

Abstract
The synthesis of 4,6-O-benzylidene protected 2-deoxy-arabino, 3-deoxy-arabino-, and 3-deoxy-ribo-thioglycosides is described and their glycosylation reactions, with activation by either 1-benzenesulfinyl piperidine/trifluoromethansulfonic anhydride or diphenyl sulfoxide/trifluoromethanesulfonic anhydride, studied. In contrast to the corresponding 4,6-O-benzylidene protected glucosyl and mannosyl donors, which are α- and β-selective, respectively, poor diastereoselectivity is observed in all cases. The reasons for this poor selectivity are discussed in terms of the interaction between the C2-O2 and C3-O3 bonds in the glucosyl and mannosyl donors and of the influence of this interaction on the ease of formation of the intermediate glycosyl oxacarbenium ions.
Introduction
One of the more intriguing aspects of the 4,6-O-benzylidene-directed β-mannopyranosylation reactions developed in this laboratory1–6 is the change in selectivity observed with the corresponding glucopyranosyl and galactopyranosyl donors, which are α-selective.4,6–9 This contrast in selectivity, originally seen with donors 1 and 2,7 on activation with trifluoromethanesulfonic anhydride in the presence of a hindered non-nucleophilic base such as 2,4,6-tri-tert-butylpyrimidine (TTBP),10 and subsequently observed with donors 3 and 4, on activation with 1-benzenesulfinyl piperidine (BSP) and triflic anhydride,4 is reflected in the reduced β-selectivity seen on going from the 3,4-O-carbonate protected 6-deoxymanno (rhamno) donor 5 to the corresponding gluco donor 6.11
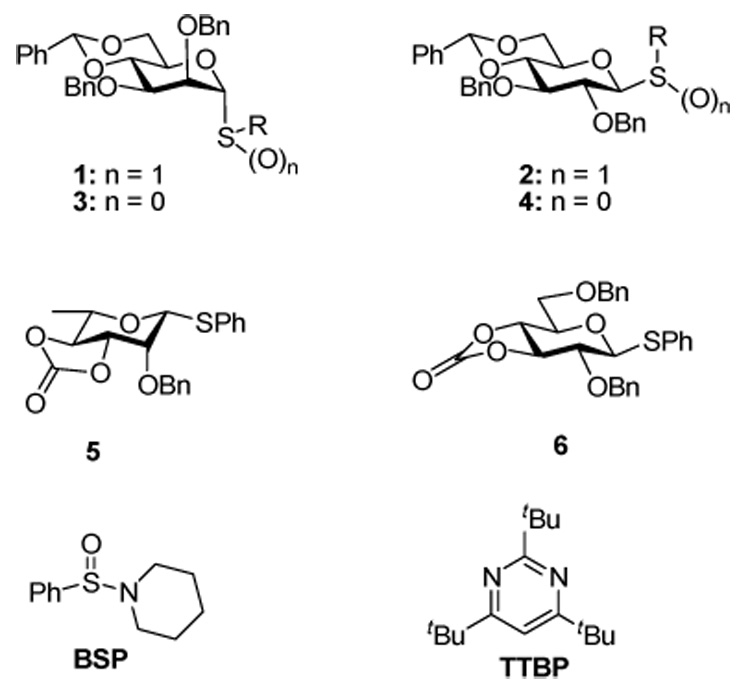
This difference in selectivity between the 4,6-O-benzylidene protected manno and glucopyranose series has been examined computationally by Nukuda and Whitfield and their coworkers, who viewed the problem in terms of the conformations of the intermediate oxacarbenium ions and of the protonated glycosides resulting from methanol attack on the oxacarbenium ions.12 However, it was recognized that the analysis was based on thermodynamic considerations and might not be relevant to kinetically determined stereoselectivity. It is well-known that the anomeric effect is higher in mannopyranosides than in glucopyranosides, leading to a greater thermodynamic preference for the α-anomers in the mannose series,13 and it is evident that the glycosylation stereochemistries obtained with donors 1–4 at reaction temperatures from −78 to −60 °C are kinetic in origin. Accordingly, while the geometries of the 4,6-O-benzylidene protected oxacarbenium ions computed by the Nukuda and Whitfield groups are taken into account in the discussion below, the rationale for glycosylation stereochemistry presented by these authors is not further considered. Differences in reactivity of mannopyranosyl and glucopyranosyl oxacarbenium ions have also been commented recently by Lucero and Woerpel,14 in the context of a broader investigation into the synthesis of C-glycosides and the influence of substituents on the stereoselectivity of oxacarbenium ion trapping.15 In the absence of a conformationally restricting group, such as the benzylidene acetal, these authors considered the tetra-O-benzylmannopyranosyl cation to exist predominantly in the 3H4 conformer but to react via the less populated 4H3 form, in a typical Curtin-Hammett kinetic scheme to afford the α-C-glycosides in high yield. The α-selective C-glycosylation of the tetra-O-benzylglucopyranosyl cation was also considered to take place through the 4H3 conformer.14 With the benzylidene acetal in place, and the 3H4 conformer of the oxacarbenium ion excluded from consideration, the problem is simplified and only the 3H4 conformer need be considered along with the somewhat related B2,5 and 4E conformers preferred by the benzylidene protected manno and glucopyranosyl cations according to the calculations of Nukuda, Whitfield, and their coworkers.12
We currently view the mechanism of the 4,6-O-benzylidene directed glycosylations, which are preformed operationally by pre-activation of the donor before addition of the acceptor, as proceeding through the intermediacy of a covalent glycosyl triflate 7, which has been demonstrated spectroscopically to have the α-configuration in both the manno and the gluco series.7,16 This intermediate, which is formed rapidly and which is stable for many hours at low temperature in CD2Cl2, acts as a reservoir for a transient contact ion pair 8 (CIP) that is the true actual glycosylating species in the β-mannosylation reaction.17 This contact ion pair is in turn in equilibrium with a solvent separated ion pair (SSIP) 9, which we presume to be the source of the α-glycosides (Scheme 1). The function of the benzylidene acetal is to shift the series of equilibria between 7 and the two ions as far as possible toward the covalent triflate 7, thereby minimizing the population of the SSIP 9 and, at least in the mannose series, the formation of α-glycosides. The benzylidene acetal exerts its influence through a combination of torsional factors that come into play as the covalent triflate chair conformer flattens to the oxacarbenium ion in 8,18,19 and by maintaining the C5-C6 bond in the most electron-withdrawing tg conformer.20 On this basis, the α-selective glucosylations arise through the SSIP 9 and the problem is reduced to one of understanding the reasons for its greater population in the glucose series. There remains the alternative possibility that the α-glucosides are formed via the intermediacy of a second contact ion pair, with the triflate counter ion shielding the β-face of the oxacarbenium ion, which would necessarily be in equilibrium with a covalent β-glycosyl triflate, much as envisaged by Lemieux in his halide-ion promoted α-glucosylations.21 While we can not exclude this mechanism conclusively, we consider it unlikely on the grounds that our NMR experiments provided no evidence for the existence of a β-glucosyl triflate.7
Scheme 1.
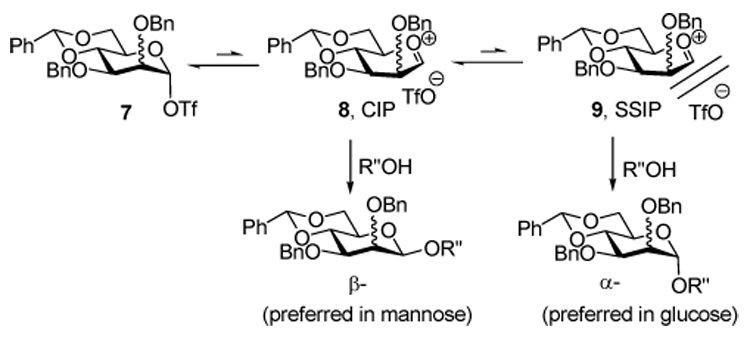
The Glycosylation Mechanism
We considered that the shift toward the SSIP 9 implied by the change from β- to α-selectivity observed on going from the manno to the gluco series might be due to greater repulsion between the O-2 substituent and the triflate counter ion in the CIP 8 in the gluco series, which would have the effect of facilitating SSIP formation (Figure 1).
Figure 1.
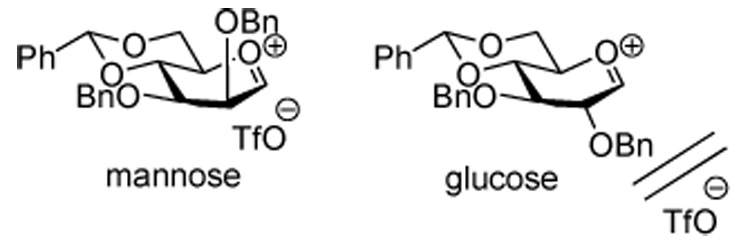
Destabilizing Interaction in the Contact Ion Pair, Illustrated for the 4,6-O-Benzylidene Protected Glucosyl Triflates.
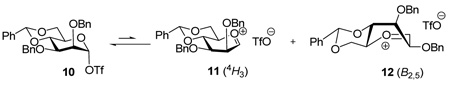
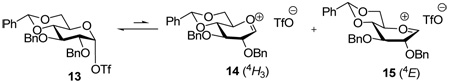
Alternatively, we were intrigued by the notion that the O2-C2-C3-O3 torsional interaction plays a significant role in the observed differing selectivities between the manno and gluco series. Effectively, as the mannosyl triflate 10 collapses to the ion pair, in either the 4H3 chair 11 or the B2,5 conformer 12 preferred by Nukuda and Whitfield,12 the O2-C2-C3-O3 torsion angle increases (Table 1), whereas when the gluco triflate 13 is transformed into either the 4H3 chair 14 or the 4E conformer 15 favored by the computations12, the corresponding torsion angle is reduced (Table 2).
Table 1.
