

Abstract
The highly stereocontrolled synthesis of the 3-amino-3-deoxy-β-mannopyranosides is achieved by means of thioglycoside donors protected with a 4,6-O-benzylidene or alkylidene acetal and a benzylidene imine group. Among the various nitrogen protecting groups investigated only the Schiff’s base was found to give high β-selectivity. N-Phthalimido and N-acetamido protected donors were found to be highly α-selective, whereas 3-azido-3-deoxy glycosyl donors gave intermediate selectivity. The reasons for the protecting group dependency are discussed in terms of the change in the O2-C2-C3-N3 torsional interaction on conversion of the covalent glycosyl triflates to the transient oxacarbenium ions.
Introduction
It is becoming increasingly apparent in the 4,6-O-benzylidene directed β-mannopyranosylation developed in our laboratories1,2 that the substituent at the mannose 3-position plays a critical role in determining the stereochemical outcome of the reaction. Thus, while the 3-O-benzyl donor 1 and related benzylic-type ethers are highly β-selective,3–5 3-O-carboxylate esters such as 2 give essentially pure α-glycosides.6,7 On the other hand intermediate selectivity, namely the formation of α/β mixtures, is seen with a broad spectrum of compounds including the 3-O-tert-butyldimethylsilyl ether 3,8 the 3-O-propargyl ether 4,9 the 3-deoxy system 5,10 and the 3-deoxy-3-fluoro system 6.11 This variation of selectivity is seen irrespective of whether a thioglycoside is activated with the combination of 1-benzenesulfinyl piperidine (BSP) and trifluoromethanesulfonic anhydride,5 with that of diphenyl sulfoxide and trifluoromethanesulfonic anhydride,12 or by oxidation to the corresponding glycosyl sulfoxide followed by activation with trifluoromethanesulfonic anhydride,3,4 and in other glycosylation systems functioning by a similar mechanism.13

With exception of the ester 2 and its analogs, for which we do not have a satisfactory explanation at present, these effects are understood in terms of our general glycosylation mechanism involving initial conversion of the donor to a demonstrable α-mannosyl triflate intermediate14 that is in equilibrium with a transient β-selective contact ion pair (CIP)15 and thereby with an α-selective solvent separated ion pair (SSIP) (Scheme 1).16 β-Selectivity is obtained when the series of equilibria is shifted as far as possible toward the covalent triflate, thereby minimizing the concentration of the α-selective CIP. The role of the 4,6-O-benzylidene acetal, or its surrogates, is to fix the C5–C6 bond of the donor in the most electron-withdrawing tg conformation thereby destabilizing any glycosyl oxacarbenium ions and favoring the covalent glycosyl triflate.17,18 As the covalent triflate, with its 4C1 conformation collapses to the CIP, with the oxacarbenium ion in the likely 4H3 conformation,19,20 the O2-C2-C3-R torsion angle is compressed from 60° to 45°.10 When R is benzyloxy this results in an increase in steric strain which further destabilizes the oxacarbenium ion and favors the covalent triflate. When R is hydrogen, fluoride, or propargyloxy the increase in steric strain is smaller and lower selectivity is the result.11 The effect of the large tert-butyldimethylsiloxy group, on the other hand, is explained by a steric buttressing interaction with the O-2 protecting group which results in increased shielding of the β-face, resulting in reduced selectivity:8 the effect of the 3-O-tert-butyldimethylsilyl ether can be thwarted when it is employed in conjunction with a sterically minimal 2-O-propargyl ether.9
Scheme 1.

Glycoslation Mechanism
In this paper we report on the synthesis and glycosylation reactions of a series of 4,6-O-alkylidene protected 3-amino-3-deoxymannopyranosyl donors and provide further insight into the range of acceptable substituents at the 3-position.
Results
Synthesis of Substrates
Methyl 3-amino-3-deoxy-α-d-mannopyranoside 8 was most expediently prepared from methyl α-d-glucopyranoside 7 by periodate cleavage followed by double Henry reaction with nitromethane, as described by Baer and Fischer,21 as improved by Richardson.22 Metal-catalyzed diazo transfer23 from trifluoromethanesulfonyl azide24 gave the corresponding azide 9, which was converted to a mixture of the peracetylated α- and β-thioglycosides 11 and 12 by standard methods. X-ray crystallography confirmed the configuration of 12, and saponification of 11 gave the triol 13 (Scheme 2).
Scheme 2.

Thioglycoside Synthesis
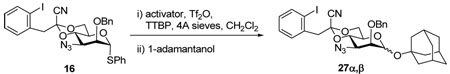
Our recent interest in the use of modified 4,6-O-acetal protecting groups, particularly those suitable for subsequent radical fission to give the 6-deoxy sugars, prompted the employment of the 1-cyano-2-(2-iodophenyl)ethylidene acetal,25 with previous work having established the β-directing ability of this group.25,26 Thus, reaction of triol 13 with triethyl 2-(2-iodophenyl)orthoacetate25 gave an unstable orthoester 14, which was converted in 62% yield to the stable cyanoacetal 15 by treatment with trimethylsilyl cyanide and BF3 ethereate.25 Benzylation then gave the donor 16 in 93% yield. Interestingly, a higher overall yield of 16 was obtained when the sequence of cyanide introduction and benzylation was inverted (Scheme 3). The complete stereochemical assignment of 15 was confirmed crystallographically.
Scheme 3.

Introduction of the 1-Cyano-2-(2-iodophenyl)ethylidene Acetal
The analogous benzylidene acetal 18 was also prepared in the standard manner via the intermediacy of 17. Treatment of 16 with triphenylphosphine in the presence of water, followed by acetylation provided an acetamide 19. Reaction of both 16 and 18 with triphenylphosphine under anhydrous conditions with subsequent reaction of the intermediate iminophosphoranes with p-trifluoromethylbenzaldehyde gave the imines 20 and 21, respectively, both with yields in the range of 60–80%.27

Finally, a phthalimido protected donor 26 was prepared by the route outlined in Scheme 4, the longer route being necessary because introduction of the phthalimido group directly on the amines derived from 16 and/or 18 failed, presumably for steric reasons. In this Scheme the use of methanolic HCl to effect cleavage of the acetate groups in 23 was dictated by incompatibility of the phthalimido group under the standard Zemplen deacetylation conditions. Likewise, the use of the trimethylsilyl ether as the protecting group for O-2 arose from difficulties in attempted benzylation reactions.
Scheme 4.

Preparation of a Phthalimido Protected Donor
Glycosylation Reactions
Our preferred candidate for the 3-position was the azide group and, thus, glycosylation studies began with donor 16. A number of experiments were carried with 1-adamantanol as acceptor, with activation by the diphenyl sulfoxide/trifluoromethanesulfonic anhydride couple12 in the presence of the hindered base 2,4,6-tri-tert-butylpyrimidine (TTBP)28 in dichloromethane. Previous work had indicated the 1-cyano-2-(2-iodophenyyl)ethylidene acetal to be somewhat disarming and to retard clean activation at −78 °C,25 thus a series of experiments were conducted in which the donor/diphenyl sulfoxide/trifluoromethanesulfonic anhydride/TTBP was warmed to different temperatures before the acceptor was added. The results presented in Table 1 indicate that warming to −50 °C before addition of the acceptor and quenching at that temperature provided the optimum results in terms of yield and selectivity (Entry 3). Using BSP in place in place of diphenyl sulfoxide gave essentially analogous results (Table 1, entry 4), and the attempted use of the nitrile effect29–33 was found to be disappointing in line with previous observations in the mannose34 and rhamnose series (Table 1, entry 5).35 Initial stereochemical assignments for 27α and β, and for all subsequent coupling products, were based on the diagnostic chemical shift (δ 3.0–3.3) of the mannose H5 resonance in the 1H NMR spectra,4 and were subsequently verified by determination of the 1JCH coupling constants for the anomeric carbon.36–38
Table 1.
Exploratory Coupling Reactions with Donor 16 and 1-Adamantanol
 | |||||
|---|---|---|---|---|---|
| Entry | Activator | Solvent | Method | Yield | α:β ratio |
| 1 | Ph2SO | CH2Cl2 | A | 97% | 1:1.7 |
| 2 | Ph2SO | CH2Cl2 | B | 94% | 1:1.5 |
| 3 | Ph2SO | CH2Cl2 | C | 98% | 1:3.3 |
| 5 | BSP | CH2Cl2 | D | 99% | 1:3.2 |
| 6 | Ph2SO | CH2Cl2/MeCN (95/5) |
E | 65% | 1:1.2 |
A) Activation at −78 °C followed, after 30 min at −78 °C, by the addition of adamantanol then warming to 0 °C before quenching. B) Activation at −78 °C followed, after 30 min at −78 °C, by the addition of adamantanol then quenching at −78 °C. C) Activation at −65 °C, followed by warming to −50 °C over 30 before addition of 1-adamantanol, and final quenching at −50 °C after 1 h. D) Same as C except that BSP was used as the activator in place of diphenyl sulfoxide. E) Activation at −65 °C, followed by warming to −30 °C over 30 min before addition of 1-adamantanol, and final quenching at −30 °C after 1 h
Donors 16, 19–21, and 26 were then coupled to 1-adamantanol with activation by the diphenyl sulfoxide/trifluoromethanesulfonic anhydride protocol with activation at −65 °C and warming to −50 °C before addition of the acceptor (Table 2).
Table 2.
Investigation of Different N-3 Protecting Groups
| Donor | Method | Product | Yield, α:β ratio | |
|---|---|---|---|---|
| 1 | 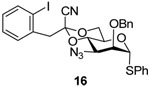 |
C | 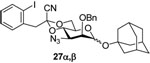 |
98%, 1:3.3 |
| 2 | 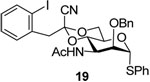 |
C | 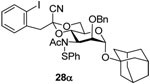 |
56%, α only |
| 3 | 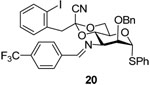 |
F | 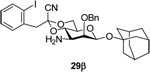 |
83%, β only |
| 4 | 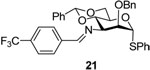 |
F | 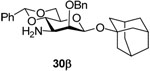 |
78%, β only |
| 5 | 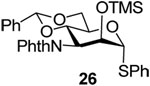 |
F | 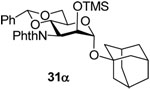 |
77%, α only |
C) Activation at −65 °C, followed by warming to −50 °C over 30 before addition of 1-adamantanol, and final quenching at −50 °C after 1 h. F) As in C except that the reaction mixture was allowed to warm to −10 °C before quenching.
