, CD4+25+Foxp3+ cells were indeed generated from Thy1.2-selected, CD25-depleted βgalTCR T cells by Ag stimulation and present in the inocula, where they could contribute to the resistance to EAU. Since cells with the Treg phenotype appeared within 48 h of activation, proliferation of a small number of contaminating Tregs could not account for the 10-fold increase.
Figure 10.
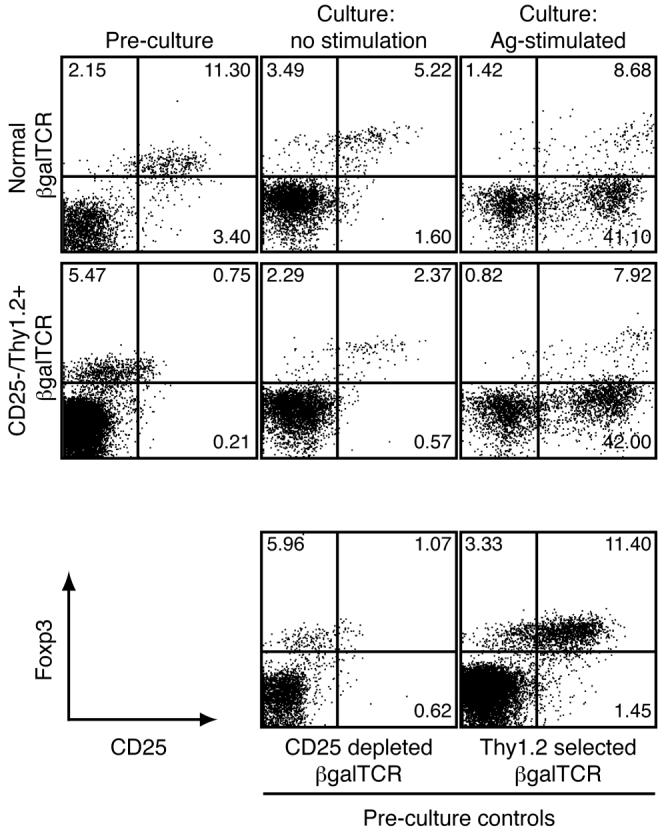
Production of T cells with the Treg phenotype in Ag-stimulated cultures. Normal and selected βgalTCR T cells were analyzed for CD25+Foxp3+ cells prior to culturing and after 48 h in culture with or without Ag. FACS plots are gated on CD4+ lymphocytes.
Discussion
We have created TCR-Tg mice (βgalTCR) specific for the neo-self Ag βgal in order to investigate factors leading to autoimmune pathogenesis resulting from recognition of CNS Ags that originate from retinal photoreceptor cells (neurons) by naïve and activated class II MHC-restricted CD4+ T cells. Although spontaneous autoimmune disease was not observed in arrβgal × βgalTCR double Tg mice, our results demonstrated that βgalTCR T cells were capable of in vivo CNS antigen recognition, but autoimmune disease in retina was found under a very limited set of activation conditions. Only a single strategy, lymphopenia-induced lymphoproliferation of CD25-depleted, naïve βgalTCR T cells, generated conditions that resulted in a high incidence of severe EAU in arrβgal mice.
T cells from TCR-Tg mice that recognize either endogenous self-Ags or neo-self Ags have been instrumental in advancing the understanding of the multifaceted processes of self-tolerance and autoimmunity (31). Both regulatory and immunopathogenic mechanisms were recently reported in two related models of retinal Ag-specific, TCR-Tg mice. Membrane-bound retinal expression of HEL under control of the IRBP promoter, in conjunction with the 3A9 TCR-Tg T cells specific for HEL, gave a high incidence of spontaneous, severe EAU (10). The absence of Ag presentation, due to the sequestration of Ag in a neural environment, was proposed to limit the generation of peripheral T cell anergy, and Tregs were not found. These conditions were proposed to be responsible for the high degree of spontaneous HEL-targeted retinal autoimmune disease in the HEL/3A9 double Tg mice, despite the significant level of negative selection due to aire-dependent thymic expression of HEL (10). In a related model, HEL with a membrane anchor was expressed on the retinal rhodopsin promoter and studied in double Tg mice also made with 3A9 T cells (9). In this model, severe, spontaneous disease was also found in the double Tg mice, but there was also clear evidence of substantial negative selection due to thymic HEL expression. The authors proposed that the robust disease was due to low affinity, HEL-specific T cells that escaped thymic deletion, although the source of these cells in the TCR-Tg T cell population was not identified. The tolerance induced by HEL expression in single Tg mice was proposed to be largely due to negative selection. It is possible that the photoreceptor cell damage associated with this form of HEL Tg expression in the photoreceptor cells altered the local environment, and subsequent immunological response to the Ag (9).