Torsional angle values for mannosyl triflate (10), and the 4H3 (11) and B2,5 (12) conformers of mannosyl cation.22
 | ||
|---|---|---|
| O2-C2-C3-O3 | O2-C2-C1-O5 | |
| Torsion angle | Torsion angle | |
| 10 | 60 ° | 60 ° |
| 11 | 45 ° | 135 ° |
| Δ (10 → 11) | −15 ° | +75 ° |
| 12 | 60 ° | 180 ° |
| Δ (10 → 12) | 0 ° | +120 ° |
Table 2.
Torsional angle values for glucosyl triflate (13), and the 4H3 (14) and 4E (15) conformers of glucosyl cation.
 | ||
|---|---|---|
| O2-C2-C3-O3 | O2-C2-C1-O5 | |
| Torsion angle | Torsion angle | |
| 13 | 60 ° | 180 ° |
| 14 | 75 ° | 105 ° |
| Δ (13 → 14) | +15 ° | −75 ° |
| 15 | 90 ° | 120 ° |
| Δ (13 → 15) | +30 ° | −60 ° |
Examination of the change in the O2-C2-C1-O5 torsion angle with formation of the oxacarbenium ion is also instructive. In mannose (Table 1) there is an increase in this torsion angle on going from the covalent triflate 10 to the 4H3 oxacarbenium ion 11, and maximization of this torsion angle in the B2,5 conformer 12 of the oxacarbenium ion. On the other hand, in glucose the O2-C2-C1-O5 torsion angle, which begins at the maximum of 180 ° in the covalent triflate 13, undergoes a significant reduction regardless of whether the oxacarbenium ion exists in the 4H3 or 4E conformations, 14 and 15, respectively (Table 2). By analogy with Bols’ rationale for the benzylidene effect,20 it might be inferred that the higher O2-C2-C1-O5 torsion angle in the mannose oxacarbenium ions 12 and 13 maximizes the electron-withdrawing effect of the C2-O2 bond, as compared to that in the glucosyl oxacarbenium ions 14 and 15. This in turn would contribute toward a destabilization of the mannosyl oxacarbenium ions with respect to their glucosyl counterparts, which translates into a longer lived glucosyl oxacarbenium ion and a concomitant loss of selectivity.
The effect of the O2-O3 interaction on the rate of acid catalyzed hydrolyzes of methyl glycopyranosides was one of several factors considered by Edward,23 Feather and Harris,24 and others25 in some of the classical studies in the area, but has subsequently been overlooked. With a view to probing these effects we have investigated the glycosylation stereoselectivity of a set of 4,6-O-benzylidene protected 2- and 3-deoxythioglycosides and describe here the results of our investigations.
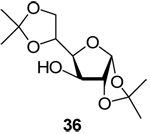
Results
Preparation of Donors
The synthesis of the 2-deoxy donor 19 was straightforward and followed the protocol outlined in Scheme 2 starting from triacetyl glucal.
Scheme 2. Synthesis of the 2-Deoxy donor 19.
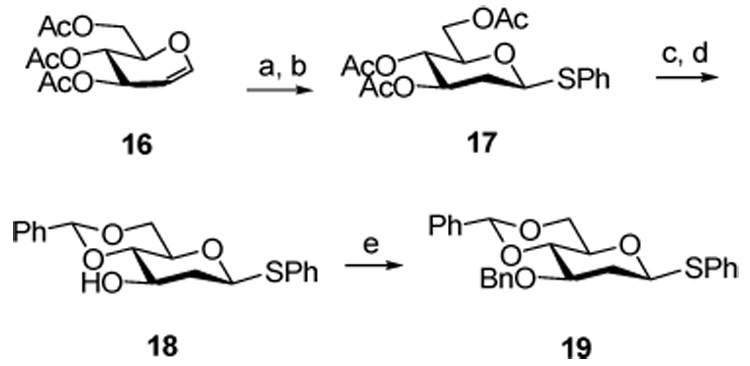
Reagents and conditions: a) HCl, toluene, 0 °C; b) PhSH, (i-Pr)2EtN, toluene, r.t., 47% over two steps; c) NaOMe, MeOH; d) PhCH(OMe)2, p-TsOH, DMF, 50 °C, 60% over two steps; e) NaH, BnBr, THF, reflux, 73%.
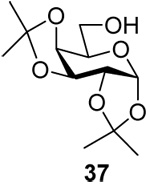
Obvious routes to the 3-deoxy donors beginning with the Barton deoxygenation of 1,2;5,6-di-O-acetoneglucofuranose26 and proceeding through 3-deoxy-β-d-ribo-hexopyranose and 1,2,4,6-tetra-O-acetyl-3-deoxy-β-d-ribo-hexopyranose were not satisfactory owing to formation of furanose isomers in significant amounts.27 Eventually, a synthesis was devised in which 1,2;5,6-di-O-acetoneglucofuranose was converted to the known 3-deoxy-3-iodo-allofuranose derivative 20,28,29 which, on deprotection in acidic media and subsequent acetylation gave mainly the desired pyranose form. Radical deiodination then afforded 2127 in good yield. Treatment with thiophenol and boron trifluoride afforded a 1:4 separable mixture of the anomeric thioglycosides 22 and 23, of which only the major β-isomer was processed through the subsequent routine steps of the synthesis to give the 3-deoxy ribo donor 25 (Scheme 3).
Scheme 3. Preparation of the 3-Deoxy ribo Donor 25.
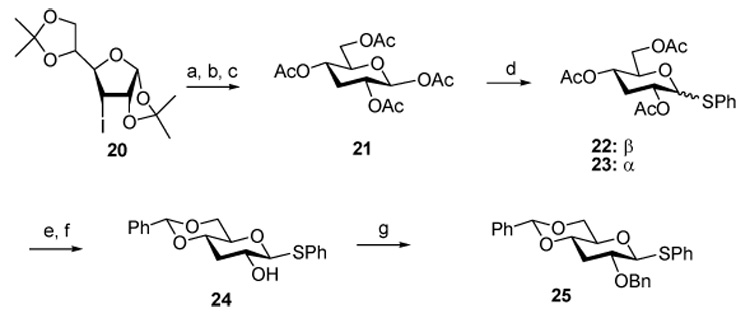
Reagents and conditions: a) IR-120 (H+), H2O, 80 °C; b) Ac2O, Pyridine, DMAP, r.t., 85% for two steps; c) Bu3SnH, AIBN, toluene, reflux, 78%; d) PhSH, BF3·Et2O, DCM, 0 °C, 97%, α:β = 1:4; e) NaOMe, MeOH; f) PhCH(OMe)2, p-TsOH, DMF, 50 °C, 96% for two steps; g) NaH, BnBr, THF, reflux, 93%.
Adapting a classical scheme for the formation of β-mannopyranosides from the corresponding β-glucopyrnaosides,30–35 Dess-Martin oxidation36 of 24 gave the corresponding ketone, which was stereoselectively reduced with L-selectride® to give the 3-deoxy arabino product 26. Benzylation then afforded the desired donor 27 (Scheme 4).
Scheme 4. Preparation of the 3-Deoxy arabino Donor 27.
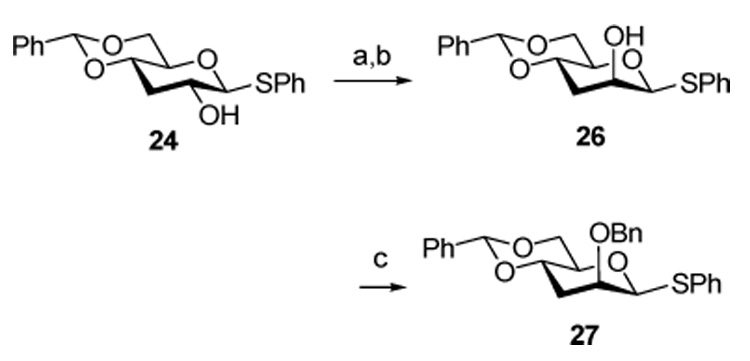
Reagents and conditions: a) Dess-Martin periodinane, DCM, r.t.; b) L-selectride, THF, −78 °C, 81% for two steps; c) NaH, BnBr, THF, reflux, 94%.