From the results presented in Table 2 it is evident that the Schiff’s base is the ideal protecting group for N-3 in these glycosylation reactions, being readily formed from the azide in a single step, giving outstanding β-selectivity, and being hydrolyzed directly to the amine in the course of the work up. Equally it is apparent that the nature of the acetal spanning O4 and O6 has little effect on the yield or stereochemical outcome of the reaction, as had been anticipated from previous work.25 One interesting feature of this series of reactions was the sulfenylation of the amide nitrogen of donor 19, ultimately leading to glycoside 28α, whose structure rests on HRMS data, and the absence of an NH resonance in the 1H NMR spectrum. This N-acetylsulfenamide arises from the reaction of the amide group with the byproducts of the thioglycoside activation process.
On the basis of these results, further reactions were carried out in which the more typical glycosyl acceptors methyl 2,3-O-isopropylidene-α-l-rhamnopyranoside 32 and methyl 2,3,4-tri-O-benzyl-α-d-glucopyranoside 33 were coupled to donors 20 and 21 under the optimium conditions, resulting in the highly β-selective formation of the products in each case (Table 3).
Table 3.
Further Couplings to Donors 20 and 21
| Donor | Acceptor | Product | Yield, α:β ratio |
|---|---|---|---|
 |
32 | 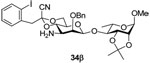 |
76%, β only |
 |
32 | 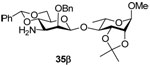 |
82%, β only |
 |
33 | 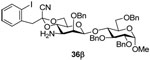 |
82%, β only |
 |
33 | 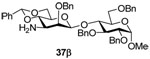 |
92%, β only |
Overall, the picture that emerges for the 3-amino-3-deoxymannopyranoside series closely follows that established by the mannopyranosides themselves. Thus, the optimum protecting group for N3 is a benzylidene imine group that mimics the steric properties of a benzyl ether (steric A value = 1.39).9 It is appropriate to point out here that no attempt was made to prepare or conduct glycosylation reactions with the reduced version of the benzylidene imines, namely the N-benzylamines, as it was considered that these would be protonated under the reaction conditions. The smaller azide (steric A value = 0.45–0.62),39,40 like fluoride (steric A value = 0.25–0.42),39,41–45 is insufficiently bulky for the compression of the O2-C2-C3-X torsion angle to impact significantly the covalent triflate-CIP equilibrium (Scheme 1) resulting in a loss of β-selectivity. The acetamide and phthalimido protected systems behave exactly as the 3-O-carboxylate esters and lead to essentially complete α-selectivity. We note that the bulk of the 2-O-trimethylsilyl ether in donor 26 is unlikely to be a major factor in the a-selectivity of this system as related 2-O-silyl-3-O-benzylmannopyranosyl donors have been shown previously to exhibit good β-selectivity.4,8
Radical Fragmentation Reactions
In contrast to the clean radical fragmentations observed previously with glycosides 38,25,26 attempted radical fragmentations of the various glycosides prepared in this study with the 1-cyano-2-(2-iodophenyl)ethylidene acetals were disappointing. Typically relatively complex reaction mixtures were observed and significant amounts of initiator were required to drive the reactions to completion irrespective of the nitrogen protecting group. By way of example the 6-deoxy glycoside 39 could only be isolated in 20% yield even though TLC indicated it to be the major product from the reaction of 29β with tributyltin hydride and AIBN in toluene at reflux. In addition to the radical fragmentation sequence 29β has undergone an O to N shift of the (2-cyano)phenylacetate group which is the obligatory product of the radical reaction. To prevent this migration 29β was converted to the N-Boc derivative 40 in good yield by standard means, but subsequent radical reactions continued to afford complex mixtures with the isolation of the premature reduction product 41 in 10% yield, along with the desired deoxy sugar 42 in yields ranging from 10–15%. Unfortunately, the reason for the disappointing nature of the radical fragmentation reactions remains unclear at this time.

Conclusion
Among the various protecting groups investigated for the 3-amino group in the 3-amino-3-deoxy mannopyranoside series the p-trifluoromethylbenzylidene imine was by far the best, affording high yields of coupled product and excellent β-selectivity. The comparable steric bulk of this benzylidene imine to the 3-O-benzyl ethers typically employed at the 3-position in the mannopyranoside series provides considerable support for our hypothesis of the importance of the O2-C2-C3-R3 torsional interaction in the stereocontrolled synthesis of β-mannopyranosides and related compounds. The benzylidene imine methodology should facilitate the introduction of 3-amino-3-deoxy-β-mannopyranosides into antibiotics, an area of current interest,46,47 and other molecules. Unfortunately, at least at the present stage of development, the unexpectedly poor results in the radical fragmentation mean that the method is not suitable for the introduction of the β-mycosamine (3-amino-3,6-dideoxy) unit into macrolide antibiotics such as the amphotericins, the nystatins, and the rimocidins.48–50
Experimental Section
Methyl 3-amino-3-deoxy-α-d-mannopyranoside hydrochloride (8)
Sodium metaperiodate (55 g) was slowly added in 30 min to a stirred solution of methyl α-d-glucopyranoside (25 g) in iced water (30 mL) at <10 °C. The mixture was then stirred for 1 h, and the released formic acid was carefully neutralized with sodium bicarbonate (10 g). The mixture, from which much sodium iodate had crystallized, was then poured into ethanol (150 mL) and the precipitate was filtered off and washed with ethanol (100 mL). The filtrate was condensed under vacuum at <10 °C and the residal syrup, which contained solid, was taken up with ethanol. The solid was filtered off and washed with ethanol and the filtrate was again condensed under vacuum at < 10 °C, and the same procedure was repeated 4 to 6 times until a clear syrup (not contaminated with visible solid) was obtained. Nitromethane (7.0 mL) was added to the solution of the syrup in dry methanol (75 mL), followed by the addition of a solution of sodium methoxide (30 mL, 25% w in methanol) at −10 °C. The stirred mixture was brought to rt slowly over 2 h, and was kept at rt for another 6 h. Amberlite-120H ion exchange resin was added to neutralize the reaction mixture, then was removed by filtration and the filtrate was condensed to give an orange oil. This oil was shaken under hydrogen (50 psi) with Raney Nickel (2.0 g) as the catalyst in ethanol (50 mL) for 24 h. The catalyst was removed by filtration and the filtrate was condensed to approx. 30 mL. To this brown solution was added HCl in ether (70 mL, 2M) with vigorously stirring. Ether was removed under vacuum and the residue was cooled in a refrigerator overnight. The so formed crystals were collected by filtration to give the product as a white powder (10–12 g, 30–40%): [α]D = + 68.4 (c, 1.0, H2O), Mp: 208 °C (decomp.); lit:21,22 [α]D = + 60 (c, H2O), Mp: 210–240 °C (decomp.); 1H NMR (500 MHz, D2O) δ 4.66 (s, 1H), 3.96 (m, 1H), 3.78 (d, J = 12.0 Hz, 1H), 3.63–3.72 (m, 2H), 3.55–3.60 (m, 1H), 3.38 (dd, J = 10.0, 2.5 Hz, 1H), 3.30 (s, 3H); 13C NMR (125 MHz, D2O) δ 99.7, 72.2, 66.6, 63.3, 60.3, 54.7, 53.2.
Methyl 3-azido-3-deoxy-α-d-mannopyranoside (9)
To a stirred mixture of salt 8 (3.4 g, 15 mmol), Na2CO3 (3.2 g, 30 mmol) and CuSO4.5H2O (50 mg) in dry methanol (100 mL) was added a solution of trifluoromethanesulfonyl azide in dichloromethane, prepared from sodium azide (8 g, 123 mmol) and triflic anhydride (4.1 mL, 25 mmol) in dichloromethane (50 mL) according to Vasella’s protocol,24 in 6 h by syringe pump. When the addition was complete, the mixture was stirred overnight before the solvent was removed under vacuum and the residue was purified by column chromatography on silica gel (dichloromethane/MeOH: 10/1) to give a colorless oil (3.2 g, 97%). [α]D = + 16 (c, 1.0, MeOH); 1H NMR (500 MHz, MeOH-d4) δ 4.60 (d, J = 1.5 Hz, 1H), 3.78–3.86 (m, 3H), 3.72 (dd, J = 11.5, 6 Hz, 1H), 3.53 (ddd, J = 9.5, 6.0, 2.0 Hz, 1H), 3.43 (dd, J = 10, 3.5 Hz, 1H), 3.38 (s, 3H); 13C NMR (125 MHz, MeOH-d4) δ 100.6, 73.3, 69.9, 64.2, 63.4, 61.3, 53.8; HRMS(ESI): m/z calcd for C7H13N3NaO5 [M+Na]+ 242.0748, found 242.0743.
Phenyl 2,4,6-tri-O-acetyl-3-azido-3-deoxy-α-1-thio-d-mannopyranoside (11) and Phenyl 2,4,6-tri-O-acetyl-3-azido-3-deoxy-β-1-thio-d-mannopyranoside (12)
A mixture of triol 9 (1.15g, 5.25 mmol), Ac2O (6.0 mL) and conc. H2SO2 (0.5 mL) was stirred at rt for 12 h. Then ice (15 g) was added followed by cold water (30 mL). The resulting mixture was extracted with ether (40 mL×3). The combined extracts were washed with saturated NaHCO3 until the aqueous layer was neutral, then with brine and dried over Na2SO4. The solvent was removed under reduced pressure and the residue was purified by column chromatography on silica gel (hexanes/ethyl acetate: 2/1) to give a mixture of isomers 10 (1.54 g, 75%) as an oil. The major component of this mixture was the α-isomer, which had the following characteristics: 1H NMR (500 MHz, CDCl3) δ 6.07 (d, J = 1.5 Hz, 1H), 5.34 (t, J = 10.5 Hz, 1H), 5.20 (m, 1H), 4.26 (dd, J = 12.5, 5.0 Hz, 1H), 3.99 (m, 1H), 3.85 (dd, J = 10.5, 3.5 Hz, 1H), 2.19 (s, 3H), 2.16 (s, 3H), 2.15 (s, 3H), 2.10 (s, 3H); 13C NMR (125 MHz, CDCl3) δ 170.7, 169.6, 169.5, 167.8, 89.9, 70.5, 69.5, 66.3, 62.1, 58.7, 20.83, 20.76, 20.72. To this mixture of isomers (1.54 g, 4.0 mmol) and thiophenol (0.6 mL, 6.0 mmol) in dry dichloromethane (20 mL) was added drop wise BF3·OEt2 (1.0 mL, 7.8 mmol) at 0 °C in 10 min. The reaction mixture was stirred overnight and monitored by TLC, before cold saturated NaHCO3 (30 mL) was added to quench the reaction. The organic layer was separated, and the aqueous layer was extracted with dichloromethane (30 mL×2). The combined dichloromethane layers were dried over Na2SO4 and concentrated under vacuum. Silica gel chromatography (hexanes/ethyl acetate: 5/2) was applied to the residue to give two compounds: α-1-thio-d-mannopyranoside 11 (1.1 g, 60%) and β-1-thio-d-mannopyranoside 12 (0.44 g, 26%). 11: oil, [α]D = + 109.2 (c, 1.0, ethyl acetate); 1H NMR (500 Mz, CDCl3) δ 7.47 (m, 2H), 7.32 (m, 3H), 5.50 (s, 1H), 5.42 (dd, J = 3.0, 1.0 Hz, 1H), 5.33 (td, J = 5.0, 2.5 Hz, 1H), 4.50 (m, 1H), 4.28 (dd, J = 12.5, 6.0 Hz, 1H), 4.12 (dd, J = 12.0, 2.5 Hz, 1 H), 3.81 (dd, J = 10.5, 3.0 Hz, 1H), 2.17 (s, 3H), 2.16 (s, 3H), 2.05 (s, 3H); 13C NMR (125 MHz, CDCl3) δ 170.6, 169.8, 169.6, 132.25, 132.17, 129.3, 128.3, 85.5 (1JCH = 168.8 Hz), 72.2, 69.4, 67.1, 62.4, 59.5, 20.9, 20.8, 20.7; HRMS(ESI): m/z calcd for C18H21N3NaO7S [M+Na]+ 446.0998; found 446.1005. 12: White solid. [α]D = − 62.4 (c, 1.0, ethyl acetate); Mp: 121 °C; 1H NMR (500 Mz, CDCl3) δ 7.52 (m, 2H), 7.31 (m, 3H), 5.70 (dd, J = 7.5, 0.5 Hz, 1H), 5.21 (t, J = 10.0 Hz, 1H), 4.85 (d, J = 1.5 Hz, 1H), 4.26 (dd, J = 12.0, 6.5 Hz, 1H), 4.17 (dd, J = 12.0, 2.5 Hz, 1H), 3.68 (ddd, J = 10.0, 6.5, 2.5 Hz, 1H), 3.63 (dd, J = 10.0, 3.5 Hz, 1H); 13C NMR (125 MHz, CDCl3) δ 170.6, 170.0, 169.6, 133.0, 132.1, 129.1, 128.2, 86.0 (1JCH = 152.5 Hz), 70.8, 66.9, 63.0, 62.8, 20.8, 20.7, 20.6. Anal. Calcd for C18H21N3O7S: C, 51.06; H, 5.00. Found: C, 50.94; H, 4.93.