The results from both of these models differ from ours in several ways. First, we found no evidence for thymic βgal expression. There were minimal differences in the number, survival, phenotype, or qualitative responses of βgalTCR T cells that resided and matured in B10.A and arrβgal hosts that would be associated with thymic expression. Conversely, the low level systemic and thymic expression of βgal in ROSA26 mice strongly affected the development and function of βgalTCR T cells. Further, whether by βgalTCR-BM transfer, or transfer of mature, naïve βgalTCR T cells, or analysis of T cells from double Tg mice, there was little evidence of recognition by T cells of photoreceptor cell-derived βgal in untreated recipients, as no reliable differences in cell-surface phenotype or function were detected. Secondly, the double Tg mice were highly resistant to spontaneous EAU, even though the TCRβgal T cells were highly pathogenic under LIP conditions.
Findings from CNS studies consistent with our results were found using double Tg mice that express OVA in brain oligodendrocytes and generate OVA-specific OT-II TCR Tg T cells. There was no evidence for recognition of cognate Ag by the T cells and no spontaneous autoimmune disease (43). A naïve phenotype was also observed in influenza virus hemagglutinin (HA) Ag-specific CD4+ T cells derived from HA-specific TCR-Tg mice crossed with mice expressing HA in glial cells (44). The lack of spontaneous autoimmune disease in arrβgal × βgalTCR double Tg mice also bears a superficial resemblance to experimental autoimmune encephalomyelitis (EAE) models using various well-known myelin basic protein (MBP)-specific TCR Tg mice, especially those whose T cells are specific for the MBP 1-11 epitope in which administration of adjuvants is required for disease onset (32). Several additional important factors further distinguish our results from those. First, the use of adjuvants did not reverse the resistance to disease induction in arrβgal mice. Second, compared to the widely expressed MBP, there is very little total βgal in arrβgal mice (300 ng/mouse) and its expression is highly restricted to photoreceptor cells of the retina and to subnanogram levels in the pineal gland. Third, the MBP epitopes are also contained in the golli form of the protein that is highly expressed, even in cells of the immune system including dendritic cells (45, 46). This leads to selection of the T cell repertoire, resulting in the generation of pathogenic and regulatory T cells that recognize MBP (47-49). Fourth, the βgal epitope is immunodominant, implying the efficient generation and persistence of stable peptide-MHC class II complexes. It has been reported that MBP 1-11-MHC class II complexes are unstable (50) and that there is competition for MHC class II binding between MBP 1-11 and flanking golli and MBP sequences (51). Finally, the CNS has clear pathways of lymphatic drainage (2, 3), whereas the retina contains only occasional LYVE-1+ microglia (52), which does not constitute draining lymphatics in the normal retina.
The mechanisms that yield the remarkable resistance to autoimmune disease to an Ag expressed in photoreceptor cells appear to be multiple. Evidence supporting the antigenic ignorance hypothesis by Lambe et al. (10) is found from results showing no differences between naive βgalTCR T cells existing in βgal Tg versus B10.A mice, suggesting diminished peripheral access to the Ag.
However, βgalTCR T cells clearly crossed the blood-retinal barrier, gained access to their target Ag in photoreceptor cells, and created the severe retinal pathology shown in lymphopenic recipients. Activated T cells, even with no particular specificity for an Ag within the retina or brain, penetrate the blood-brain barrier and the blood-retinal barrier (53, 54). The outcome of that penetration appears to result from an important, but undefined, local factor in susceptibility. The partial reversal of this state of ignorance by systemic treatment with IFNγ and PTx found in the arrβgal mice is further consistent with maintenance of antigenic ignorance. However, this state should leave the mice, as proposed by Lambe et al. (10), hypersensitive to attack by adoptive transfer of activated, autoreactive T cells. This is clearly not the case with the arrβgal Tg mice. While attack on βgal+ astrocytes by the T cells was promoted by in vitro activation protocols, no significant susceptibility was observed in the arrβgal mice. Obviously, there remains an important local barrier to pathogenesis in these mice that is unrelated to sequestration, as the retina appears to pose no barrier to activated T cells (54).