Investigation of Key Intermediates
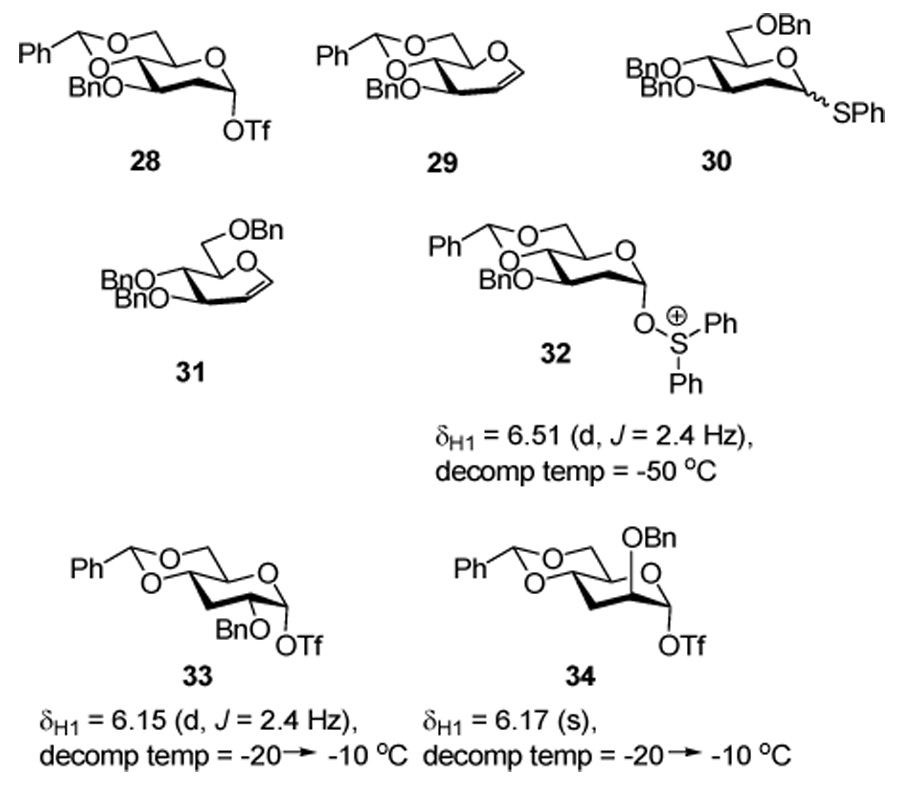
Exploratory experiments conducted in CD2Cl2 by variable temperature 1H-NMR spectroscopy revealed the 2-deoxy donor 19 to be fully activated by the BSP/Tf2O combination within minutes at −60 °C. However, only minor signals were observed in the region anticipated16 (δ 6.0–6.3) for the anomeric signal of the putative glycosyl triflate intermediate 28. On warming to −30 °C, one major compound, the glycal 29,37 was formed, as determined from key resonances at δ 6.38 (dd, J = 6.0, 1.2 Hz) and δ 4.86 (dd, J = 6.0, 2.0 Hz) indicative of the olefinic hydrogens, along with other unidentified byproducts. The result suggested the triflate 28 to be too unstable for the glycosylations in the 2-deoxy series. This behavior appears to be typical of the 2-deoxy series as a parallel experiment with donor 3038 also failed to afford an observable glycosyl triflate but provided instead tribenzyl glucal 31 on warming to −40 °C. Activation of thioglycoside 19 with 2 equivalents of diphenyl sulfoxide39,40 and 1.1 equivalents of triflic anhydride was also rapid and provided a putative intermediate glycosyl sulfonium ion 32, of the type observed by Gin in his studies on the activation of anomeric hemiacetals with diphenyl sulfoxide and triflic anhydride.41 On warming, decomposition of this intermediate was complete by −50 °C. In the 3-deoxy series, the chemistry reverted to more typical pattern observed previously in our laboratory4,7,11,16,42 with clean activation of both 25 and 27 by the BSP/Tf2O couple at −60 °C in CD2Cl2 and the formation of the putative glycosyl triflates 33 and 34, respectively, neither of which underwent decomposition until the temperature was raised to −10 °C.
Glycosylation Reactions
In view of the variable temperature NMR experiments, couplings to the 2-deoxy donor 19 were conducted at −60 °C with preactivation by a combination of 2 equivalents of diphenyl sulfoxide and 1.1 equivalents of triflic anhydride in the presence of TTBP, before addition of the acceptor alcohol. Satisfactory yields were obtained in this manner but selectivities were poor, approaching a maximum of 4:1 β:α with a primary alcohol (Table 3). As determined by NMR spectroscopy of the crude reaction mixtures, comparable results were obtained when diphenyl sulfoxide was replaced by BSP, but isolated yields were reduced owing to difficulties in purification and are hence not reported.
Table 3.
Glycosylation of the 2-Deoxy Donor 19.
| Acceptor | Product | α/β ratioa | Yield, % |
|---|---|---|---|
 |
 |
1:1.5 | 75 |
 |
 |
1:1.7 | 75 |
 |
 |
1:4 | 61 |
 |
 |
1:1.9 | 30 |
determined by 1H-NMR spectroscopy on the reaction mixture.
Glycosylation reactions with the 3-deoxy donors 25 and 27 were conducted with preactivation by the BSP/Tf2O couple in the presence of TTBP before addition of the acceptor, and are thought to proceed via the intermediacy of the glycosyl triflates 33 and 34, respectively, on the basis of the variable temperature experiments. The results of these experiments, all of which proceeded with good yield but modest selectivity, are reported in Table 4 and Table 5.
Table 4.
Glycosylation of the 3-Deoxy Donor 25.
| Acceptor | Product | α/β ratioa | Yield, % |
|---|---|---|---|
 |
 |
1.2:1 | 95 |
 |
 |
4.7:1 | 76 |
 |
 |
1:1.8 | 92 |
determined by 1H-NMR spectroscopy on the reaction mixture.
Table 5.
Glycosylation of the 3-Deoxy Donor 27.
| Acceptor | Product | α/β ratioa | Yield, % |
|---|---|---|---|
 |
 |
1.9:1 | 85 |
 |
 |
1.3:1 | 80 |
 |
 |
3.6:1 | 83 |
determined by 1H-NMR spectroscopy on the reaction mixture.
While the assignment of stereochemistry in the 2-deoxy series (Table 3) and in the 3-deoxy ribo series (Table 4) was straightforward based on the 3JH1, H2 coupling constants, that in the 3-deoxy arabino series relies on the magnitude of the 1JC1,H1 coupling constants.43,44
Discussion
The lack of stereoselectivity observed with the 2-deoxy donor 19 (Table 3) is not surprising in view of the well-known difficulties in the stereoselective formation of 2-deoxyglycopyranosidic linkages in general,45–47 and is somewhat anticipated on the basis of the variable temperature experiments and the manifest instability of the intermediates. These problems obviously arise from the greater lability of the 2-deoxy systems, which is the result of the absence of the electron-withdrawing C2-O2 bond and the greater facility of oxacarbenium ion formation that this affords.24,25,48–50 Accordingly, no further account is taken here of the results presented in Table 3. We do note, however, the reduced α-selectivity seen with the donor 19 as compared to the high α-selectivity seen by Kahne and coworkers with a series of 2,6-dideoxy glycosyl sulfoxides in the course of their syntheses of ciclamycin:51,52 evidently the 4,6-O-benzylidene acetal is not without influence over the course of these reactions. In a similar vein, we note the β-selective glycosylations reported by Hashimoto on activation of the 2-deoxyglycosyl phosphite 49, and related phosphates, with trimethylsilyl triflate at −94 °C, which were considered to take place via intermediate glycosyl triflates, something that can not be excluded on the basis of the experiments presented here.53

The results obtained with the 3-deoxy donors 25 and 27 (Table 4 and Table 5) are more germane to the question in hand. Various 3-deoxyglycopyranosyl donors have been described in the literature but, with rare exceptions,54 all carried ester protecting groups on O2 to direct the glycosylation stereochemistry55–58 and are not relevant to the present discussion. Interestingly, although the coupling was carried out under very different conditions to the ones employed here, the benzyl protected 3-deoxyglucosyl chloride 50 gave an approximately 1:1 mixture of two anomers on coupling to methyl 2,3,6-tri-O-benzyl-β-d-glucopyranoside by the Lemieux chloride ion-catalyzed protocol.21,54 Classical work on the acid-catalyzed hydrolysis of simple glycopyranosides revealed that removal of the C3-O3 bond has only a relatively minor accelerating effect,23–25,59 as might also be inferred from more recent work on the disarming effect of ester protecting groups and their location on the pyranoside ring.
In the 3-deoxy ribo series (Table 4) there is a significant preference toward the formation of the β-glycosides, away from the α-selectivity previously observed with the glucosyl donors 2 and 4. This does not accord with the hypothesis of Figure 1 in which repulsion between the 2-O-benzyl group and the triflate counter ion is responsible for a general loosening of the ion pair in the glucose series. Possibly, the move away from α-selectivity with donor 25 is a reflection of a minor change in conformation of the oxacarbenium ion in the resulting CIP which facilitates β-face attack. As noted below, conformation effects have been shown to play a major role in the stereoselectivity of these glycosylation reactions.
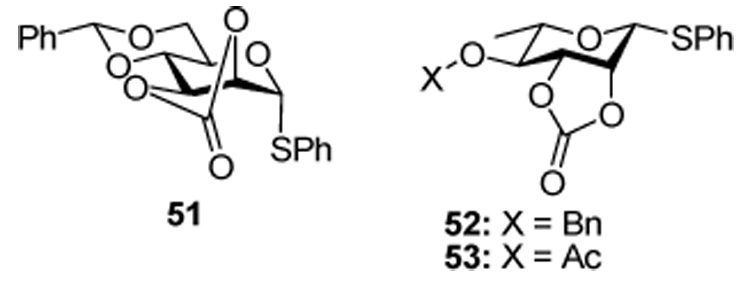
In the 3-deoxy-arabino series (Table 5) there is a strong shift away from the high β-selectivity typically observed with donors 1 and 3. Obviously the absence of the electron-withdrawing C3-O3 bond can not be totally neglected here, but it seems apparent that the loss of the compression of the O2-C2-C3-O3 torsion angle seen in the mannose series (Table 1) on going from the covalent triflate to the oxacarbenium ion leads to a higher population of CIP and consequently of the SSIP with an overall reduction in β-stereoselectivity. In support of this argument, we recall the mannosyl donor 51 and its rhamnosyl counterparts 52 and 53, which adopt a half-chair conformation in which the O2-C2-C3-O3 torsion angle is 25–30 °, as opposed to the 60 ° in 1 and 3, and which are highly α-selective in homogeneous glycosylation reactions:60–62 subtle conformational effects play a major role in these glycosylation reactions, even to the point of overriding powerful electronic effects.
Finally, we return to the possibility of the formation of the α-glucosides via the intermediacy of covalent β-glucosyl triflate and an associated transient β-face contact ion pair. While this mechanism can not be ruled out as a contributing factor to the overall α-selectivity seen in the glucose series,4,6–9 the poor selectivity seen with the 3-deoxy donors 25 and 27, irrespective of the configuration at C2, suggests that it is not a major pathway.
Experimental Section
General coupling protocol for S-Phenyl 3-O-benzyl-4,6-O-benzylidene-2-deoxy-β-d-arabino-thiohexopyranoside (19) using Ph2SO (method A)
To a stirred solution of 19 (100 mg, 0.23 mmol) in CH2Cl2 (6 mL) containing Ph2SO (93 mg, 0.46 mmol), TTBP (114 mg, 0.46 mmol), and activated 3 Å powdered molecular sieves, Tf2O (42.6 µL, 0.25 mmol) was added at −60 °C under argon atmosphere. After 5 minutes, a solution of acceptor (0.28 mmol) in CH2Cl2 (2.4 mL) was added and the reaction mixture was stirred for an additional 2 min at −60 °C, then warmed to room temperature, filtered, washed (saturated aqueous NaHCO3, brine), and dried (Na2SO4). The solvent was removed under reduced pressure. The crude was filtered through a pad of silica gel (with ethyl acetate as an eluent) and chromatographed using SiO2.