Phenyl 3-azido-3-deoxy-α-1-thio-d-mannopyranoside (13)
To a stirred solution of 11 (1.06 g, 2.5 mmol) in methanol (20 mL) was added one drop of NaOMe in methanol (25% w/w). The reaction mixture was followed by TLC and, after 4 h, Amberlite I-120H ion exchange resin was added to neutralize the base and then removed by filtration. Methanol was removed under vacuum to give the triol as a syrup (0.72 g, 100%). [α]D = + 254.6 (c, 0.5, MeOH); 1H NMR (500 MHz, MeOH-d4) δ 7.51–7.54 (m, 2H), 7.26–7.33 (m, 3H), 5.39 (d, J = 1.0 Hz, 1H), 4.15 (dd, J = 3.0, 1.5 Hz, 1H), 4.08 (ddd, J = 10.0, 5.0, 2.5 Hz, 1H), 3.95 (t, J = 10.0 Hz, 1H), 3.76–3.83 (m, 2H), 3.47 (dd, J = 10.0, 3.0 Hz, 1H); 13C NMR (125 MHz, MeOH-d4) δ 134.0, 131.7, 128.8, 127.3, 88.7 (1JCH = 166.3 Hz), 74.4, 71.5, 65.4, 63.8, 61.0; HRMS(ESI): m/z calcd for C12H15N3NaO4S [M+Na]+ 320.0676, found 320.0668.
Phenyl 3-azido-2-O-benzyl-3-deoxy-4,6-O-[1-cyano-2-(2-iodophenyl)]ethylidene-α-1-thio-d-mannopyranoside (16)
A mixture of triol 13 (0.72 g, 2.42 mmol), triethyl 2-iodophenylorthoacetate (1.5 equiv about 50% purity), prepared from 2-iodophenyl acetonitrile according to Crich’s protocol,25 and camphorsufonic acid (20 mg) in dry dichloromethane (20 mL) was stirred at rt for 12 h. Triethylamine (drops) was added to quench the reaction before removal of the solvent under vacuum to afford an oily residue, which on silica gel chromatography (hexanes/ethyl acetate: 10/3) gave a mixture of orthoester isomers 14 (1.1g, 80%) as an oil. The major component had the following characteristics: 1H NMR (500 MHz, CDCl3) δ 7.83 (dd, J = 8.0, 1.0 Hz, 1H), 7.52 (dd, J = 8.0, 2.0 Hz, 1H), 7.40–7.43 (m, 2H), 7.27–7.33 (m, 4H), 6.91 (td, J = 8.0, 2.0 Hz, 1H), 5.48 (s, 1H), 4.40 (t, J = 10.0 Hz, 1H), 4.22 (td, J = 3.0, 1.0 Hz, 1H), 4.10–4.15 (m, 1H), 4.02 (t, J = 10.0 Hz, 1H), 3.80 (dd, J = 10.5, 3.0 Hz, 1H), 3.77 (dd, J = 9.5, 5.0 Hz, 1H), 3.69 (q, J = 7.0 Hz, 2H), 3.34 (d, J = 15.0 Hz, 1H), 3.31 (d, J = 15.0 Hz, 1H), 2.74 (d, J = 3.0 Hz, 1H), 1.26 (t, J = 7.5 Hz, 3H); 13C NMR (125 MHz, CDCl3) δ 139.4, 138.2, 133.1, 131.6, 131.0, 129.3, 128.3, 127.9, 113.3, 102.7, 88.0 (1JCH = 167.5 Hz), 71.9, 69.9, 64.8, 61.4, 61.0, 59.1, 45.5, 15.1.
Two sequences of transformations were applied to orthoester 14 to obtain donor 16
Sequence A
To a stirred mixture of 14 (1.0 g, 1.76 mmol) and TMSCN (1.2 ml, 8.8 mmol) in dry dichloromethane (15 mL) was added BF3·OEt2 (111 uL, 0.88 mmol) under Argon at 0 °C. After 2 h stirring, the reaction was quenched by saturated aqueous NaHCO3 (10 mL), washed with water (20 mL) and brine (20 mL), dried over Na2SO4, condensed under vacuum to give an oily residue. Purification by silica gel chromatography (hexanes/ethyl acetate: 10/3) afforded 15 as white crystals (0.60 g, 62%). [α]D = 184.2 (c, 0.5, CHCl3); Mp: 191–192 °C; 1H NMR (500 MHz, CDCl3) δ 7.89 (d, J = 8.0 Hz, 1H), 7.45 (m, 3H), 7.33 (m, 4H), 6.99 (td, J = 8.0, 1.5 Hz, 1H), 5.51 (s, 1H), 4.37 (t, J = 10.0 Hz, 1H), 4.28 (td, J = 10.0, 5.0 Hz, 1H), 4.24 (s, 1H), 4.12 (dd, J = 11.0, 5.0 Hz, 1H), 4.02 (t, 10.5 Hz, 1H), 3.92 (dd, J = 9.5, 3.0 Hz, 1H), 3.55 (s, 3H), 2.54 (d, J = 3.0 Hz, 1H); 13C NMR (125 MHz, CDCl3) δ 140.0, 135.2, 132.6, 131.7, 131.6, 129.6, 129.4, 128.3, 128.1, 114.1, 102.8, 97.1, 87.8,75.2, 71.8, 65.7, 63.8, 60.7, 48.7; Anal. Calcd for C21H19IN4O4S: C, 45.83; H, 3.48; N, 10.18; S, 5.83. Found: C, 45.84; H, 3.51; N, 10.04; S, 5.70. A mixture of 15 (0.41 g, 0.745 mmol), benzyl bromide (260 uL, 2.2 mmol) and 60 % NaH (90 mg, 2.2 mmol) in dry THF (20 mL) was heated to reflux under Argon for 2 h. The reaction mixture was cooled to rt before it was passed through a short silica gel pad. Removal of the solvent then afforded an oily residue which was purified by silica gel chromatography (hexanes/ethyl acetate: 10/1) to give thiomannopyranoside 16 (0.45 g, 93%) as an oil. [α]D = + 100.6 (c,1.0, ethyl acetate); 1H NMR (500 MHz, CDCl3) δ 7.89 (dd, J = 8.0, 1.0 Hz, 1H), 7.47 (dd, J = 7.5, 1.5 Hz, 1H), 7.30–7.35 (m, 6H), 6.90 (td, J = 7.5, 1.5 Hz, 1H), 5.38 (d, J = 1.0 Hz, 1H), 4.70 (d, J = 12.0 Hz, 1H), 4.60 (d. J = 12.0 Hz, 1H), 4.47 (t, J = 10.0 Hz, 1H), 4.19 (td, J = 10.0, 4.5 Hz, 1H), 4.11 (dd, J = 10.5, 5.0 Hz, 1H), 4.02 (t, J = 10.0 Hz, 1H), 4.01 (dd, J = 3.0, 1.0 Hz, 1H), 3.71 (dd, J = 10.5, 3.0 Hz, 1H), 3.55 (s, 2H); 13C (125 MHZ, CDCl3) δ 140.0, 136.8, 135.3, 133.0, 131.7, 131.6, 129.5, 129.3, 128.6, 128.3, 128.2, 128.0, 114.0, 102.8, 97.0, 86.5 (1JCH = 167.5 Hz), 78.8, 74.2, 73.4, 65.6, 64.2, 59.4, 48.7; HRMS(ESI): m/z calcd for C28H25IN4NaO4S [M+Na]+ 663.0534, found 663.0518.
Sequence B
A mixture of 14 (2.0 g, 3.6 mmol), benzyl bromide (0.64 mL, 6.4 mmol) and 60% NaH (200 mg, 5.0 mmol) in dry THF (30 mL) was heated to reflux for 3 h under Argon, then cooled down to rt. The reaction mixture was filtered through a thin silica gel pad, the filtrate was concentrated to afford a crude product, which was used directly for next reaction without further purification. To a stirred mixture of this crude product and TMSCN (1.2 ml, 9.0 mmol) in dry dichloromethane (20 mL) was added BF3·OEt2 (50 uL, 0.4 mmol) under Argon at 0 °C. The reaction was kept at 0 °C for 2 h before quenched by NaHCO3 (20 mL), washed with water and brine, dried (Na2SO4). Evaporation of the solvent under vacuum furnished an oily residue which was purified by silica gel chromatography (hexanes/ethyl acetate: 10/1) afford 16 (2.1 g 90% for two steps).