In a recent report using the MBP model for EAE, transfer of naive, TCR Tg, MBP-specific CD4 T cells into Rag(-/-) or TCRα(-/-) deficient mice was found to rapidly produce severe signs of EAE within 5 days of transfer (42). Pre-transfer of normal spleen cells provided sufficient regulatory T cells to prevent disease. In our LIP strategy, it is worth noting that disease required 8-9 weeks to develop in the arrβgal × Rag(-/-) mice, but then progressed to total destruction of the target cells in the retina. Recovery of the T cells from Rag(-/-) and arrβgal × Rag(-/-) recipient mice showed that their Ag-stimulated cytokine production was similar, but much different from naïve βgalTCR T cells that had not undergone LIP.
While adoptive transfer of activated, autoreactive CD4+ T cells is a common strategy to induce experimental autoimmune disease in the retina, use of Tg mice that express a target Ag in photoreceptor cells, in conjuction with CD4+ TCR Tg T cells, revealed the presence of multiple mechanisms of resistance to EAU. Transfer of naive, CD25-depleted, βgalTCR T cells into arrβgal mice on the Rag(-/-) background was the only procedure we have found to date that induced a high frequency of severe retinal disease. Since Rag(-/-) recipients have no endogenous Tregs that can limit autoimmunity, it suggests that such Tregs may be another source of resistance to EAU in arrβgal mice. Surprisingly, CD25-depletion of the donor βgalTCR mice prior to harvesting the T cells, and then depleting the CD25+ cells again prior to in vitro activation with APC and Ag still did not confer significant pathogenicity, even in lymphopenic recipients. Since the activation of CD25-depleted T cells prior to transfer induced Foxp3+ expression, additional evidence for peripheral induction of Tregs was found. In light of these results, we propose that the limitation on retinal disease acted via at least two possible mechanisms: 1) the CD25-depleted donor T cell population was still able to give rise to regulatory cells and 2) interaction of the transferred T cells with a local retinal cell population limited disease induction and/or progression. LIP-induced activation and in vitro Ag/APC stimulation both resulted in the appearance of CD4+CD25+Foxp3+ T cells, but significantly more disease was found in the lymphopenic recipients. We are pursuing the possibility that regulatory cells induced by Ag stimulation are more protective than those that appear during LIP.
Acknowledgements
The authors thank Heidi Roehrich for histology, Thien N. Sam for technical assistance, and Kathleen M. Pierson for animal care and genetic analysis. The authors have no conflicting financial interest.
Footnotes
This study was supported by NIH grants EY011542 and EY016376; Research to Prevent Blindness; The Anna Heilmaier Foundation, St. Paul, Minnesota; and the Minnesota Lions and Lionesses Clubs.
- CNS
- central nervous system
- PC
- photoreceptor cells
- Tg
- transgenic
- βgal
- Escherichia coli beta-galactosidase
- arrβgal
- photoreceptor expression of βgal
- GFAPβgal
- astrocyte expression of βgal
- βgalTCR
- βgal-specific TCR Tg T cells
- Treg
- regulatory T cell
- EAU
- experimental autoimmune uveoretinitis
- EAE
- experimental autoimmune encephalomyelitis
- HEL
- hen egg lysozyme
References
- 1.Carson MJ, Doose JM, Melchior B, Schmid CD, Ploix CC. CNS immune privilege: hiding in plain sight. Immunol. Rev. 2006;213:48–65. doi: 10.1111/j.1600-065X.2006.00441.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Cserr HF, Harling-Berg CJ, Knopf PM. Drainage of brain extracellular fluid into blood and deep cervical lymph and its immunological significance. Brain Path. 1992;2:269–276. doi: 10.1111/j.1750-3639.1992.tb00703.x. [DOI] [PubMed] [Google Scholar]