General coupling protocol for S-Phenyl 2-O-benzyl-4,6-O-benzylidene-3-deoxy-β-d-ribo-thiohexopyranoside (25) and S-Phenyl 2-O-benzyl-4,6-O-benzylidene-3-deoxy-β-d-arabino-thiohexopyranoside (27) using BSP (method B)
To a stirred solution of 25 or 27 (50 mg, 0.12 mmol) in CH2Cl2 (3 mL) containing BSP (24 mg, 0.12 mmol), TTBP (57 mg, 0.23 mmol), and activated 3 Å powdered molecular sieves, Tf2O (21.3 µL, 0.13 mmol) was added at −60 °C under argon atmosphere. After 5 minutes, a solution of acceptor (0.14 mmol) in CH2Cl2 (1.2 mL) was added and the reaction mixture was stirred for an additional 2 min at −60 °C, then warmed to room temperature, filtered, washed (saturated aqueous NaHCO3, brine), and dried (Na2SO4). The solvent was removed under reduced pressure. The crude was filtered through a pad of silica gel (with ethyl acetate as an eluent) and chromatographed using SiO2.
Methyl 4-O-(3’-O-benzyl-4’,6’-O-benzylidene-2’-deoxy-α-d-arabino-thiohexopyranosyl)-2,3-O-isopropylidene-α-l-rhamnoside (39α) and Methyl 4-O-(3’-O-benzyl-4’,6’-O-benzylidene-2’-deoxy-β-d-arabino-thiohexopyranosyl)-2,3-O-isopropylidene-α-l-rhamnoside (39β)
Prepared by method A, purified by means of radial chromatography, eluting consecutive plates with haxanes to 1:9 diethyl ether:hexanes, and then with hexanes to 1:19 ethyl acetate:hexanes. Combined yield 94 mg (75%, 1:1.5 α/β). 39α. [α]24d +50.0 (c 0.53, CHCl3); 1H NMR (500 MHz, CDCl3) δ: 7.52 – 7.57 (m, 2H), 7.27 – 7.43 (m, 8H), 5.64 (s, 1H), 5.00 (d, J = 3.5 Hz, 1H), 4.87 (d, J = 11.9 Hz, 1H), 4.86 (s, 1H), 4.70 (d, J = 11.9 Hz, 1H), 4.29 (dd, J = 10.0, 5.0 Hz, 1H), 4.07 – 4.15 (m, 3H), 4.03 (ddd, J = 11.2, 9.1, 5.1 Hz, 1H), 3.75 (t, J = 10.1 Hz, 1H), 3.71 (t, J = 9.4 Hz, 1H), 3.65 (dd, J = 10.0, 6.3 Hz, 1H), 3.37 (s, 3H), 3.31 – 3.36 (m, 1H), 2.27 (dd, J = 13.2, 5.1 Hz, 1H), 1.82 (ddd, J = 13.3, 11.2, 3.9 Hz, 1H), 1.52 (s, 3H), 1.34 (s, 3H), 1.26 (d, J = 6.4 Hz, 3H); 13C NMR (125 MHz, CDCl3) δ: 138.7, 137.7, 128.8, 128.4, 128.2, 127.7, 127.6, 126.1, 109.1, 101.3, 98.6, 98.0, 84.0, 80.3, 76.9, 75.9, 73.0, 72.8, 69.1, 64.8, 63.0, 54.8, 36.6, 28.2, 26.4, 17.5; ESIHRMS Calcd for C30H38NaO9 [M+Na]+: 565.2414. Found 565.2409. 39β. White solid. M.p. 108–111 °C; [α]24d −40.3 (c 0.31, CHCl3); 1H NMR (500 MHz, CDCl3) δ: 7.49 – 7.54 (m, 2H), 7.27 – 7.42 (m, 8H), 5.61 (s, 1H), 5.04 (dd, J = 9.9, 2.0 Hz, 1H), 4.86 (s, 1H), 4.81 (d, J = 12.1 Hz, 1H), 4.72 (d, J = 12.3 Hz, 1H), 4.28 (dd, J = 10.5, 5.0 Hz, 1H), 4.13 – 4.18 (m, 1H), 4.09 (d, J = 5.5 Hz, 1H), 3.83 (t, J = 10.3 Hz, 1H), 3.77 (ddd, J = 11.2, 9.0, 5.1 Hz, 1H), 3.58 – 3.70 (m, 3H), 3.37 (s, 3H), 3.30 – 3.36 (m, 1H), 2.36 (ddd, J = 12.9, 5.1, 2.1 Hz, 1H), 1.65 (ddd, J = 12.8, 11.2, 9.9 Hz, 1H), 1.51 (s, 3H), 1.35 (s, 3H), 1.28 (d, J = 5.9 Hz, 3H); 13C NMR (125 MHz, CDCl3) δ: 138.5, 137.6, 128.9, 128.4, 128.2, 127.7, 127.6, 126.1, 109.3, 101.3, 98.7, 97.9, 83.2, 78.5, 77.8, 76.1, 74.6, 72.4, 69.0, 66.6, 64.1, 54.8, 37.8, 27.9, 26.4, 17.5; ESIHRMS Calcd for C30H38NaO9 [M+Na]+: 565.2414. Found 565.2411.
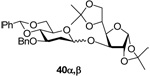
3-O-(3’-O-benzyl-4’,6’-O-benzylidene-2’-deoxy-α-d-arabino-thiohexopyranosyl)-1,2:5,6-di-O-isopropylidene-α-d-glucose (40α) and 3-O-(3’-O-benzyl-4’,6’-O-benzylidene-2’-deoxy-β-d-arabino-thiohexopyranosyl)-1,2:5,6-di-O-isopropylidene-α-d-glucose (40β)
Prepared by method A, purified by means of radial chromatography, eluting consecutive plates with hexanes to 1:9 ethyl acetate:hexanes, and then with 7:13 or 1:4 diethyl ether:hexanes for 40α and 40β, respectively. 40α yield 39 mg (29%), 40β yield 62 mg (46%). 40α. [α]24d +38.4 (c 0.25, CHCl3); 1H NMR (500 MHz, CDCl3) δ: 7.50 – 7.57 (m, 2H), 7.28 – 7.45 (m, 8H), 5.89 (d, J = 3.7 Hz, 1H), 5.66 (s, 1H), 5.21 (d, J = 3.7 Hz, 1H), 4.87 (d, J = 11.7 Hz, 1H), 4.69 (d, J = 12.5 Hz, 1H), 4.56 (d, J = 3.7 Hz, 1H), 4.33 (dd, J = 9.5, 4.4 Hz, 1H), 4.24 (d, J = 2.9 Hz, 1H), 4.09 – 4.18 (m, 2H), 3.96 – 4.09 (m, 3H), 3.78 – 3.90 (m, 2H), 3.73 (t, J = 8.8 Hz, 1H), 2.29 (dd, J = 13.2, 5.1 Hz, 1H), 1.78 – 1.86 (m, 1H), 1.50 (s, 3H), 1.41 (s, 3H), 1.33 (s, 3H), 1.32 (s, 3H); 13C NMR (125 MHz, CDCl3) δ: 138.6, 137.4, 129.0, 128.4, 128.3, 127.6, 126.0, 112.1, 109.3, 105.3, 101.3, 99.2, 84.1, 83.8, 81.4, 80.2, 73.1, 72.6, 72.4, 69.0, 67.9, 63.9, 36.1, 26.9, 26.3, 25.4; ESIHRMS Calcd for C32H40NaO10 [M+Na]+: 607.2519. Found 607.2524. 40β. [α]24d −15.8 (c 0.36, CHCl3); 1H NMR (500 MHz, CDCl3) δ: 7.47 – 7.54 (m, 2H), 7.27 – 7.44 (m, 8H), 5.92 (d, J = 3.7 Hz, 1H), 5.61 (s, 1H), 4.83 (d, J = 12.1 Hz, 1H), 4.67 – 4.73 (m, 2H), 4.49 (d, J = 3.9 Hz, 1H), 4.28 – 4.38 (m, 4H), 4.00 – 4.07 (m, 2H), 3.82 (t, J = 10.3 Hz, 1H), 3.67 – 3.77 (m, 2H), 3.34 (td, J = 13.9, 5.0 Hz, 1H), 2.26 (ddd, J = 13.2, 4.8, 2.2 Hz, 1H), 1.69 (ddd, J = 13.2, 10.2 Hz, 1H), 1.50 (s, 3H), 1.44 (s, 3H), 1.38 (s, 3H), 1.32 (s, 3H); 13C NMR (125 MHz, CDCl3) δ: 138.4, 137.4, 129.0, 128.4, 128.3, 127.7, 126.0, 111.9, 108.5, 105.1, 101.3, 98.1, 82.84, 82.76, 80.3, 79.4, 74.2, 73.4, 72.6, 68.8, 66.8, 66.0, 37.5, 26.8, 26.6, 26.3, 25.5; ESIHRMS Calcd for C32H40NaO10 [M+Na]+: 607.2519. Found 607.2524.