Phenyl 3-azido-4,6-O-benzylidene-3-deoxy-α-1-thio-d-mannopyranoside (17)
A mixture of 11 (550 mg, 1.85 mmol), benzaldehyde dimethylacetal (300 mg, 1.97 mmol) and camphorsufonic acid (10 mg) in dry dichloromethane (10 mL) was stirred at rt overnight. Triethylamine (drops) then was added before the solvent was removed under vacuum. The residue was purified by silica gel chromatography (hexanes/ethyl acetate/dichloromethane: 10/3/2) to give 17 (605 mg, 85%) as a white solid. [α]D = + 221 (c, 0.5, CHCl3); Mp:134 °C ; 1H NMR (500 MHz, CDCl3) δ 7.45–7.52 (m, 4H), 7.30–7.41 (m, 6H), 5.67 (s, 1H), 5.53 (s, 1H), 4.41 (td, J = 10.0, 5.0 Hz, 1H), 4.20–4.27 (m, 3H), 4.05 (dd, J = 10.5, 3.0 Hz, 1H), 3.88 (t, 10.0 Hz, 1H); 13C NMR (125 MHz, CDCl3) δ 136.9, 132.9, 131.9, 129.3, 129.1, 128.3, 128.0, 125.9, 101.7, 87.9, 77.9, 71.7, 68.6, 64.9, 60.9; HRMS(ESI): m/z calcd for C19H19N3NaO4S [M+Na]+ 408.0989, found 408.0978.
Phenyl 3-azido-2-O-benzyl-4,6-O-benzylidene-3-deoxy-α-1-thio-d-mannopyranoside (18)
A solution of 17 (542 mg, 1.41 mmol) and benzyl bromide (0.7g, 4.09 mmol) together with 60% NaH (112 mg, 2.8 mmol) in dry THF (30 mL) was heated to reflux for 3 h. After cooling to rt, the solution was passed through a thin silica gel pad then concentrated. The residue was purified by silica gel chromatography (hexanes/ethyl acetate 10/1) to give 18 (670 mg, 100%) as a crystalline solid. [α]D = + 143 (c, 0.5, ethyl acetate); Mp 118 °C; 1H NMR (500 MHz, CDCl3) δ 7.51–7.53 (m, 2H), 7.30–7.40 (m, 13H), 5.67 (s, 1H), 5.48 (s, 1H), 4.71 (s, 2H), 4.23–4.38 (m, 3H), 4.05 (d, J = 2.5 Hz, 1H), 3.84–3.91 (m, 2H); 13C NMR (125 MHz, CDCl3) δ 137.0, 133.2, 131.9, 129.3, 129.1, 128.6, 128.3, 128.22, 128.16, 128.0, 126.0, 101.7, 86.3, 79.0, 76.9, 73.3, 68.5, 65.3, 59.6. Anal. Calcd for C26H25N3O4S: C, 65.67; H, 5.30; N, 8.84; S, 6.74. Found: C, 65.68; H, 5.27; N, 8.69; S, 6.54.
Phenyl 3-acetamido-2-O-benzyl-3-deoxy-4,6-O-[1-cyano-2-(2-iodophenyl)]ethylidene-α-1-thio-d-mannopyranoside (19)
A mixture of azide 16 (157 mg, 0.25 mmol), triphenylphosphine (77 mg, 0.29 mmol) and water (10 uL, 0.56 mmol) in THF (5 mL) was stirred at rt overnight, after which Ac2O (53 uL, 0.56 mmol) was added, followed by pyridine (50 uL, 0.62 mmol). The mixture was allowed to stir for 12 h at rt before the solvent was removed to afford an oily residue which was purified by silica gel chromatography (hexanes/ethyl acetate: 10/3) to give donor 19 (120 mg, 75%) as an oil. [α]D = + 71.4 (c, 1.0, ethyl acetate); 1H NMR (500 MHz, CDCl3) δ 7.86 (dd, J = 8.0, 1.0 Hz, 1H), 6.93–7.43 (m, 12H), 6.95 (td, J = 8.0, 1.5 Hz, 1H), 5.60 (d, J = 9.5 Hz, 1H), 5.53 (s, 1H), 4.71 (d, J = 11.5 Hz, 1H), 4.56 (td, J = 9.5, 3.5 Hz, 1H), 4.37 (d, J = 12.0 Hz, 1H), 4.29 (td, J = 9.5, 4.5 Hz, 1H), 4.10 (dd, J = 11.0, 5.0 Hz, 1H), 3.93–4.04 (m, 3H), 3.40 (d, J = 14.5 Hz, 1H), 3.20 (d, J = 14.0 Hz, 1H), 1.86 (s, 3H); 13C (125 MHZ, CDCl3) δ 169.7, 139.8, 136.7, 135.4, 133.4, 132.4, 131.6, 129.5, 129.3, 128.9, 128.6, 128.4, 128.0, 127.9, 114.3, 102.8, 96.8, 85.6 (1JCH = 166.5 Hz), 78.9, 74.3, 72.7, 65.6, 64.3, 48.4, 48.2, 23.4; HRMS(ESI): m/z calcd for C30H29IN2NaO5S [M+Na]+ 679.0734, found 679.0720.
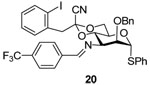
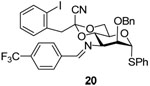
Phenyl 2-O-benzyl-4,6-O-(1-cyano-2-[2-iodophenyl])ethylidene-3-(4-trifluoromethyl) benzylideneimino-3-deoxy-α-1-thio-d-mannopyranoside (20)
A solution of azide 16 (128 mg, 0.2 mmol) and triphenylphosphine (58 mg, 0.22 mmol) in dry dichloromethane (6 mL) was stirred at rt overnight before 4-trifluoromethylbenzaldehyde (52 mg, 0.30 mmol) was added. The reaction mixture then was stirred for 24 h before the solvent was evaporated on rotary bath. The residue was purified by flash chromatography on neutral alumina (hexanes/ethyl acetate/triethylamine: 100/10/1) to give 20 (60–80%) as an oil. [α]D = + 87.3 (c, 1.0, ethyl acetate); 1H NMR (500 MHz, CDCl3) δ 8.39 (s, 1H), 7.86 (d, J = 8.0 Hz, 2H), 7.70–7.73 (m, 2H), 7.40–74.1 (m, 2H), 7.23–7.34 (m, 9H), 7.01 (dd, J = 2.5, 1.5 Hz, 1H), 6.80 (dd, J = 2.5, 1.5 Hz, 1H), 5.53 (d, J = 1.0 Hz, 1H), 4.80 (d, J = 12.0 Hz, 1H), 4.68 (d, J = 11.5 Hz, 1H), 4.66 (t, 9.5 Hz, 1H), 4.29 (td, J = 10.0, 5.0 Hz, 1H), 4.16 (dd, J = 11.0, 5.0 Hz, 1H), 4.11 (t, J = 10.5 Hz, 1H), 4.03 (dd, J = 3.0, 1.0 Hz, 1H), 3.93 (dd, J = 10.0, 2.5 Hz, 1H), 3.47(d, J = 14.5 Hz, 1H), 3.46 (d, J = 14.5 Hz, 1H); 13C NMR (125 MHz, CDCl3) δ 162.5, 139.7, 138.9, 137.6, 135.6, 133.7, 131.4, 131.3, 129.9,129.3, 129.2, 128.8, 128.4, 128.0, 127.94, 127.88, 127.7, 125.5, 114.5, 102.7, 96.9, 87.4, 79.5, 73.9, 69.0, 66.0, 64.3, 48.7; HRMS(ESI): m/z calcd for C36H30F3IN2NaO4S [M+Na]+ 793.0816, found 793.0800.
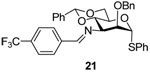
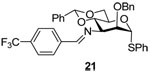
Phenyl 3-azido-2-O-benzyl-4,6-O-benzylidene-3-deoxy-3-(4-trifloromethyl)benzylideneimino-α-1-thio-d-mannopyranoside (21)
The protocol the preparation of 20 was applied to azide 18 to give imine 21 (60–80%) as an oil. [α]D = + 72.0 (c, 0.5, ethyl acetate); 1H NMR (500 MHz, CDCl3) δ 8.47 (s, 1H), 7.91 (d, J = 3.0 Hz, 2H), 7.70 (d, J = 3.0 Hz, 2H), 7.26–7.45 (m, 15 H), 5.62 (s, 1H), 5.60 (s, 1H), 4.84 (d, J = 12.0 Hz, 1H), 4.77 (d, J = 12.0 Hz, 1H), 4.41–4.50 (m, 2H), 4.29 (dd, J = 10.5, 4.5 Hz, 1H), 3.99–4.02 (m, 2H), 3.94 (t, J = 10.0, 1H); 13C NMR (125 MHz, CDCl3) δ 162.6, 139.1, 137.7, 137.4, 134.0, 131.6, 129.2, 128.9, 128.7, 128.4, 128.2, 128.1, 127.9, 127.7, 126.0, 125.6, 101.5, 87.2, 80.0, 76.7, 73.7, 69.5, 68.7, 65.5; HRMS(ESI): m/z calcd for C34H31F3NO4S [M+H]+ 606.19204, found 606.19200.
Methyl 2,4,6-tri-O-acetyl-3-deoxy-3-phthalimido-α-d-mannopyranoside (22)
A mixture of 8 (1.15 g, 5 mmol), N-ethoxyphthalimide (1.15 g, 5.25 mmol) and triethylamine (0.55 g, 5.4 mmol) in dry DMF (10 mL) was stirred at 100 °C for 12 h. The solvent was removed under vacuum and the residue was mixed with acetic anhydride (2.3 g, 22.8 mmol) and pyridine (3.5 g, 45.4 mmol) and stirred at rt for 12 h. Cold water (20 mL) was added to the reaction mixture and the mixture was stirred for 3 h before it was extracted with dichloromethane (30 mL×3). The combined organic layers was washed by aqueous HCl (1.0 M), and saturated NaHCO3, dried over Na2SO4, and concentrated to give an oily residue which was purified by silica gel chromatography (hexanes/ethyl acetate: 1/1) to give 22 (2.2 g, 98%) as a colorless oil. [α]D = +23.2 (c, 1.0, ethyl acetate); 1H NMR (500 MHz, CDCl3) δ 7.79–7.83 (m, 2H), 7.70–7.72 (m, 2H), 6.21 (t, J = 10.5 Hz, 1H), 5.17 (dd, J = 3.0, 1.5 Hz, 1H), 4.91 (dd, J = 11.5, 3.0 Hz, 1H), 4.70 (d, J = 1.5 Hz, 1H), 4.27 (dd, J = 12.0, 6.0 Hz, 1H), 4.19 (dd, J = 12.0, 3.0 Hz, 1H), 3.99 (ddd, J = 12.0, 6.0, 3.0 Hz, 1H), 3.45 (s, 3H), 2.11 (s, 3H), 2.06 (s, 3H), 1.86 (s, 3H); 13C NMR (125 MHz, CDCl3) δ 170.7, 169.2, 167.8, 134.3, 131.4, 123.5, 98.4, 71.0, 70.0, 64.1, 63.2, 55.2, 50.9, 21.0, 20.8, 20.6; HRMS(ESI): m/z calcd for C21H23NNaO10 [M+Na]+ 472.1215, found 472.1217.