- 3.Yamada S, DePasquale M, Patlak SC, Cserr HF. Albumin outflow into deep cervical lymph from different regions of rabbit brain. Am. J. Physiol. 1991;261:H1197–H1204. doi: 10.1152/ajpheart.1991.261.4.H1197. [DOI] [PubMed] [Google Scholar]
- 4.Anderson MS, Venanzi ES, Klein L, Chen Z, Berzins S, Turley SJ, Von Boehmer H, Bronson R, Dierich A, Benoist C, Mathis D. Projection of an immunological self-shadow within the thymus by the aire protein. Science. 2002;298:1395–1401. doi: 10.1126/science.1075958. [DOI] [PubMed] [Google Scholar]
- 5.DeVoss J, Hou Y, Johannes K, Lu W, Liou GI, Rinn J, Chang H, Caspi R, Fong L, Anderson MS. Spontaneous autoimmunity prevented by thymic expression of a single self-antigen. J. Exp. Med. 2006;203:2727–2735. doi: 10.1084/jem.20061864. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Egwuagu CE, Charukamnoetkanok P, Gery I. Thymic expression of autoantigens correlates with resistance to autoimmune disease. J.Immunol. 1997;159:3109–3112. [PubMed] [Google Scholar]
- 7.McPherson SW, Gregerson DS. In: Nussenblatt RB, Whitcup SM, Caspi RR, Gery I, editors. Nucleotide sequences homologous to retinal S-antigen in the rat thymus lack a known EAU inducing epitope; Sixth International Symposium on the Immunology and Immunopathology of the Eye; Elsevier Sciences B.V., Bethesda, MD, USA. 1994.pp. 67–70. [Google Scholar]
- 8.Avichezer D, Grajewski RS, Chan CC, Mattapallil MJ, Silver PB, Raber JA, Liou GI, Wiggert B, Lewis GM, Donoso LA, Caspi RR. An immunologically privileged retinal antigen elicits tolerance: major role for central selection mechanisms. J. Exp. Med. 2003;198:1665–1676. doi: 10.1084/jem.20030413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Ham DI, Kim SJ, Chen J, Vistica BP, Fariss RN, Lee RS, Wawrousek EF, Takase H, Yu CR, Egwuagu CE, Chan CC, Gery I. Central immunotolerance in transgenic mice expressing a foreign antigen under control of the rhodopsin promoter. Invest. Ophthalmol. Vis. Sci. 2004;45:857–862. doi: 10.1167/iovs.03-1028. [DOI] [PubMed] [Google Scholar]
- 10.Lambe T, Leung JC, Ferry H, Bouriez-Jones T, Makinen K, Crockford TL, Jiang HR, Nickerson JM, Peltonen L, Forrester JV, Cornall RJ. Limited peripheral T cell anergy predisposes to retinal autoimmunity. J. Immunol. 2007;178:4276–4283. doi: 10.4049/jimmunol.178.7.4276. [DOI] [PubMed] [Google Scholar]
- 11.Grajewski RS, Silver PB, Agarwal RK, Su SB, Chan CC, Liou GI, Caspi RR. Endogenous IRBP can be dispensable for generation of natural CD4+CD25+ regulatory T cells that protect from IRBP-induced retinal autoimmunity. J. Exp. Med. 2006;203:851–856. doi: 10.1084/jem.20050429. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Fling SP, Donoso LA, Gregerson DS. In vitro unresponsiveness to autologous sequences of the immunopathogenic autoantigen, S-antigen. J. Immunol. 1991;147:483–489. [PubMed] [Google Scholar]
- 13.Gregerson DS. Peripheral expression of ocular antigens in regulation and therapy of ocular autoimmunity. Int. Rev. Immunol. 2002;21:101–121. doi: 10.1080/08830180212062. [DOI] [PubMed] [Google Scholar]
- 14.Gregerson DS, Xiao J. Failure of memory (CD44 high) CD4 T cells to recognize their target antigen in retina. J. Neuroimmunol. 2001;120:34–41. doi: 10.1016/s0165-5728(01)00406-4. [DOI] [PubMed] [Google Scholar]