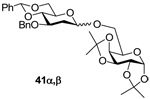
6-O-(3’-O-benzyl-4’,6’-O-benzylidene-2’-deoxy-α-d-arabino-thiohexopyranosyl)-1,2:3,4-di-O-isopropylidene-α-d-galactose (41α) and 6-O-(3’-O-benzyl-4’,6’-O-benzylidene-2’-deoxy-β-darabino-thiohexopyranosyl)-1,2:3,4-di-O-isopropylidene-α-d-galactose (41β)
Prepared by method A, purified by means of radial chromatography, eluting consecutive plates with hexanes to 3:17 ethyl acetate:hexanes, and then with 1:3 to 3:7 diethyl ether:hexanes. Combined yield 82 mg (61%, 1:4 α/β). 41α. [α]24d +16.9 (c 0.14, CHCl3); 1H NMR (400 MHz, CDCl3) δ: 7.48 – 7.54 (m, 2H), 7.23 – 7.41 (m, 8H), 5.62 (s, 1H), 5.53 (d, J = 5.3 Hz, 1H), 4.97 (d, J = 3.5 Hz, 1H), 4.85 (d, J = 12.0 Hz, 1H), 4.68 (d, J = 12.0 Hz, 1H), 4.63 (dd, J = 7.9, 2.3 Hz, 1H), 4.32 (dd, J = 5.0, 2.3 Hz, 1H), 4.23 – 4.29 (m, 2H), 4.05 (ddd, J = 11.1, 9.1, 5.3 Hz, 1H), 3.97 (td, J = 6.6, 1.8 Hz, 1H), 3.87 (td, J = 9.9, 4.7 Hz, 1H), 3.64 – 3.79 (m, 4H), 2.32 (dd, J = 13.2, 5.3 Hz, 1H), 1.80 (ddd, J = 13.4, 11.1, 3.8 Hz, 1H), 1.55 (s, 3H), 1.45 (s, 3H), 1.35 (s, 3H), 1.34 (s, 3H); 13C NMR (100 MHz, CDCl3) δ: 138.8, 137.6, 128.9, 128.3, 128.2, 127.6, 127.5, 126.1, 109.4, 108.6, 101.3, 98.1, 96.4, 83.9, 73.0, 71.0, 70.6, 69.1, 65.9, 65.8, 63.1, 36.5, 26.2, 26.0, 25.0, 24.6; FABHRMS Calcd for C32H40NaO10 [M+Na]+: 607.2519. Found 607.2515. 41β. [α]24d −51.4 (c 0.21, CHCl3); 1H NMR (500 MHz, CDCl3) δ: 7.48 – 7.53 (m, 2H), 7.26 – 7.42 (m, 8H), 5.59 (s, 1H), 5.55 (d, J = 5.0 Hz, 1H), 4.79 (d, J = 12.3 Hz, 1H), 4.71 (d, J = 12.1 Hz, 1H), 4.66 (dd, J = 9.8, 2.1 Hz, 1H), 4.60 (dd, J = 8.0, 2.5 Hz, 1H), 4.29 – 4.34 (m, 2H), 4.21 (dd, J = 7.9, 1.8 Hz, 1H), 3.97 – 4.05 (m, 2H), 3.82 (t, J = 10.3 Hz, 1H), 3.64 – 3.77 (m, 3H), 3.35 (td, J = 9.6, 5.0 Hz, 1H), 2.44 (ddd, J = 13.0, 5.0, 2.1 Hz, 1H), 1.69 – 1.78 (m, 1H), 1.54 (s, 3H), 1.45 (s, 3H), 1.34 (s, 3H),1.31 (s, 3H); 13C NMR (125 MHz, CDCl3) δ: 138.7, 137.7 129.1, 128.6, 128.4, 127.9, 127.8, 126.3, 109.6, 108.9, 101.5, 101.3, 96.5, 83.2, 74.6, 72.5, 71.7, 70.9, 70.6, 69.4, 69.1, 68.0, 66.8, 37.7, 26.3, 26.2, 25.2, 24.6; ESIHRMS Calcd for C32H40NaO10 [M+Na]+: 607.2519. Found 607.2521.
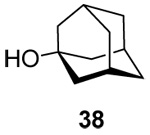
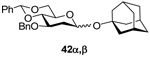
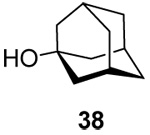
(1-Adamantyl) 3-O-benzyl-4,6-O-benzylidene-2’-deoxy-α-d-arabino-hexopyranoside (42α) and (1-Adamantyl) 3-O-benzyl-4,6-O-benzylidene-2’-deoxy-β-d-arabino-hexopyranoside (42β)
Prepared by method A, purified by means of radial chromatography, eluting consecutive plates with hexanes to 1:39 ethyl acetate:hexanes, and then with 1:9 diethyl ether:hexanes. Combined yield 33 mg (30%, 1:1.9 α/β). 42α. White solid. M.p. 160–162 °C; [α]24d +54.7 (c 0.086, CHCl3); 1H NMR (500 MHz, CDCl3) δ: 7.50 – 7.55 (m, 2H), 7.22 – 7.42 (m, 8H), 5.63 (s, 1H), 5.35 (d, J = 3.3 Hz, 1H), 4.87 (d, J = 11.7 Hz, 1H), 4.69 (d, J = 11.7 Hz, 1H), 4.22 (dd, J = 10.3, 5.0 Hz, 1H), 4.01 – 4.15 (m, 2H), 3.73 (t, J = 10.4 Hz, 1H), 3.67 (t, J = 9.3 Hz, 1H), 2.08 – 2.18 (m, 4H),1.73 – 1.86 (m, 7H), 1.56 – 1.68 (m, 6H); 13C NMR (125 MHz, CDCl3) δ: 139.0, 137.8, 128.8, 128.3, 128.2, 127.6, 127.5, 126.0, 101.2, 91.2, 84.4, 74.2, 73.4, 73.0, 69.2, 62.7, 42.5, 38.1, 36.3, 30.6; ESIHRMS Calcd for C30H36NaO5 [M+Na]+: 499.2461. Found 499.2472. 42β. Slightly yellow solid. M.p. 86–88 °C; [α]24d −11.5 (c 0.19, CHCl3); 1H NMR (400 MHz, CDCl3) δ: 7.47 – 7.55 (m, 2H), 7.24 – 7.43 (m, 8H), 5.59 (s, 1H), 4.89 (dd, J = 9.9, 2.3 Hz, 1H), 4.80 (d, J = 12.0 Hz, 1H), 4.72 (d, J = 12.0 Hz, 1H), 4.27 (dd, J = 10.5, 5.0 Hz, 1H), 3.83 (t, J = 10.4 Hz, 1H), 3.70 – 3.79 (m, 1H), 3.67 (t, J = 8.9 Hz, 1H), 3.35 (td, J = 14.0, 5.0 Hz, 1H), 2.11 – 2.22 (m, 4H), 1.71 – 1.88 (m, 7H), 1.56 – 1.70 (m, 6H); 13C NMR (100 MHz, CDCl3) δ: 138.6, 137.6, 128.9, 128.4, 128.2, 127.63, 127.56, 126.1, 101.3, 93.1, 82.9, 75.0, 74.9, 72.3, 69.1, 66.5, 42.5, 39.1, 36.2, 30.6; ESIHRMS Calcd for C30H36NaO5 [M+Na]+: 499.2461. Found 499.2452.
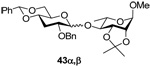
Methyl 4-O-(2’-O-benzyl-4’,6’-O-benzylidene-3’-deoxy-α-d-ribo-thiohexopyranosyl)-2,3-O-isopropylidene-α-l-rhamnoside (43α) and Methyl 4-O-(2’-O-benzyl-4’,6’-O-benzylidene-3’-deoxy-β-d-ribo-thiohexopyranosyl)-2,3-O-isopropylidene-α-l-rhamnoside (43β)
Prepared by method B. Purified by means of radial chromatography, eluting with hexanes to 3:17 diethyl ether : hexanes to give 43α (33 mg, 53%) and 43β (26 mg, 42%). 43α. White solid. M.p. 138–142 °C; [α]22d +31.1 (c 0.074, CHCl3); 1H NMR (500 MHz, CDCl3) δ: 7.48 – 7.52 (m, 2 H) 7.28 – 7.40 (m, 8H), 5.50 (s, 1H), 4.93 (d, J = 3.3 Hz, 1H), 4.86 (s, 1H), 4.67 (d, J = 12.1 Hz, 1H), 4.56 (d, J = 12.3 Hz, 1H), 4.21 – 4.28 (m, 2H), 4.12 (d, J = 5.5 Hz, 1H), 4.06 (td, J = 9.8, 5.0 Hz, 1H), 3.75 – 3.82 (m, 1H), 3.57 – 3.67 (m, 2H), 3.49 (ddd, J = 11.6, 9.5, 4.0 Hz, 1H), 3.39 (dd, J = 10.1, 7.5 Hz, 1H), 3.36 (s, 3H), 2.31 (ddd, J = 11.0, 4.3, 4.1 Hz, 1H), 2.04 (ddd, J1 = J2 = J3 =11.7 Hz, 1H), 1.52 (s, 3H), 1.37 (d, J = 6.2 Hz, 3H), 1.35 (s, 3H); 13C NMR (125 MHz, CDCl3) δ: 137.8, 137.5, 129.1, 128.5, 128.4, 127.9, 127.7, 126.3, 109.1, 101.8, 97.9, 81.1, 76.1, 74.3, 71.1, 69.5, 64.9, 64.3, 54.7, 29.81, 29.75, 28.2, 26.5, 17.3; FABHRMS Calcd for C30H38NaO9 [M+Na]+: 565.2414. Found 565.2390. 43β. [α]22d −43.9 (c 0.098, CHCl3); 1H NMR (500 MHz, CDCl3) δ: 7.45 – 7.49 (m, 2H), 7.27 – 7.39 (m, 8H), 5.49 (s, 1H), 5.03 (d, J = 7.5 Hz, 1H), 4.87 (s, 1H), 4.82 (d, J = 12.1 Hz, 1H), 4.65 (d, J = 12.1 Hz, 1H), 4.23 – 4.29 (m, 2H), 4.11 (d, J = 5.7 Hz, 1H), 3.63 – 3.76 (m, 3H), 3.51 (ddd, J = 11.7, 8.9, 4.3 Hz, 1H), 3.31 – 3.43 (m, 5H), 2.40 (dt, J = 12.0, 4.7 Hz, 1H), 1.79 (ddd, J1 = J2 = J3 = 11.7 Hz, 1H), 1.53 (s, 3H), 1.36 (s, 3H), 1.30 (d, J = 5.9 Hz, 3H); 13C NMR (125 MHz, CDCl3) δ: 138.6, 137.4, 129.1, 128.4, 127.7, 127.6, 126.2, 109.3, 103.1, 101.7, 98.1, 78.2, 77.9, 77.6, 76.3, 76.0, 75.5, 72.8, 70.0, 69.3, 64.2, 55.0, 34.9, 27.9, 26.4, 17.7; FABHRMS Calcd for C30H38NaO9 [M+Na]+: 565.2414. Found 565.2398.