Phenyl 2,4,6-tri-O-acetyl-3-deoxy-3-phthalimido-α-1-thio-d-mannopyranoside (23)
To a solution of 22 (2.25 g, 5 mmol) in acetic anhydride (10 mL) was added HClO4 (two drops) at 0 °C, followed by stirring at rt for 24 h before cold water (50 mL) was added. The resulting mixture was allowed to stir for 2 h before it was extracted with dichloromethane (50 ml×3). The combined organic layers were washed with water (50 mL), aqueous NaHCO3 (50 mL), and brine (50 mL), dried over Na2SO4, and concentrated under vacuum to afford a crude product as an oil. BF3·Et2O (2 mL, 18.6 mmol) and PhSH (0.8 ml, 7.8 mmol) were added to this crude product in dry dichloromethane (30 mL) at 0 °C and the reaction mixture was stirred overnight before NaHCO3 (30 mL) was added. The dichloromethane phase was separated and washed with brine (20 mL), dried over Na2SO4, and concentrated to a residue which was purified by silica gel chromatography (hexanes/ethyl acetate/dichloromethane: 10/10/3) to give 23 (2.0 g 75 %) as a white solid. [α]D = + 82.4 (c, 0.5, ethyl acetate); Mp: 195 °C; 1H NMR (500 MHz, CDCl3) δ 7.85 (dd, J = 5.5, 3.0 Hz, 2H), 7.74 (dd, J = 5.5, 3.0 Hz, 2H), 7.30–7.35 (m, 2H), 7.30–7.35 (m, 3H), 6.32 (t, J = 10.5 Hz, 1H), 5.52 (d, 1.0 Hz, 1H), 5.45 (dd, J = 2.5, 1.0 Hz, 1H), 4.92 (dd, J = 11.0, 2.5 Hz, 1H), 4.58 (m, 1H), 4.31 (dd, J = 12.0, 6.5 Hz, 1H), 4.21 (dd, J = 12.0, 2.5 Hz, 1H), 2.07 (s, 3H), 2.06 (s, 3H), 1.93 (s, 3H); 13C (125 MHZ, CDCl3) δ 170.62, 170.55, 169.2, 167.7, 134.4, 132.6, 132.2, 131.3, 129.2, 128.1, 123.7, 85.9 (1JCH = 168.8 Hz), 72.5, 71.2, 64.2, 63.1, 51.9, 21.0, 20.8, 20.7; HRMS(ESI): m/z calcd for C26H25NNaO9S [M+Na]+ 550.1543, found 550.1545.
Phenyl 3-deoxy-3-phthalimido-α-1-thio-d-mannopyranoside (24)
Peracetyl thioglycoside 23 (1.24 g, 2.35 mmol) and HCl in ether (3 ml, 2M) were mixed in dry methanol (30 mL) at rt and stirred overnight. The solvent was removed by rotary evaporation at rt and the residue was purified by silica gel chromatography (dichloromethane/MeOH: 5/1) to give triol 24 (0.92 g, 98%) as a solid. [α]D = + 82.8 (c, 1.0, CHCl3); Mp: 95 °C; 1H NMR (300 MHz, MeOH-d4) δ 7.76–7.89 (m, 4H), 7.54–7.59 (m, 2H), 7.27–7.36 (m, 3H), 5.45 (d, J = 0.9 Hz, 1H), 5.13 (dd, J = 11.1, 9.9 Hz, 1H), 4.51 (dd, J = 11.1, 3.0 Hz, 1H), 4.14–4.20 (m, 2H), 3.85 (m, 2H); 13C NMR (125 MHz, MeOH-d4) δ 168.9, 134.0, 133.9, 131.9, 131.8, 128.8, 127.3, 122.7, 88.8, 75.6, 72.2, 61.2, 60.8, 57.0; HRMS(ESI): m/z calcd for C20H19NNaO6S [M+Na]+ 424.0826, found 424.0815.
Phenyl 4,6-O-benzylidene-3-deoxy-3-phthalimido-α-1-thio-d-mannopyranoside (25)
Triol 24 (305 mg, 0.76 mmol), benzaldehyde dimethylacetal (127 mg, 125 uL) and camphorsufonic acid (5 mg) were stirred in dry dichloromethane (10 mL) overnight before triethylamine (1 drop) was added. The solvent was removed under reduced pressure and the residue was purified by silica gel chromatography (hexanes/ethyl acetate: 1/1) to give compound 25 (298 mg, 80%) as an oil. [α]D = + 41.2 (c, 1.0, ethyl acetate); 1H NMR (500 MHz, CDCl3) δ 7.85–7.87 (m, 2H), 7.72–7.75 (m, 2H), 7.51–7.53 (m, 2H), 7.26–7.37 (m, 8H), 5.59 (d, J = 1.5 Hz, 1H), 5.58 (s, 1H), 4.92–5.01 (m, 2H), 4.52 (td, J = 10.0, 5.0 Hz, 1H), 4.39–4.41 (m, 1H), 4.31 (d, J = 4.5 Hz, 1H), 4.25 (dd, J = 10.5, 5.5 Hz, 1H), 3.93 (t, 10.0 Hz, 1H); 13C NMR (125 MHz, CDCl3) δ 137.1, 134.5, 133.2, 132.0, 129.2, 129.1, 128.2, 127.8, 126.3, 123.8, 102.1, 89.2, 73.7, 72.7, 68.6, 67.0, 53.4; HRMS(ESI): m/z calcd for C27H23NNaO6S [M+Na]+ 512.1139, found 512.1126.
Phenyl 4,6-O-benzylidene-3-deoxy-3-phthalimido-2-O-trimethylsilyl-α-1-thio-d-mannopyranoside (26)
TMSOTf (130 uL, 0.72 mmol) was added to a mixture of 25 (178 mg, 0.36 mmol) and sym-collidine (142 uL, 1.08 mmol) in dichloromethane (4 mL) at 0 °C. The resulting mixture was stirred for 30 min at rt before it was washed with aqueous NaHCO3, brine, dried over Na2SO4, and condensed under vacuum. Purification of the residue on silica gel (hexanes/ethyl acetate/triethylamine 100/30/0.5) gave 26 (168 mg, 83%) as an oil. [α]D = + 20.8 (c, 0.5, ethyl acetate); 1H NMR (500 MHz, CDCl3) δ 7.83 (m, 2H), 7.72 (m, 2H), 7.54 (m, 2H), 7.28–7.43 (m, 8H), 5.70 (s, 1H), 5.43 (dd, J = 11.5, 9.5 Hz, 1H), 5.33 (s, 1H), 4.72 (dd, J = 11.5, 3.0 Hz, 1H), 4.42 (td, J = 9.5, 4.5 Hz, 1H), 4.36 (d, J = 3.0 Hz, 1H), 4.26 (dd, J = 10.5, 5.0 Hz, 1H), 3.97 (t, 10.0 Hz, 1H), − 0.10 (s, 9H); 13C NMR (125 M Hz, CDCl3) δ 168.8, 167.6, 137.4, 134.2, 133.9, 133.4, 132.4, 129.3, 129.0, 128.2, 127.9, 126.3, 123.2, 101.8, 89.4 (1JCH = 166.0 Hz), 72.5, 72.3, 68.7, 66.8, 54.3, − 0.4; HRMS(ESI): m/z calcd for C30H31NNaO6SSi [M+Na]+ 584.1534, found 584.1535.
General procedure for coupling of donors 16, 19 and 26 with adamantanol
A mixture of donor 16, 19, or 26 (0.10 mmol), 1-benzenesulfinyl piperidine or diphenyl sulfoxide (0.11 mmol), and 2,4,6-tri-tert-butylpyrimidine (50 mg, 0.20 mmol) in dry dichloromethane (4 mL) was stirred together with 4Å MS (30 mg) at rt for 30 min, then cooled to −78 °C or −65 °C. Trifluoromethanesulfonic anhydride (20 uL, 0.12 mmol) was added in one portion under Argon and the reaction was kept at this temperature for 30 min. 1-Adamantanol (30 mg, 0.20 mmol) in dichloromethane (1 mL) was added at either −78 °C or −50 °C, and after being stirred for the requisite time the reaction mixture was quenched with saturated NaHCO3 (2 mL), and extracted with dichloromethane (10 mL×3). The combined organic phases were washed with water (10 mL), and brine (10 mL), dried over Na2SO4, and concentrated under reduced pressure, and the resulting residue was purified by silica gel chromatography.
1-Adamantanyl 3-azido-2-O-benzyl-3-deoxy-4,6-O-[1-cyano-2-(2-iodophenyl)]ethylidene-α-d-mannopyranoside (27α) and 1-Adamantanyl 3-azido-2-O-benzyl-3-deoxy-4,6-O-[1-cyano-2-(2-iodophenyl)ethylidene-β-d-mannopyranoside (27β)
Coupling of 16 with adamantanol, after column chromatography on silica gel (hexanes/ethyl acetate: 10/1), to gave two stereoisomers: 27α and 27β. 27α: oil, [α]D = + 39.8 (c, 1.0, ethyl acetate); 1H NMR (500 MHz, CDCl3) δ 7.88 (dd, J = 8.0, 1.0 Hz, 1H), 7.48 (dd, J = 8.0, 1.5 Hz, 1H), 7.25–7.55 (m, 6H), 6.80 (td, J = 8.0, 1.5 Hz, 1H), 4.96 (d, J = 1.5 Hz, 1H), 4.79 (d, 12.0 Hz, 1H), 4.59 (d, J = 12.0 Hz, 1H), 4.35 (t, J = 10.0 Hz, 1H), 4.07 (dd, J = 10, 5.0 Hz, 1H), 3.96 (t, 10.5 Hz, 1H), 3.87 (td, J = 10.0, 5.0 Hz, 1H), 3.78 (dd, J = 10.5, 3.0 Hz, 1H), 3.55 (dd, J = 3.0, 2.0 Hz, 1H), 3.53 (s, 2H), 2.10 (s, 3H), 1.52–1.61 (m, 12H); 13C NMR (125 MHz, CDCl3) δ 139.9, 137.4, 135.5, 131.7, 129.4, 128.6, 128.4, 128.2, 128.1, 114.2, 102.8, 97.0, 91.6 (1JCH = 167.5 Hz), 78.6, 75.3, 74.6, 73.8, 66.1, 62.8, 59.1, 48.8, 42.1, 36.1, 30.5; HRMS(ESI): m/z calcd for C32NaH35IN4O5 [M+Na]+ 705.1545, found 705.1527. 27β: oil, [α]D = − 16.7 (c, 1.0, ethyl acetate); 1H NMR (500 MHz, CDCl3) δ 7.86 (dd, J = 8.0, 1.5 Hz, 1H), 7.42–7.45 (m, 3H), 7.28–7.35 (m, 4H), 6.97 (td, J = 8.0, 1.5 Hz, 1H), 4.97 (d, J = 12.0 Hz, 1H), 4.82 (d, J = 12.0 Hz, 1H), 4.78 (s, 1H), 4.34 (t, J = 10.0 Hz, 1H), 4.05–4.16 (m, 2H), 3.69 (d, 3.0 Hz, 1H), 3.51 (s, 2H), 3.39 (dd, J = 10.5, 3.0 Hz, 1H), 3.28 (td, J = 10.0, 5.0 Hz, 1H), 2.16 (s, 3H), 1.57–1.82 (m, 12 H); 13C NMR (125 MHz, CDCl3) δ 139.9, 137.6, 135.5, 131.7, 129.4, 128.8, 128.1, 127.8, 114.2, 102.9, 96.7, 94.9 (1JCH = 151.3 Hz), 77.9, 75.8, 75.4, 73.6, 66.9, 65.9, 61.8, 48.6, 42.3, 36.1, 30.6; HRMS(ESI): m/z calcd for C32H35IN4NaO5 [M+Na]+ 705.1545, found 705.1528.