- 15.Agarwal RK, Kang Y, Zambidis E, Scott DW, Chan CC, Caspi RR. Retroviral gene therapy with an immunoglobulin-antigen fusion construct protects from experimental autoimmune uveitis. J. Clin. Invest. 2000;106:245–252. doi: 10.1172/JCI9168. see comments. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.McPherson SW, Roberts JP, Gregerson DS. Systemic expression of rat soluble retinal antigen induces resistance to experimental autoimmune uveoretinitis. J. Immunol. 1999;163:4269–4276. [PubMed] [Google Scholar]
- 17.Xu H, Wawrousek EF, Redmond TM, Nickerson JM, Wiggert B, Chan CC, Caspi RR. Transgenic expression of an immunologically privileged retinal antigen extraocularly enhances self tolerance and abrogates susceptibility to autoimmune uveitis. Eur. J. Immunol. 2000;30:272–278. doi: 10.1002/1521-4141(200001)30:1<272::AID-IMMU272>3.0.CO;2-X. [DOI] [PubMed] [Google Scholar]
- 18.Silver PB, Agarwal RK, Su SB, Suffia I, Grajewski RS, Luger D, Chan CC, Mahdi RM, Nickerson JM, Caspi RR. Hydrodynamic vaccination with DNA encoding an immunologically privileged retinal antigen protects from autoimmunity through induction of regulatory T cells. J. Immunol. 2007;179:5146–5158. doi: 10.4049/jimmunol.179.8.5146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Gregerson DS, Torseth JW, McPherson SW, Roberts JP, Shinohara T, Zack DJ. Retinal expression of a neo-self antigen, beta-galactosidase, is not tolerogenic and creates a target for autoimmune uveoretinitis. J. Immunol. 1999;163:1073–1080. [PubMed] [Google Scholar]
- 20.Gregerson DS, Dou C. Spontaneous induction of immunoregulation by an endogenous retinal antigen. Invest. Ophthalmol. Vis. Sci. 2002;43:2984–2991. [PubMed] [Google Scholar]
- 21.McPherson SW, Yang J, Chan CC, Dou C, Gregerson DS. Resting CD8 T cells recognize beta-galactosidase expressed in the immune-privileged retina and mediate autoimmune disease when activated. Immunology. 2003;110:386–396. doi: 10.1046/j.1365-2567.2003.01750.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Kimura A, Singh D, Wawrousek EF, Kikuchi M, Nakamura M, Shinohara T. Both PCE-1/RX and OTX/CRX interactions are necessary for photoreceptor-specific gene expression. J. Biol. Chem. 2000;275:1152–1160. doi: 10.1074/jbc.275.2.1152. [DOI] [PubMed] [Google Scholar]
- 23.Kikuchi T, Raju K, Breitman ML, Shinohara T. The proximal promoter of the mouse arrestin gene directs gene expression in photoreceptor cells and contains an evolutionarily conserved retinal factor-binding site. Mol. Cell. Biol. 1993;13:4400–4408. doi: 10.1128/mcb.13.7.4400. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Johnson WB, Ruppe MD, Rockenstein EM, Price J, Sarthy VP, Verderber LC, Mucke L. Indicator expression directed by regulatory sequences of the glial fibrillary acidic protein (GFAP) gene: In vivo comparison of distinct GFAP-lacZ transgenes. Glia. 1995;13:174–184. doi: 10.1002/glia.440130304. [DOI] [PubMed] [Google Scholar]
- 25.Gregerson DS, Obritsch WF, Donoso LA. Oral tolerance in experimental autoimmune uveoretinitis. Distinct mechanisms of resistance are induced by low dose vs high dose feeding protocols. J. Immunol. 1993;151:5751–5761. [PubMed] [Google Scholar]
- 26.McPherson SW, Heuss ND, Roehrich H, Gregerson DS. Bystander killing of neurons by cytotoxic T cells specific for a glial antigen. Glia. 2006;53:457–466. doi: 10.1002/glia.20298. [DOI] [PubMed] [Google Scholar]
- 27.Caspi RR. Th1 and Th2 responses in pathogenesis and regulation of experimental autoimmune uveoretinitis. Int. Rev. Immunol. 2002;21:197–208. doi: 10.1080/08830180212063. [DOI] [PubMed] [Google Scholar]
- 28.McPherson SW, Roberts JP, Gregerson DS. Peripheral expression of rod photoreceptor arrestin induces an epitope-specific, protective response against experimental autoimmune uveoretinitis. Curr. Eye Res. 2005;30:491–502. doi: 10.1080/02713680590956270. [DOI] [PubMed] [Google Scholar]