3-O-(2’-O-benzyl-4’,6’-O-benzylidene-3’-deoxy-α-d-ribo-thiohexopyranosyl)-1,2:5,6-di-O-isopropylidene-α-d-glucose (44α) and 3-O-(2’-O-benzyl-4’,6’-O-benzylidene-3’-deoxy-β-d-ribo-thiohexopyranosyl)-1,2:5,6-di-O-isopropylidene-α-d-glucose (44β)
Prepared by method B. Purified by means of radial chromatography, eluting with hexanes to 1:4 diethyl ether: hexanes, followed by normal phase HPLC, eluting with hexanes to 3:2 ethyl acetate: haxanes. Combined yield 51 mg (76%, 4.7:1 α/β). 44α. [α]22d +38.7 (c 0.20, CHCl3); 1H NMR (400 MHz, CDCl3) δ: 7.45 – 7.52 (m, 2H), 7.27 – 7.41 (m, 8H), 5.96 (d, J = 3.5 Hz, 1H), 5.52 (s, 1H), 5.21 (d, J = 3.2 Hz, 1H), 4.60 – 4.67 (m, 3H), 4.53 (dt, J = 8.6, 5.4 Hz, 1H), 4.27 – 4.34 (m, 2H), 4.10 (dd, J = 8.5, 2.6 Hz, 1H), 4.06 (d, J = 5.6 Hz, 2H), 3.75 – 3.83 (m, 1H), 3.70 (t, J = 10.2 Hz, 1H), 3.61 (ddd, J = 11.7, 4.2, 3.9 Hz, 1H), 3.49 – 3.57 (m, 1H), 2.29 (ddd, J = 11.3, 4.2, 4.1 Hz, 1H), 2.04 (ddd, J1 = J2 = J3 = 11.7 Hz, 1H), 1.51 (s, 3H), 1.43 (s, 3H), 1.33 (s, 3H), 1.28 (s, 3H); 13C NMR (100 MHz, CDCl3) δ: 137.9, 137.2, 129.2, 128.5, 128.4, 127.8, 127.4, 126.1, 112.0, 109.2, 105.2, 101.7, 97.5, 84.1, 81.4, 80.1, 73.9, 72.1, 70.9, 69.3, 67.3, 65.0, 29.8, 27.1, 26.9, 26.4, 25.4; FABHRMS Calcd for C32H40NaO10 [M+Na]+: 607.2519. Found 607.2524. 44β. [α]22d −50.0 (c 0.042, CHCl3); 1H NMR (500 MHz, CDCl3) δ: 7.45 – 7.49 (m, 2H), 7.28 – 7.40 (m, 8H), 5.84 (d, J = 3.7 Hz, 1H), 5.49 (s, 1H), 4.66 (d, J = 11.7 Hz, 1H), 4.63 (d, J = 12.1 Hz, 1 H), 4.54 – 4.58 (m, 2H), 4.33 – 4.39 (m, 3H), 4.29 (dd, J = 10.5, 5.0 Hz, 1H), 4.04 – 4.09 (m, 2H), 3.72 (t, J = 10.3 Hz, 1H), 3.54 (ddd, J = 16.2, 4.6, 4.5 Hz, 1H), 3.31 – 3.43 (m, 2H), 2.47 (dt, J = 12.0, 4.7 Hz, 1H), 1.75 (ddd, J1 = J2 = J3 = 11.9 Hz, 1H), 1.50 (s, 3H), 1.45 (s, 3H), 1.38 (s, 3H), 1.28 (s, 3H); 13C NMR (125 MHz, CDCl3) δ: 137.9, 137.2, 129.2, 128.5, 128.4, 127.9, 127.5, 126.1, 111.9, 108.6, 105.1, 103.1, 101.6, 82.6, 80.5, 80.2, 75.7, 75.3, 73.4, 72.6, 70.4, 69.0, 66.0, 34.6, 26.7, 26.6, 26.2, 25.6; FABHRMS Calcd for C32H40NaO10 [M+Na]+: 607.2519. Found 607.2509.
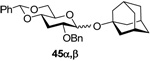
(1-Adamantyl) 2-O-benzyl-4,6-O-benzylidene-3-deoxy-α-d-ribo-hexopyranoside (45α) and (1-Adamantyl) 2-O-benzyl-4,6-O-benzylidene-3-deoxy-β-d-ribo-hexopyranoside (45β)
Prepared by method B. Purified by means of radial chromatography, eluting consecutive plates with hexanes to 1:19 diethyl ether:hexanes, and then with hexanes to 1:39 ethyl acetate:hexanes. Combined yield 50.4 mg (92%, 1:1.8 α/β). 45α. White solid. M.p. 119–122 °C; [α]22d +43.6 (c 0.078, CHCl3); 1H NMR (400 MHz, CDCl3) δ: 7.45 – 7.53 (m, 2H), 7.27 – 7.43 (m, 8H), 5.50 (s, 1H), 5.28 (d, J = 3.5 Hz, 1H), 4.62 (d, J = 12.3 Hz, 1H), 4.58 (d, J = 12.3 Hz, 1H), 4.20 (dd, J = 10.2, 4.7 Hz, 1H), 4.00 (td, J = 9.9, 4.8 Hz, 1H), 3.63 (t, J = 10.4 Hz, 1H), 3.53 – 3.59 (m, 1H), 3.49 (ddd, J = 11.4, 9.4, 4.4 Hz, 1H), 2.08 – 2.26 (m, 5H), 1.82 – 1.96 (m, 6H), 1.59 – 1.70 (m, 6H); 13C NMR (100 MHz, CDCl3) δ: 138.3, 137.5, 129.0, 128.5, 128.3, 127.7, 127.6, 126.2, 101.7, 89.7, 74.5, 74.0, 70.6, 69.6, 63.7, 42.6, 36.3, 30.7, 30.1; FABHRMS Calcd for C30H36NaO5 [M+Na]+: 499.2461. Found 499.2447. 45β. [α]22d −19.2 (c 0.052, CHCl3); 1H NMR (500 MHz, CDCl3) δ: 7.43 – 7.51 (m, 2H), 7.27 – 7.41 (m, 8H), 5.47 (s, 1H), 4.85 (d, J = 12.1 Hz, 1H), 4.78 (d, J = 7.5 Hz, 1H), 4.68 (d, J = 12.1 Hz, 1H), 4.26 (dd, J = 10.5, 5.0 Hz, 1H), 3.74 (t, J = 10.3 Hz, 1H), 3.51 (ddd, J = 11.7, 8.9, 4.3 Hz, 1H), 3.35 – 3.44 (m, 2H), 2.40 (dt, J = 12.0, 4.6 Hz, 1H), 2.15 – 2.22 (m, 3H), 1.90 – 1.97 (m, 3H), 1.81 – 1.88 (m, 3H), 1.76 (ddd, J1 = J2 = J3 = 11.7 Hz, 1H), 1.60 – 1.71 (m, 6H); 13C NMR (125 MHz, CDCl3) δ: 138.6, 137.4, 129.1, 128.3, 127.8, 127.6, 126.2, 101.6, 98.2, 76.2, 75.43, 75.37, 73.0, 70.0, 69.3, 42.8, 36.3, 35.3, 30.7; FABHRMS Calcd for C30H36NaO5 [M+Na]+: 499.2461. Found 499.2447.
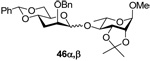
Methyl 4-O-(2’-O-benzyl-4’,6’-O-benzylidene-3’-deoxy-α-d-arabino-thiohexopyranosyl)-2,3-O-isopropylidene-α-l-rhamnoside (46α) and Methyl 4-O-(2’-O-benzyl-4’,6’-O-benzylidene-3’-deoxy-β-d-arabino-thiohexopyranosyl)-2,3-O-isopropylidene-α-l-rhamnoside (46β)
Prepared by method B. Purified by means of radial chromatography, eluting with hexanes to 3:17 diethyl ether : hexanes. Combined yield 53.0 mg (85%, 1.9:1 α/β). Analytical samples of both isomers were obtained by reverse phase HPLC separation, eluting with 1:1 water:acetonitrile to acetonitrile. 46α. White solid. M.p. 152 – 155 °C; [α]22d +54.6 (c 0.108, CHCl3); 1H NMR (400 MHz, CDCl3) δ: 7.48 – 7.54 (m, 2H), 7.27 – 7.41 (m, 8H), 5.59 (s, 1H), 4.84 (s, 1H), 4.79 (s, 1H), 4.67 (d, J = 12.3 Hz, 1H), 4.56 (d, J = 12.3 Hz, 1H), 4.23 (dd, J = 10.1, 4.8 Hz, 1H), 4.06 – 4.15 (m, 3H), 3.98 – 4.06 (m, 1H), 3.78 (t, J = 9.9 Hz, 1H), 3.65 – 3.69 (m, 1H), 3.54 – 3.63 (m, 1H), 3.33 – 3.41 (m, 4H), 2.23 (dt, J = 13.1, 3.3 Hz, 1H), 1.92 – 2.01 (m, 1H), 1.52 (s, 3H), 1.34 (s, 3H), 1.13 (d, J = 6.4 Hz, 3H); 13C NMR (100 MHz, CDCl3) δ: 137.8, 137.7, 129.0, 128.5, 128.3, 127.9, 127.8, 126.2, 109.2, 102.0, 98.5, 98.0 (1JCH = 166.5), 80.3, 76.9, 76.0, 74.6, 74.2, 71.3, 69.4, 65.1, 64.8, 54.8, 29.3, 28.2, 26.5, 17.3; ESIHRMS Calcd for C30H39O9 [M+H]+: 543.25886. Found 543.25986. 46β. White solid. M.p. 158–162 °C; [α]22d −67.1 (c 0.082, CHCl3); 1H NMR (500 MHz, CDCl3) δ: 7.46 – 7.51 (m, 2H), 7.26 – 7.42 (m, 8H), 5.56 (s, 1H), 5.10 (d, J = 0.9 Hz, 1H), 4.88 (s, 1H), 4.84 (d, J = 12.5 Hz, 1H), 4.68 (d, J = 12.5 Hz, 1H), 4.25 (dd, J = 10.5, 5.0 Hz, 1H), 4.14 – 4.18 (m, 1H), 4.10 (d, J = 5.5 Hz, 1H), 4.04 (ddd, J = 11.8, 9.2, 4.5 Hz, 1H), 3.91 (t, J = 10.4 Hz, 1H), 3.80 – 3.82 (m, 1H), 3.63 – 3.73 (m, 2H), 3.45 (td, J = 9.7, 4.9 Hz, 1H), 3.40 (s, 3H), 2.31 (dt, J = 13.1, 3.9 Hz, 1H), 1.72 – 1.79 (m, 1H), 1.54 (s, 3H), 1.33 – 1.37 (m, 6H); 13C NMR (125 MHz, CDCl3) δ: 138.7, 137.6, 129.0, 128.32, 128.28, 127.7, 127.6, 126.1, 109.3, 101.9, 100.9 (1JCH = 160.0), 97.9, 78.5, 77.7, 76.1, 74.6, 74.1, 73.3, 71.2, 69.1, 64.3, 54.9, 33.9, 27.9, 26.5, 17.7; FABHRMS Calcd for C30H38NaO9 [M+Na]+: 565.2414. Found 565.2408.