1-Adamantanyl 3-acetamido-2-O-benzyl-3-deoxy-4,6-O-[1-cyano-2-(2-iodophenyl)]ethylidene-α-d-mannopyranoside (28α)
Coupling of 19 with adamantanol gave 28α (56%), which was separated by silica gel chromatography (hexanes/ethyl acetate: 10/3): oil, [α]D = + 31.4 (c, 0.5, ethyl acetate); 1H NMR (500 MHz, CDCl3, −40 °C) ä 7.77 (d, J = 7.5 Hz, 1H), 6.93–7.42 (m, 13H), 5.09 (s, 1H), 4.96 (d, J = 11.0 Hz, 1H), 4.72 (t, J = 10.5 Hz, 1H), 4.65 (d, J = 12.5 Hz, 1H), 4.36 (d, J = 12.0 Hz, 1H), 3.85–3.99 (m, 2H), 3.78 (t, J = 10.5 Hz, 1H), 3.72 (s, 1H), 2.59 (d, J = 14.0 Hz, 1H), 2.37 (d, J = 14.5, 1H), 2.10 (m, 3H), 2.05 (s, 3H), 1.40–1.66 (m, 12H); 13C (125 MHZ, CDCl3) δ 177.7, 139.9, 139.7, 137.3, 135.7, 131.6, 129.1, 128.8, 128.72, 128.65, 128.2, 128.0, 125.5, 122.0, 113.9, 102.6, 96.6, 91.0 (1JCH = 165.0 Hz), 75.1, 73.3, 73.0, 66.3, 62.8, 56.4, 47.6, 42.2, 36.2, 30.6, 22.4; HRMS(ESI): m/z calcd for C40H43IN2NaO6S [M+Na]+ 829.1779, found 829.1777.
1-Adamantanyl 4,6-O-benzylidene-3-deoxy-3-phthalimido-2-O-trimethylsilyl-α-d-mannopyranoside (31α)
Coupling of 26 with adamantanol gave 31α (77%), which was separated by silica gel chromatography (hexanes/ethyl acetate: 10/3). 31α: oil. [α]D = −30.0 (c, 0.5, ethyl acetate); 1H NMR (500 MHz, CDCl3) δ 7.80–7.82 (m, 2H), 7.67–7.70 (m, 2H), 7.40–7.42 (m, 2H), 7.26–7.29 (m, 3H), 5.68 (s, 1H), 5.33 (dd, J = 11.5, 9.5 Hz, 1H), 5.04 (d, J = 1.0 Hz, 1H), 4.74 (dd, J = 11.0, 3.0 Hz, 1H), 4.25 (dd, J = 10.0, 5.0 Hz, 1H), 4.16 (td, J = 10.0, 5.0, 1H), 3.93 (m, 1H), 3.90 (t, J = 10.5 Hz, 1H), 2.17 (br. s, 3H), 1.81–1.89 (m, 6H), 1.61–1.69 (m, 6H), −0.08 (s, 9H); 13C NMR (125 MHz, CDCl3) δ 137.7, 134.0, 128.9, 128.1, 126.3, 123.0, 101.7, 94.1 (1JCH = 166.3 Hz), 74.7, 73.0, 72.5, 69.1, 65.1, 53.8, 42.4, 36.3, 30.6, −0.4; HRMS(ESI): m/z calcd for C34H42NO7Si [M+H]+ 604.2725, found 604.2726.
General procedure of coupling reaction of donors 20, 21 with acceptors 32 and 33 and adamantanol
A mixture of donor 20 or 21 (0.10 mmol), diphenyl sulfoxide (0.11 mmol), and 2,4,6-tri-tert-butylpyrimidine (50 mg, 0.20 mmol) in dry dichloromethane (4 mL)was stirred together with 4Å MS (30 mg) at rt for 30 min, then cooled to −78 °C. Trifluoromethanesulfonic anhydride (20 uL, 0.12 mmol) then was added in one portion under Argon. Then the reaction mixture was raised to −65 °C over 15 min before the acceptor (0.20 mmol for adamantanol, 0.15 mmol for 32 and 33) in dichloromethane (1.5 mL) was added. The resulting mixture was allowed to rise to −10 °C gradually before it was quenched with NaHCO3. The resulting mixture was extracted with dichloromethane and the extracts concentrated to give a residue, which was mixed with silica gel (1.5 g) then loaded onto a silica gel column. Elution of the products was achieved with a solution of 5% ethyl acetate in hexanes (100 mL), then a solution of 20% ethyl acetate in hexanes (100 mL), and then a mixture of hexanes/ethyl acetate/MeOH (10/5/1).
1-Adamantanyl 3-amino-2-O-benzyl-3-deoxy-4,6-O-[1-cyano-2-(2-iodophenyl)]ethylidene-β-d-mannopyranoside (29β)
Coupling of 20 with 1-adamantanol gave 29β (83%) as an oil. [α]D = − 129.2 (c, 0.25, ethyl acetate); 1H NMR (500 MHz, CDCl3) δ 7.85 (d, J = 7.5 Hz, 1H), 7.26–7.41 (m, 7H), 6.96 (td, J = 7.5, 1.5 Hz, 1H), 5.09 (d, J = 11.5 Hz, 1H), 4.84 (s, 1H), 4.64 (d, J = 12.0 Hz, 1H), 4.11 (dd, J = 11.0, 5.0 Hz, 1H), 4.01(t, J = 10.5 Hz, 1H), 3.79 (t, 9.5 Hz, 1H), 3.69 (d, 3.0 Hz, 1H), 3.47 (s, 2H), 3.15 (td, J = 10.0, 5.0 Hz, 1H), 2.78 (dd, J = 10.0, 3.0 Hz, 1H), 2.16 (s, 3H), 1.76–1.86 (m, 6H), 1.59–1.67 (m, 6H); 13C NMR δ (125 MHz, CDCl3) δ 139.9, 138.4, 135.8, 131.8, 129.3, 128.5, 128.4, 128.0, 127.8, 114.7, 102.8, 96.8, 95.9 (1JCH = 151.3 Hz), 80.4, 76.0, 75.4, 67.2, 65.9, 54.4, 48.7, 42.4, 36.2, 30.6; HRMS(ESI): m/z calcd for C32H38IN2O5 [M+H]+ 657.1820, found 657.1830.
1-Adamantanyl 3-amino-2-O-benzyl-4,6-O-benzylidene-3-deoxy-β-mannopyranoside (30β)
Coupling of 21 with 1-adamantanol gave 30β (78%) as an oil. [α]D = − 50.0 (c, 1.0, ethyl acetate); 1H NMR (500 MHz, CDCl3) δ 7.27–7.51 (m, 10H), 5.50 (s, 1H), 5.16 (d, J = 12.0 Hz, 1H), 4.91 (s, 1H), 4.65 (d, 11.5 Hz, 1H), 4.25 (dd, J = 10.0, 5.0 Hz, 1H), 3.85 (t, J = 10.0 Hz, 1H), 3.73 (d, J = 3.0 Hz, 1H), 3.61 (t, J = 9.5 Hz, 1H), 3.35 (td, J = 10.0, 5.0 Hz, 1H), 2.91 (dd, J = 10.0, 3.0 Hz, 1H), 2.19 (s, 3H), 1.89 (d, J = 11.5 Hz, 3H), 1.83 (d, J = 11.0 Hz, 3H), 1.62–1.70 (m, 6H); 13C NMR (125 MHz, CDCl3) δ 138.8, 137.6, 129.1, 128.5, 128.3, 127.7, 126.2, 102.0, 95.8 (1JCH = 151.3 Hz), 81.1, 80.3, 76.2, 75.2, 68.9, 68.4, 54.7, 42.4, 36.2, 30.6; HRMS(ESI): m/z calcd for C30H37NNaO5 [M+Na]+ 514.2564, found 514.2561.
Methyl 4-O-{3-amino-2-O-benzyl-3-deoxy-4,6-O-[1-cyano-2-(2-iodophenyl)]ethylidene-β-d-mannopyranosyl}-(1→4)-2,3-O-isopropylidene-α-l-rhamnopyranoside (34β)
Coupling of 20 with 32 gave 34β (76%) as an oil. [α]D = − 52.8 (c, 1.0, ethyl acetate); 1H NMR (500 MHz, CDCl3) δ 7.86 (d, J = 7.5 Hz, 1H), 7.41 (d, J = 7.5 Hz, 1H), 7.26–7.34 (m, 6H), 6.97 (t, J = 7.5 Hz, 1H), 5.04 (s, 1H), 4.96 (d, 11.0 Hz, 1H), 4.86 (s, 1H), 4.56 (d, J = 12.0 Hz, 1H), 4.10–4.18 (m, 3H), 4.01 (t, J = 11.0 Hz, 1H), 3.86 (d, J = 3.0 Hz, 1H), 3.77 (t, J = 9.5 Hz, 1H), 3.63 (m, 2H), 3.47 (m, 2H), 3.37 (s, 3H), 3.15 (td, J = 10.0, 5.0 Hz, 1H), 2.82 (dd, J = 10.0, 3.5 Hz, 1H), 1.51 (s, 3H), 1.35 (s, 3H), 1.29 (d, J = 5.5 Hz, 3H); 13C NMR (125 MHz, CDCl3) δ 139.9, 138.3, 135.7, 131.8, 129.4, 128.4, 128.14, 128.10, 127.8, 114.7, 109.4, 102.7, 101.4 (1JCH = 157.5 Hz), 97.9, 96.9, 79.1, 78.32, 78.27, 77.7, 76.1, 75.8, 67.4, 65.9, 64.1, 54.9, 54.1, 48.7, 28.0, 26.4, 17.7; HRMS(ESI): m/z calcd for C32H40IN2O9 [M+H]+ 723.1779, found 723.1773.