- 29.Tang J, Zhu W, Silver PB, Su SB, Chan CC, Caspi RR. Autoimmune uveitis elicited with antigen-pulsed dendritic cells has a distinct clinical signature and is driven by unique effector mechanisms: initial encounter with autoantigen defines disease phenotype. J. Immunol. 2007;178:5578–5587. doi: 10.4049/jimmunol.178.9.5578. [DOI] [PubMed] [Google Scholar]
- 30.Peng Y, Han G, Shao H, Wang Y, Kaplan HJ, Sun D. Characterization of IL-17+ interphotoreceptor retinoid-binding protein-specific T cells in experimental autoimmune uveitis. Invest. Ophthalmol. Vis. Sci. 2007;48:4153–4161. doi: 10.1167/iovs.07-0251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Lafaille JJ. T-cell receptor transgenic mice in the study of autoimmune diseases. J. Autoimmun. 2004;22:95–106. doi: 10.1016/j.jaut.2003.10.007. [DOI] [PubMed] [Google Scholar]
- 32.Goverman J, Woods A, Larson L, Weiner LP, Hood L, Zaller DM. Transgenic mice that express a myelin basic protein-specific T cell receptor develop spontaneous autoimmunity. Cell. 1993;72:551–560. doi: 10.1016/0092-8674(93)90074-z. [DOI] [PubMed] [Google Scholar]
- 33.Caspi RR, Silver PB, Chan CC, Sun B, Agarwal RK, Wells J, Oddo S, Fujino Y, Najafian F, Wilder RL. Genetic susceptibility to experimental autoimmune uveoretinitis in the rat is associated with an elevated Th1 response. J. Immunol. 1996;157:2668–2675. [PubMed] [Google Scholar]
- 34.Amadi-Obi A, Yu CR, Liu X, Mahdi RM, Clarke GL, Nussenblatt RB, Gery I, Lee YS, Egwuagu CE. TH17 cells contribute to uveitis and scleritis and are expanded by IL-2 and inhibited by IL-27/STAT1. Nat. Med. 2007;13:711–718. doi: 10.1038/nm1585. [DOI] [PubMed] [Google Scholar]
- 35.Furuzawa-Carballeda J, Vargas-Rojas MI, Cabral AR. Autoimmune inflammation from the Th17 perspective. Autoimmun. Rev. 2007;6:169–175. doi: 10.1016/j.autrev.2006.10.002. [DOI] [PubMed] [Google Scholar]
- 36.Dick AD, Forrester JV, Liversidge J, Cope AP. The role of tumour necrosis factor (TNF-alpha) in experimental autoimmune uveoretinitis (EAU) Prog. Retin. Eye Res. 2004;23:617–637. doi: 10.1016/j.preteyeres.2004.06.005. [DOI] [PubMed] [Google Scholar]
- 37.Su SB, Grajewski RS, Luger D, Agarwal RK, Silver PB, Tang J, Tuo J, Chan CC, Caspi RR. Altered chemokine profile associated with exacerbated autoimmune pathology under conditions of genetic interferon-gamma deficiency. Invest. Ophthalmol. Vis. Sci. 2007;48:4616–4625. doi: 10.1167/iovs.07-0233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Fujimoto C, Yu CR, Shi G, Vistica BP, Wawrousek EF, Klinman DM, Chan CC, Egwuagu CE, Gery I. Pertussis toxin is superior to TLR ligands in enhancing pathogenic autoimmunity, targeted at a neo-self antigen, by triggering robust expansion of Th1 cells and their cytokine production. J. Immunol. 2006;177:6896–6903. doi: 10.4049/jimmunol.177.10.6896. [DOI] [PubMed] [Google Scholar]
- 39.Krupica T, Jr., Fry TJ, Mackall CL. Autoimmunity during lymphopenia: a two-hit model. Clin. Immunol. 2006;120:121–128. doi: 10.1016/j.clim.2006.04.569. [DOI] [PubMed] [Google Scholar]
- 40.Knoechel B, Lohr J, Kahn E, Bluestone JA, Abbas AK. Sequential development of interleukin 2-dependent effector and regulatory T cells in response to endogenous systemic antigen. J. Exp. Med. 2005;202:1375–1386. doi: 10.1084/jem.20050855. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Abbas AK, Lohr J, Knoechel B. Balancing autoaggressive and protective T cell responses. J. Autoimmun. 2007;28:59–61. doi: 10.1016/j.jaut.2007.02.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Cabbage SE, Huseby ES, Sather BD, Brabb T, Liggitt D, Goverman J. Regulatory T cells maintain long-term tolerance to myelin basic protein by inducing a novel, dynamic state of T cell tolerance. J. Immunol. 2007;178:887–896. doi: 10.4049/jimmunol.178.2.887. [DOI] [PubMed] [Google Scholar]