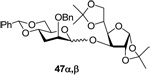
3-O-(2’-O-benzyl-4’,6’-O-benzylidene-3’-deoxy-α-d-arabino-thiohexopyranosyl)-1,2:5,6-di-O-isopropylidene-α-d-glucose (47α) and 3-O-(2’-O-benzyl-4’,6’-O-benzylidene-3’-deoxy-β-d-arabino-thiohexopyranosyl)-1,2:5,6-di-O-isopropylidene-α-d-glucose (47β)
Prepared by method B. Purification by means of radial chromatography, eluting with hexanes to 1:3 diethyl ether : hexanes, gave crude products. Further purification of crude 47α and 47β by means of normal phase HPLC, eluting with hexanes to 1:1 ethyl acetate : hexanes and hexanes to 3:2 ethyl acetate:hexanes respectively, afforded pure products (53.5 mg, combined 80% yield, 1.3:1 α/β). 47α. White solid. M.p. 145–148 °C; [α]22d +37.6 (c 0.492, CHCl3); 1H NMR (500 MHz, CDCl3) δ:7.48 – 7.52 (m, 2H), 7.28 – 7.40 (m, 8H), 5.88 (d, J = 3.5 Hz, 1H), 5.61 (s, 1H), 5.15 (s, 1H), 4.64 (d, J = 12.3 Hz, 1H), 4.57 – 4.62 (m, 2H), 4.34 (d, J = 2.4 Hz, 1H), 4.28 – 4.31 (m, 1H), 4.00 – 4.15 (m, 5H), 3.81 – 3.89 (m, 2H), 3.71 – 3.75 (m, 1H), 2.24 (dt, J = 13.0, 3.3 Hz, 1H), 1.96 (td, J = 12.5, 3.0 Hz, 1H), 1.51 (s, 3H), 1.42 (s, 3H), 1.33 (s, 6H); 13C NMR (125 MHz, CDCl3) δ: 137.7, 137.5, 129.1, 128.5, 128.4, 127.9, 127.6, 126.1, 112.1, 109.4, 105.3, 101.9, 98.1 (1JCH = 172.0), 84.1, 81.5, 79.9, 74.5, 74.1, 72.5, 71.3, 69.3, 67.9, 66.0, 29.4, 26.9, 26.3, 25.5; EIHRMS Calcd for C32H40O10 [M]+: 584.2622. Found 584.2659. 47β. [α]22d − 30.3 (c 0.330, CHCl3); 1H NMR (400 MHz, CDCl3) δ: 7.44 – 7.51 (m, 2H), 7.27 – 7.40 (m, 8H), 5.92 (d, J = 3.8 Hz, 1H), 5.56 (s, 1H), 4.72 (d, J = 12.6 Hz, 1H), 4.69 (s, 1H), 4.64 (d, J = 12.6 Hz, 1H), 4.40 – 4.47 (m, 2H), 4.33 – 4.38 (m, 2H), 4.28 (dd, J = 10.4, 4.8 Hz, 1H), 4.15 – 4.20 (m, 1H), 4.02 – 4.11 (m, 2H), 3.87 (t, J = 10.4 Hz, 1H), 3.71 – 3.75 (m, 1H), 3.46 (td, J = 9.6, 5.0 Hz, 1H), 2.33 (dt, J = 13.2, 4.2 Hz, 1H), 1.68 – 1.78 (m, 1H), 1.51 (s, 3H), 1.45 (s, 3H), 1.36 (s, 3H), 1.32 (s, 3H); 13C NMR (125 MHz, CDCl3) δ: 138.0, 137.4, 129.1, 128.43, 128.36, 127.8, 126.1, 112.0, 108.6, 105.1, 101.8, 100.2 (1JCH = 155.4), 82.7, 80.5, 80.2, 73.8, 73.6, 73.3, 72.6, 71.0, 69.0, 66.1, 32.8, 26.8, 26.6, 26.3, 25.5; EIHRMS Calcd for C32H40O10 [M]+: 584.2622. Found 584.2602.
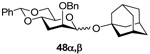
(1-Adamantyl) 2-O-benzyl-4,6-O-benzylidene-3-deoxy-α-d-arabino-hexopyranoside (48α) and (1-Adamantyl) 2-O-benzyl-4,6-O-benzylidene-3-deoxy-β-d-arabino-hexopyranoside (48β)
Prepared by method B. Purification by means of radial chromatography, eluting with hexanes to 1:19 ethyl acetate:hexanes, gave crude products. Further purification of crude 48α and 48β by means of normal phase HPLC, eluting with hexanes to 3:2 ethyl acetate:hexanes, afforded pure 48α (35.4 mg, 65%) and 48β (9.6 mg, 18%). 48α. White solid. M.p. 110–114 °C; 22d +61.9 (c 0.690, CHCl3); 1H NMR (500 MHz, CDCl3) δ: 7.49 – 7.53 (m, 2H), 7.28 – 7.41 (m, 8H), 5.59 (s, 1H), 5.15 (s, 1H), 4.67 (d, J = 12.1 Hz, 1H), 4.59 (d, J = 12.1 Hz, 1H), 4.19 (dd, J = 10.3, 4.8 Hz, 1H), 3.96 – 4.10 (m, 2H), 3.78 (t, J = 10.2 Hz, 1H), 3.54 – 3.57 (m, 1H), 2.21 (dt, J = 12.8, 3.5 Hz, 1H), 2.11 – 2.17 (m, 3H), 2.06 (td, J = 12.2, 2.9 Hz, 1H), 1.74 – 1.83 (m, 6H), 1.57 – 1.68 (m, 6H); 13C NMR (125 MHz, CDCl3) δ: 138.1, 137.8, 129.0, 128.5, 128.3, 127.8, 127.7, 126.2, 101.9, 91.0 (1JCH = 166.5), 76.7, 74.6, 74.5, 71.2, 69.5, 64.9, 42.5, 36.3, 30.6, 29.2; FABHRMS Calcd for C30H36NaO5 [M+Na]+: 499.2461. Found 499.2492. 48β. [α]22d −33.2 (c 0.196, CHCl3); 1H NMR (400 MHz, CDCl3) δ: 7.41 – 7.50 (m, 4H), 7.25 – 7.40 (m, 6H), 5.55 (s, 1H), 4.88 (d, J = 13.1 Hz, 1H), 4.85 (d, J = 0.9 Hz, 1H), 4.79 (d, J = 12.9 Hz, 1H), 4.24 (dd, J = 10.4, 4.8 Hz, 1H), 4.02 (ddd, J = 11.8, 9.2, 4.4 Hz, 1H), 3.88 (t, J = 10.4 Hz, 1H), 3.57 – 3.62 (m, 1H), 3.45 (td, J = 9.6, 5.0 Hz, 1H), 2.30 (ddd, J = 13.0, 3.8, 3.7 Hz, 1H), 2.14 – 2.20 (m, 3H), 1.58 – 1.91 (m, 13H); 13C NMR (100 MHz, CDCl3) δ: 138.8, 137.7, 129.0, 128.3, 128.2, 127.9, 127.4, 126.2, 101.9, 95.3 (1JCH = 152.6), 75.4, 74.9, 74.0, 73.0, 70.9, 69.3, 42.4, 36.3, 34.1, 30.6; FABHRMS Calcd for C30H36NaO5 [M+Na]+: 499.2461. Found 499.2413.
Supplementary Material
Details of the preparation of all donors and copies of NMR spectra for all compounds. This material is available free of charge via the Internet at http://pubs.acs.org
Acknowledgment
We thank Dr. Tom Hutton for helpful discussions and the NIH (GM 62160) for support of this work.