Methyl (3-amino-2-O-benzyl-4,6-O-benzylidene-3-deoxy-β-d-mannopyranosyl)-(1→4)-2,3-O-isopropylidene-α-l-rhamnopyranoside (35β)
Coupling of 21 with 32 gave 35β (82%) as an oil. [α]D = −86.8 (c, 1.0, ethyl acetate); 1H NMR (500 MHz, CDCl3) δ 7.45–7.50 (m, 2H), 7.30–7.40 (m, 8H), 5.52 (s, 1H), 5.13 (s, 1H), 5.02 (d, J = 11.5Hz, 1H), 4.88 (s, 1H), 4.60 (d, J = 11.5 Hz, 1H), 4.26 (dd, J = 10.5, 5.0 Hz, 1H), 4.21 (dd, J = 7.0, 5.5 Hz, 1H), 4.11 (d, J = 5.5 Hz, 1H), 3.90 (d, J = 5.0 Hz, 1H), 5.87 (t, J = 10.0 Hz, 1H), 3.72 (dd, J = 10.0, 2.5 Hz, 1H), 3.66 (m, 1H), 3.61 (t, J = 10.0 Hz, 1H), 3.39 (s, 3H), 3.34 (m, 1H), 2.93 (dd, J 10.0, 3.0 Hz, 1H), 1.54 (s, 3H), 1.36 (s, 3H), 1.33 (d, J = 6.0 Hz, 3H); 13C NMR (125 MHz, CDCl3) δ 138.7, 137.5, 129.1, 128.4, 128.3, 128.1, 127.8, 126.2, 109.4, 102.0, 101.3 (1JCH = 157.5 Hz), 97.9, 80.6, 79.6, 78.4, 77.9, 76.1, 75.8, 68.7,68.6, 64.2, 54.9, 54.4, 27.9, 26.5, 17.7; HRMS(ESI): m/z calcd for C30H40NO9 [M+H]+ 558.2698, found 558.2694.
Methyl 2,3,6-tri-O-benzyl-4-O-{3-amino-2-O-benzyl-3-deoxy-4,6-O-[1-cyano-2-(2-iodophenyl)]ethylidene-β-d-mannopyranosyl}-α-d-glucopyranoside (36β)
Coupling of 20 with 33 gave 36β (82%) as an oil. [α]D = + 0.8 (c, 1.0, ethyl acetate); 1H NMR (500 MHz, CDCl3) δ 7.43 (dd, J = 8.0, 1.5 Hz, 1H), 7.24–7.39 (m, 22H), 7.00 (td, J = 8.0, 1.5 Hz, 1H), 4.95 (d, J = 11.0 Hz, 1H), 4.94 (d, J = 10.5 Hz, 1H), 4.86 (d, J = 12.0 Hz, 1H), 4.75 (d, J = 12.0 Hz, 1H), 4.74 (d, J = 10.0, 1H), 4.68 (d, J = 12.0 Hz, 1H), 4.64 (d, J = 3.5 Hz, 1H), 4.57 (d, J = 11.5 Hz, 1H), 4.33 (d, J = 12.0 Hz, 1H), 4.28 (s, 1H), 3.92 (t, 9.0 Hz, 1H), 3.80–3.87 (m, 2H), 3.45–3.69 (m, 9H), 3.42 (s, 3H), 2.73 (td, J = 10.0, 5.0 Hz, 1H), 2.36 (dd, J = 10.0, 5.0 Hz, 1H); 13C NMR (125 MHz, CDCl3) δ 139.9, 139.1, 138.4, 138.1, 137.5, 135.9, 132.1, 129.5, 128.7, 128.6, 128.4, 128.3, 128.13, 128.09, 128.0, 127.9, 127.8, 127.5, 114.8, 102.9, 102.2 (1JCH = 155.0 Hz), 98.5, 96.7, 80.2, 79.3, 79.0, 77.0, 76.0, 75.6, 73.7, 69.7, 68.1, 67.0, 65.7, 55.5, 53.9, 48.7; HRMS(ESI): m/z calcd for C50H54IN2O10 [M+H]+ 969.2823, found 969.2827.
Methyl (3-amino-2-O-benzyl-4,6-O-benzylidene-3-deoxy-β-d-mannopyranosyl)-(1→4)-2,4,6-tri-O-benzyl-α-d-glucopyranoside (37β)
Coupling of 21 with 33 gave 37β (92%) as a colorless oil. [α]D = − 36.1 (c, 1.0, CHCl3); 1H NMR (500 MHz, CDCl3) δ 7.25–7.47 (m, 25H), 5.43 (s, 1H), 5.07 (d, J = 11.0 Hz, 1H), 5.00 (d, J = 11.0 Hz, 1H), 4.82 (d, 12.0 Hz, 1H), 4.53–4.75 (m, 6H), 4.42 (d, J = 12.0 Hz, 1H), 4.11 (dd, J = 10.5, 5.0 Hz, 1H), 3.98 (t, J = 9.0 Hz, 1H), 3.85 (t, 9.0 Hz, 1H), 3.64–3.71 (m, 4H), 3.55 (dd, J = 10.0, 4.0 Hz, 1H), 3.49 (dd, J = 18.0, 10.5 Hz, 2H), 3.41 (s, 3H), 3.07 (td, J = 10.0, 5.0 Hz, 1H), 2.63 (dd, J = 10.5, 3.0 Hz, 1H); 13C NMR (125 MHz, CDCl3) δ 139.4, 138.5, 138.4, 137.5, 129.2, 128.7, 128.41, 128.36, 128.2, 128.1, 127.82, 127.76, 127.3, 126.2, 102.5, 102.0 (1JCH = 162.5 Hz), 98.5, 80.4, 80.2, 79.7, 78.9, 77.5, 76.0, 75.4, 73.7, 69.8, 68.7, 68.4, 68.3, 55.4, 54.2; HRMS(ESI): m/z calcd for C48H54NO10 [M+H]+ 804.3742, found 804.3727.
1-Adamantanyl 3-N-(tert-butyloxycarbonylamino)-2-O-benzyl-3-deoxy-4,6-O-[1-cyano-2-(2-iodophenyl)]ethylidene-β-d-mannopyranoside (40)
To a solution of amine 29β (75 mg, 0.11 mmol) and (Boc)2O (30 mg, 0.14 mmol) was added triethylamine (30 uL, 0.23 mmol) in dry THF (5 mL). The reaction mixture was stirred at rt for 4 h, then concentrated under vacuum to give a residue, which was purified by chromatography on silica gel (hexanes/ethyl acetate: 10/1) to give 40 (89 mg, 100%) as a white solids. [α]D = − 42.6 (c, 0.5, ethyl acetate); Mp 202–203 C; 1H NMR (500 MHz, CDCl3) δ 7.84 (dd, J = 8.0, 1.0 Hz, 1H), 7.40, (dd, J = 8.0, 1.5 Hz, 1H), 7.24–7.37 (m, 6H), 6.94 (td, J = 8.0, 1.5 Hz, 1H), 5.01 (d, J = 12.0 Hz, 1H), 4.87 (s, 1H), 4.79 (d, J = 11.0 Hz, 1H), 4.66 (d, J = 12.0 Hz, 1H), 4.11 (dd, J = 11.0, 6.0 Hz, 1H), 4.00 (t, J = 10.5 Hz, 1H), 3.83–3.92 (m, 2H), 3.67 (d, 2.5 Hz, 1H), 3.48 (d, J = 14.5 Hz, 1H), 3.43 (d, J = 14.5 Hz, 1H), 3.28 (m, 1H), 2.17 (s, 3H), 1.84 (d, J = 10.5 Hz, 3H), 1.76 (d, J = 11.5 Hz, 3H), 1.6 (d, J = 12.5 Hz, 3H), 1.61 (d, J = 12.0 Hz, 3H), 1.42 (s, 9H); 13C NMR (125 MHz, CDCl3) δ 155.3, 139.8, 138.0, 135.7, 131.8, 129.3, 128.9, 128.6, 128.0, 114.5, 102.9, 96.8, 95.3 (1JCH = 156.3 Hz), 79.6, 78.5, 75.6, 73.9, 67.3, 65.9, 52.5, 48.7, 42.4, 36.2, 30.6, 28.4; HRMS(ESI): m/z calcd for C37H46IN2O7 [M+H]+ 757.2344, found 757.2347.
General procedure for radical fragmentation
A solution of substrate (0.006 M) in dry toluene or xylenes was deoxygenated by sparging with Argon (from a Argon balloon) at rt with stirring for 2 h then was heated to reflux with stirring under Argon while a mixture of initiator (azoisobutyronitrile or 1,1’-azobis(cyanocyclohexane) (0.2 equiv) and hydrogen donor (tributyltin hydride or tris(trimethylsilylsilane) (1.6 equiv) in the same deoxygenated solvent (0.025 M) was added by syringe pump in 2 h. The reaction mixture was cooled, the solvent was removed under vacuum, and the residue was dissolved in acetonitrile and washed with hexanes (5 times). The acetontrile phase was concentrated under vacuum and the residue subjected to silica gel chromatography.
1-Adamantanyl 2-O-benzyl-3-deoxy-3-N-(2-cyanophenyl)acetyl-β-d-rhamnopyranoside (39)
With 29β as substrate the radical reaction was executed in xylenes, with addition of the reagents over 2 h. Silica gel chromatography (hexanes/ ethyl acetate /MeOH: 10/5/1) then preparative HPLC were applied to give compound 39 in 20% yield: [α]D = −59.8 (c, 0.5, ethyl acetate); 1H NMR (500 MHz, CDCl3) δ 7.58 (d, J = 8.0 Hz, 1H), 7.51 (td, J = 7.5, 1.5Hz, 1H), 7.30–7.42 (m, 7H), 6.01 (d, J = 8.0 Hz, 1H), 5.08 (d, J = 12.5 Hz, 1H), 4.82 (s, 1H), 4.58 (d, J = 12.0 Hz, 1H), 3.87–3.92 (m, 1H), 3.56 (d, J = 3.0 Hz, 1H), 3.49 (d, J = 16.0 Hz, 1H), 3.41 (d, J = 15.5 Hz, 1H), 3.27–3.34 (m, 2H), 2.79 (br. s, 1H), 2.18 (s, 3H), 1.89 (d, J = 11.0 Hz, 3H), 1.81 (d, J = 11.0 Hz, 3H), 1.61–1.68 (m, 6H), 1.33 (d, J = 6.0 Hz, 3H); 13C NMR (500M, CDCl3) δ 170.4, 138.6, 138.2, 133.1, 132.8, 130.6, 128.9, 128.7, 128.2, 127.8, 117.8, 112.9, 94.3 (1JCH = 151.3 Hz), 77.9, 75.2, 73.5, 73.4, 55.2, 42.5, 41.3, 36.3, 30.7, 17.9; HRMS(ESI): m/z calcd for C32H39N2O5 [M+H]+ 531.2854, found 531.2855.