- 43.Cao Y, Toben C, Na SY, Stark K, Nitschke L, Peterson A, Gold R, Schimpl A, Hunig T. Induction of experimental autoimmune encephalomyelitis in transgenic mice expressing ovalbumin in oligodendrocytes. Eur. J. Immunol. 2006;36:207–215. doi: 10.1002/eji.200535211. [DOI] [PubMed] [Google Scholar]
- 44.Cabarrocas J, Piaggio E, Zappulla JP, Desbois S, Mars LT, Lassmann H, Liblau RS. A transgenic mouse model for T-cell ignorance of a glial autoantigen. J. Autoimmun. 2004;22:179–189. doi: 10.1016/j.jaut.2004.01.001. [DOI] [PubMed] [Google Scholar]
- 45.Campagnoni AT, Pribyl TM, Campagnoni CW, Kampf K, Amur-Umarjee S, Landry CF, Handley VW, Newman SL, Garbay B, Kitamura K. Structure and developmental regulation of Golli-mbp, a 105-kilobase gene that encompasses the myelin basic protein gene and is expressed in cells in the oligodendrocyte lineage in the brain. J. Biol. Chem. 1993;268:4930–4938. [PubMed] [Google Scholar]
- 46.Pribyl TM, Campagnoni CW, Kampf K, Kashima T, Handley VW, McMahon J, Campagnoni AT. The human myelin basic protein gene is included within a 179-kilobase transcription unit: expression in the immune and central nervous systems. Proc. Natl. Acad. Sci. U S A. 1993;90:10695–10699. doi: 10.1073/pnas.90.22.10695. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Clark L, Otvos L, Jr., Stein PL, Zhang XM, Skorupa AF, Lesh GE, McMorris FA, Heber-Katz E. Golli-induced paralysis: a study in anergy and disease. J. Immunol. 1999;162:4300–4310. [PubMed] [Google Scholar]
- 48.Targoni OS, Lehmann PV. Endogenous myelin basic protein inactivates the high avidity T cell repertoire. J. Exp. Med. 1998;187:2055–2063. doi: 10.1084/jem.187.12.2055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Perchellet A, Stromnes I, Pang JM, Goverman J. CD8+ T cells maintain tolerance to myelin basic protein by ‘epitope theft’. Nat. Immunol. 2004;5:606–614. doi: 10.1038/ni1073. [DOI] [PubMed] [Google Scholar]
- 50.Fairchild PJ, Wildgoose R, Atherton E, Webb S, Wraith DC. An autoantigenic T cell epitope forms unstable complexes with class II MHC: a novel route for escape from tolerance induction. Int. Immunol. 1993;5:1151–1158. doi: 10.1093/intimm/5.9.1151. [DOI] [PubMed] [Google Scholar]
- 51.Maverakis E, Beech J, Stevens DB, Ametani A, Brossay L, van den Elzen P, Mendoza R, Thai Q, Macias LH, Ethell D, Campagnoni CW, Campagnoni AT, Sette A, Sercarz EE. Autoreactive T cells can be protected from tolerance induction through competition by flanking determinants for access to class II MHC. Proc. Natl. Acad. Sci. U S A. 2003;100:5342–5347. doi: 10.1073/pnas.0936151100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Xu H, Chen M, Reid DM, Forrester JV. LYVE-1-positive macrophages are present in normal murine eyes. Invest. Ophthalmol. Vis. Sci. 2007;48:2162–2171. doi: 10.1167/iovs.06-0783. [DOI] [PubMed] [Google Scholar]
- 53.Hickey W, Hsu B, Kimura H. T-lymphocyte entry into the central nervous system. J. Neurosci. Res. 1991;28:254–260. doi: 10.1002/jnr.490280213. [DOI] [PubMed] [Google Scholar]
- 54.Prendergast RA, Iliff CE, Coskuncan NM, Caspi RR, Sartani G, Tarrant TK, Lutty GA, McLeod DS. T cell traffic and the inflammatory response in experimental autoimmune uveoretinitis. Invest. Ophthalmol. Vis. Sci. 1998;39:754–762. [PubMed] [Google Scholar]