References
- 1.Crich D, Sun S. J. Org. Chem. 1997;62:1198–1199. [Google Scholar]
- 2.Crich D, Sun S. Tetrahedron. 1998;54:8321–8348. [Google Scholar]
- 3.Crich D, Sun S. J. Am. Chem. Soc. 1998;120:435–436. [Google Scholar]
- 4.Crich D, Smith M. J. Am. Chem. Soc. 2001;123:9015–9020. doi: 10.1021/ja0111481. [DOI] [PubMed] [Google Scholar]
- 5.Crich D, Smith M. J. Am. Chem. Soc. 2002;124:8867–8869. doi: 10.1021/ja011406u. [DOI] [PubMed] [Google Scholar]
- 6.Crich D, de la Mora M, Vinod AU. J. Org. Chem. 2003;68:8142–8148. doi: 10.1021/jo0349882. [DOI] [PubMed] [Google Scholar]
- 7.Crich D, Cai W. J. Org. Chem. 1999;64:4926–4930. doi: 10.1021/jo990243d. [DOI] [PubMed] [Google Scholar]
- 8.Bousquet E, Khitri M, Lay L, Nicotra F, Panza L, Russo G. Carbohydr. Res. 1998;311:171–181. doi: 10.1016/s0008-6215(98)00218-3. [DOI] [PubMed] [Google Scholar]
- 9.Kim KS, Kang SS, Seo YS, Kim HJ, Jeong K-S. Synlett. 2003:1311–1314. [Google Scholar]
- 10.Crich D, Smith M, Yao Q, Picione J. Synthesis. 2001:323–326. [Google Scholar]
- 11.Crich D, Jayalath P. J. Org. Chem. 2005;70:7252–7259. doi: 10.1021/jo0508999. [DOI] [PubMed] [Google Scholar]
- 12.Nukada T, Berces A, Whitfield DM. Carbohydr. Res. 2002;337:765–774. doi: 10.1016/s0008-6215(02)00043-5. [DOI] [PubMed] [Google Scholar]
- 13.Lemieux RU. In: Molecular Rearrangements, Part 2. De Mayo P, editor. New York: Wiley; 1964. pp. 709–769. [Google Scholar]
- 14.Lucero CG, Woerpel KA. J. Org. Chem. 2006;71:2641–2647. doi: 10.1021/jo0522963. [DOI] [PubMed] [Google Scholar]
- 15.Smith DM, Woerpel KA. Org. Biomol. Chem. 2006:1195–1201. doi: 10.1039/b600056h. [DOI] [PubMed] [Google Scholar]
- 16.Crich D, Sun S. J. Am. Chem. Soc. 1997;119:11217–11223. [Google Scholar]
- 17.Crich D, Chandrasekera NS. Angew. Chem. Int. Ed. 2004;43:5386–5389. doi: 10.1002/anie.200453688. [DOI] [PubMed] [Google Scholar]
- 18.Fraser-Reid B, Wu ZC, Andrews W, Skowronski E. J. Am. Chem. Soc. 1991;113:1434–1435. [Google Scholar]
- 19.Andrews CW, Rodebaugh R, Fraser-Reid B. J. Org. Chem. 1996;61:5280–5289. doi: 10.1021/jo961115h. [DOI] [PubMed] [Google Scholar]
- 20.Jensen HH, Nordstrom M, Bols M. J. Am. Chem. Soc. 2004;126:9205–9213. doi: 10.1021/ja047578j. [DOI] [PubMed] [Google Scholar]
- 21.Lemieux RU, Hendriks KB, Stick RV, James K. J. Am. Chem. Soc. 1975;97:4056–4062. [Google Scholar]
- 22.Torsion angles were determined with ring conformations fixed according to the parameters provided by Bérces and coworkers (http://www.nrc.ca/ibs/6ring.html)Bérces A, Whitfield DM, Nukuda T. Tetrahedron. 2001;57:477–491.
- 23.Edward JT. Chem. Ind. 1955:1102–1104. [Google Scholar]
- 24.Feather MS, Harris JF. J. Org. Chem. 1965;30:153–157. [Google Scholar]
- 25.Overend WG, Rees CW, Sequeira JS. J. Chem. Soc. 1962:3429–3440. [Google Scholar]
- 26.Barton DHR, McCombie SW. J. Chem. Soc. Perkin Trans. 1. 1975:1574–1585. [Google Scholar]
- 27.Withers SG, Percival MD, Street IP. Carbohydr. Res. 1989;187:43–66. [Google Scholar]
- 28.Russell RN, Weigel TM, Han O, Liu H-w. Carbohydr. Res. 1990;201:95–114. doi: 10.1016/0008-6215(90)84227-l. [DOI] [PubMed] [Google Scholar]
- 29.Classon B, Liu Z, Samuelsson B. J. Org. Chem. 1988;53:6126–6130. [Google Scholar]
- 30.Theander O. Acta Chem. Scand. 1958;12:1883–1885. [Google Scholar]
- 31.Barresi F, Hindsgaul O. In: Modern Methods in Carbohydrate Synthesis. Khan SH, O'Neill RA, editors. Amsterdam: Harwood Academic Publishers; 1996. pp. 251–276. [Google Scholar]
- 32.Pozsgay V. In: Carbohydrates in Chemistry and Biology. Ernst B, Hart GW, Sinaÿ P, editors. Vol. 1. Weinheim: Wiley-VCH; 2000. pp. 319–343. [Google Scholar]
- 33.Ito Y, Ohnishi Y. In: Glycoscience: Chemistry and Chemical Biology. Fraser-Reid B, Kuniaki T, Thiem J, editors. Vol. 2. Berlin: Springer-Verlag: 2001. pp. 1589–1619. [Google Scholar]
- 34.Lichtenthaler Fw, Lergenmüller M, Peters S, Varga Z. Tetrahedron: Asym. 2003;14:727–736. [Google Scholar]
- 35.Ekborg G, Lindberg B, Lonngren J. Acta Chem. Scand. B. 1972;26:3287–3292. doi: 10.3891/acta.chem.scand.26-2231. [DOI] [PubMed] [Google Scholar]
- 36.Dess PB, Martin JC. J. Org. Chem. 1983;48:4155–4156. [Google Scholar]
- 37.Roush WR, Sebesta DP, Bennett CE. Tetrahedron. 1997;53:8825–8836. [Google Scholar]
- 38.Crich D, Ritchie TJ. J. Chem. Soc., Perkin Trans. 1. 1990:945–954. [Google Scholar]
- 39.Codée JDC, van den Bos LJ, Litjens REJN, Overkleeft HS, van Boeckel CAA, van Boom JH, van der Marel GA. Tetrahedron. 2004;60:1057–1064. [Google Scholar]
- 40.Codée JDC, Litjens REJN, Van den Bos LJ, Overkleeft HS, Van der Marel G. Chem. Soc. Rev. 2005;34:769–782. doi: 10.1039/b417138c. [DOI] [PubMed] [Google Scholar]
- 41.Garcia BA, Gin DY. J. Am. Chem. Soc. 2000;122:4269–4279. [Google Scholar]
- 42.Crich D, Hutton TK, Banerjee A, Jayalath P, Picione J. Tetrahedron: Asym. 2005;16:105–119. [Google Scholar]
- 43.Bock K, Pedersen C. J. Chem. Soc., Perkin Trans. 2. 1974:293–297. [Google Scholar]
- 44.Duus JO, Gotfredsen CH, Bock K. Chem. Rev. 2000;100:4589–4614. doi: 10.1021/cr990302n. [DOI] [PubMed] [Google Scholar]
- 45.Thiem J, Klaffke W. Topics Curr. Chem. 1990;154:285–332. [Google Scholar]
- 46.Marzabadi CH, Franck RW. Tetrahedron. 2000;56:8385–8417. [Google Scholar]
- 47.Veyrières A. In: Carbohydrates in Chemistry and Biology. Ernst B, Hart GW, Sinaÿ P, editors. Vol. 13. Weinheim: Wiley-VCH; 2000. pp. 367–405. [Google Scholar]
- 48.Green LG, Ley SV. In: Carbohydrates in Chemistry and Biology. Ernst B, Hart GW, Sinaÿ P, editors. Vol. 1. Weinheim: Wiley-VCH; 2000. pp. 427–448. [Google Scholar]
- 49.Zhang Z, Ollmann IR, Ye X-S, Wischnat R, Baasov T, Wong C-H. J. Am. Chem. Soc. 1999;121:734–753. [Google Scholar]
- 50.Namchuk MN, McCarter JD, Becalski A, Andrews T, Withers SG. J. Am. Chem. Soc. 2000;122:1270–1277. [Google Scholar]
- 51.Raghavan S, Kahne D. J. Am. Chem. Soc. 1993;115:1580–1581. [Google Scholar]
- 52.Gildersleeve J, Smith A, Sakurai D, Raghavan S, Kahne D. J. Am. Chem. Soc. 1999;121:6176–6182. [Google Scholar]
- 53.Hashimoto S, Sano A, Sakamoto H, Nakajima M, Yanagiya Y, Ikegami S. Synlett. 1995:1271–1273. [Google Scholar]
- 54.Bock K, Pedersen H. Acta Chem. Scand. 1987;B41:617–628. doi: 10.3891/acta.chem.scand.41b-0617. [DOI] [PubMed] [Google Scholar]
- 55.van Dorst JALM, van Heusden CJ, Tikkanen JM, Kamerling JP, Vliegenthart JFG. Carbohydr. Res. 1997;297:209–227. doi: 10.1016/s0008-6215(96)00266-2. [DOI] [PubMed] [Google Scholar]
- 56.Oscarson S, Svahnberg P. Carbohydr. Res. 1998;309:207–212. doi: 10.1016/s0008-6215(98)00107-4. [DOI] [PubMed] [Google Scholar]
- 57.Oscarson S, Tedebark U. Carbohydr. Res. 1995;278:271–287. doi: 10.1016/0008-6215(95)00268-5. [DOI] [PubMed] [Google Scholar]
- 58.Kováač P, Edgar KJ. Carbohydr. Res. 1990;201:79–83. doi: 10.1016/0008-6215(90)84226-k. [DOI] [PubMed] [Google Scholar]
- 59.Richards GN. Chem. Ind. 1955:228–228. [Google Scholar]
- 60.Crich D, Cai W, Dai Z. J. Org. Chem. 2000;65:1291–1297. doi: 10.1021/jo9910482. [DOI] [PubMed] [Google Scholar]
- 61.Crich D, Vinod AU, Picione J, Wink DJ. ARKIVOC. 2005;(vi):339–344. [PMC free article] [PubMed] [Google Scholar]
- 62.Crich D, Vinod AU, Picione J. J. Org. Chem. 2003;68:8453–8458. doi: 10.1021/jo035003j. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Details of the preparation of all donors and copies of NMR spectra for all compounds. This material is available free of charge via the Internet at http://pubs.acs.org