1-Adamantanyl 2-O-benzyl-3-N-(tert-butyloxycarbonylamino)-3-deoxy-4,6-O-[2-(2-cyanophenyl)]ethylidene-β-d-mannopyranoside (41) and 1-Adamantanyl 2-O-benzyl-3-N-(tert-butyloxycarbonylamino)-3-deoxy-β-d-rhamnopyranoside (42)
Under a variety of conditions of solvent, temperature, reductant and initiator for the radical reaction, 40 gave mixtures of two isomers after silica gel chromatography (hexanes/ethyl acetate: 5/1) in low yield. Saponification of these mixtures with catalytic MeONa in methanol followed by silica gel chromatography (hexanes/ethyl acetate: 5/1 then 3/1) enabled the separation of 41 and 42. Reduction product 41: [α]D = − 20.4 (c, 0.5, ethyl acetate); 1H NMR (500 MHz, CDCl3) δ 7.60 (d, J = 8.0 Hz, 1H), 7.47 (t, J = 7.0, Hz, 1H), 7.40 (d, J = 8.0 Hz, 1H), 7.27–7.35 (m, 6H), 5.01 (d, J = 12.0 Hz, 1H), 4.86 (s, 1H), 4.76 (d, J = 9.0 Hz, 1H), 4.71 (t, J = 5.0 Hz, 1H), 4.61 (d, J = 11.5 Hz, 1H), 4.06 (dd, J = 11.0, 5.0 Hz, 1H), 3.83 (td, J = 8.0, 3.5 Hz, 1H), 3.69 (d, J = 2.0 Hz, 1H), 3.56 (t, J = 10.5 Hz, 1H), 3.44 (t, J = 10.5 Hz, 1H), 3.26 (td, J = 9.5, 5.0 Hz, 1H), 3.16 (d, J = 5.0 Hz, 1H), 2.16 (s, 3H), 1.83 (d, J = 11.5 Hz, 3H), 1.76 (d, J = 11.5 Hz, 3H), 1.63 (m, 6H), 1.41 (s, 9H); 13C NMR (125 MHz, CDCl3) δ 155.5, 140.0, 132.9, 132.6, 132.4, 131.6, 128.7, 128.5, 127.9, 127.1, 118.1, 113.4, 101.0, 95.1, 79.4, 79.0, 76.3, 75.8, 75.4, 68.4, 68.3, 52.9, 42.4, 39.2, 36.2, 30.6, 28.4; HRMS(ESI): m/z calcd for C37H46N2NaO7 [M+Na]+ 653.3203, found 653.3177. Fragmentation product 42: [α]D = − 37.9 (c, 1.0, ethyl acetate); 1H NMR (500 MHz, CDCl3) δ 7.30–7.37 (m, 5H), 5.70 (d, J = 12.5 Hz, 1H), 4.96 (d, J = 8.0 Hz, 1H), 4.79 (s, 1H), 4.63 (d, J = 12.5 Hz, 1H), 3.50–3.54 (m, 2H), 3.32 (t, J = 9.5 Hz, 1H), 3.22–3.27 (m, 1H), 2.95 (s, 1H), 2.17 (s, 3H), 1.88 (d, J = 11.5 Hz, 3H), 1.80 (d, J = 11.5 Hz, 3H), 1.63 (m, 6H), 1.38 (s, 9H), 1.33 (d, J = 5.5 Hz, 3H); 13C NMR (125 MHz, CDCl3) δ 157.5, 138.5, 128.6, 127.9, 94.6 (1JCH = 151.3 Hz), 80.1, 77.9, 75.0, 73.6, 73.4, 55.9, 42.5, 36.3, 30.7, 28.3, 17.9; HRMS(ESI): m/z calcd for C28H41NNaO6 [M+Na]+ 510.2832, found 510.2837.
Supplementary Material
Copies of spectra of all compounds, and cif files for compounds 12 and 15. This material is available free of charge via the Internet at http://pubs.acs.org.
Acknowledgment
We thank the NIH (GM57335) for support of this work and Professor D. J. Wink for the X-ray structures.
References
- 1.Crich D, Lim LBL. Org. React. 2004;64:115–251. [Google Scholar]
- 2.Crich D. In: Glycochemistry: Principles, Synthesis, and Applications. Wang PG, Bertozzi CR, editors. New York: Dekker; 2001. pp. 53–75. [Google Scholar]
- 3.Crich D, Sun S. J. Org. Chem. 1997;62:1198–1199. [Google Scholar]
- 4.Crich D, Sun S. Tetrahedron. 1998;54:8321–8348. [Google Scholar]
- 5.Crich D, Smith M. J. Am. Chem. Soc. 2001;123:9015–9020. doi: 10.1021/ja0111481. [DOI] [PubMed] [Google Scholar]
- 6.Crich D, Cai W, Dai Z. J. Org. Chem. 2000;65:1291–1297. doi: 10.1021/jo9910482. [DOI] [PubMed] [Google Scholar]
- 7.Crich D, Yao Q. J. Am. Chem. Soc. 2004;126:8232–8236. doi: 10.1021/ja048070j. [DOI] [PubMed] [Google Scholar]
- 8.Crich D, Dudkin V. Tetrahedron Lett. 2000;41:5643–5646. [Google Scholar]
- 9.Crich D, Jayalath P, Hutton TK. J. Org. Chem. 2006;71:3064–3070. doi: 10.1021/jo0526789. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Crich D, Vinogradova O. J. Org. Chem. 2006;71:8473–8480. doi: 10.1021/jo061417b. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Crich D, Li L. J. Org. Chem. 2007;72:1681–1690. doi: 10.1021/jo062294y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Codée JDC, van den Bos LJ, Litjens REJN, Overkleeft HS, van Boeckel CAA, van Boom JH, van der Marel GA. Tetrahedron. 2004;60:1057–1064. [Google Scholar]
- 13.Codée JDC, Hossain LH, Seeberger PH. Org. Lett. 2005;7:3251–3254. doi: 10.1021/ol051038p. [DOI] [PubMed] [Google Scholar]
- 14.Crich D, Sun S. J. Am. Chem. Soc. 1997;119:11217–11223. [Google Scholar]
- 15.Crich D, Chandrasekera NS. Angew. Chem. Int. Ed. 2004;43:5386–5389. doi: 10.1002/anie.200453688. [DOI] [PubMed] [Google Scholar]
- 16.As suggested by a reviewer of a recent manuscript, whom we thank, an SNi mechanism can not be completely excluded from consideration for the formation of the α-mannosides.
- 17.Jensen HH, Nordstrom M, Bols M. J. Am. Chem. Soc. 2004;126:9205–9213. doi: 10.1021/ja047578j. [DOI] [PubMed] [Google Scholar]
- 18.Torsional effects also potentially contribute to the benzylidene acetal effectFraser-Reid B, Wu ZC, Andrews W, Skowronski E. J. Am. Chem. Soc. 1991;113:1434–1435.Andrews CW, Rodebaugh R, Fraser-Reid B. J. Org. Chem. 1996;61:5280–5289. doi: 10.1021/jo961115h.
- 19.Nukada T, Berces A, Whitfield DM. Carbohydr. Res. 2002;337:765–774. doi: 10.1016/s0008-6215(02)00043-5. [DOI] [PubMed] [Google Scholar]
- 20.Nukuda T, Bérces A, Wang L, Zgierski MZ, Whitfield DM. Carbohydr. Res. 2005;340:841–852. doi: 10.1016/j.carres.2004.12.021. [DOI] [PubMed] [Google Scholar]
- 21.Baer HH, Fischer HOL. J. Am. Chem. Soc. 1960;82:3709–3713. [Google Scholar]
- 22.Richardson AC. J. Chem. Soc. 1962:373–374. [Google Scholar]
- 23.Alper PB, Hung S-C, Wong C-H. Tetrahedron Lett. 1996;37:6029–6032. [Google Scholar]
- 24.Vasella A, Witzig C, Chiara J-L, Martin-Lomas M. Helv. Chim. Acta. 1991;74:2073–2077. [Google Scholar]
- 25.Crich D, Bowers AA. J. Org. Chem. 2006;71:3452–3463. doi: 10.1021/jo0526688. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Crich D, Bowers AA. Org. Lett. 2006;8:4327–4330. doi: 10.1021/ol061706m. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Previous application of benzylidene imines as amine protecting groups in oligosaccharide synthesisMadsen R, Udodonge UE, Roberts C, Mootoo DR, Konradsson P, Fraser-Reid B. J. Am. Chem. Soc. 1995;117:1554–1565.Also see:Khiar N, Fernandez I, Araujo CS, Rodriguez JA, Suarez B, Alvarez E. J. Org. Chem. 2003;68:1433–1442. doi: 10.1021/jo026519q.
- 28.Crich D, Smith M, Yao Q, Picione J. Synthesis. 2001:323–326. [Google Scholar]
- 29.Majumdar D, Boons G-J. In: Handbook of Reagents for Organic Synthesis: Reagents for Glycoside, Nucleotide, and Peptide Synthesis. Crich D, editor. Chichester: Wiley; 2005. pp. 11–15. [Google Scholar]
- 30.Lemieux RU, Ratcliffe RM. Can. J. Chem. 1979;57:1244–1251. [Google Scholar]
- 31.Pougny J-R, Sinaÿ P. Tetrahedron Lett. 1976;17:4073–4076. [Google Scholar]
- 32.Braccini J, Derouet C, Esnault J, de Penhoat CH, Mallet J-M, Michon V, Sinaÿ P. Carbohydr. Res. 1993;246:23–41. [Google Scholar]
- 33.Ratcliffe AJ, Fraser-Reid B. J. Chem. Soc., Perkin Trans. 1990;1:747–750. [Google Scholar]
- 34.Schmidt RR, Behrendt M, Toepfer A. Synlett. 1990:694–696. [Google Scholar]
- 35.Crich D, Patel M. Carbohydr. Res. 2006;341:1467–1475. doi: 10.1016/j.carres.2006.03.036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Bock K, Pedersen C. J. Chem. Soc., Perkin Trans. 1974;2:293–297. [Google Scholar]
- 37.Tvaroska I, Taravel FR. Adv. Carbohydr. Chem. Biochem. 1995;51:15–61. doi: 10.1016/s0065-2318(08)60191-2. [DOI] [PubMed] [Google Scholar]
- 38.Duus JO, Gotfredsen CH, Bock K. Chem. Rev. 2000;100:4589–4614. doi: 10.1021/cr990302n. [DOI] [PubMed] [Google Scholar]
- 39.Schneider H-J, Hoppen V. J. Org. Chem. 1978;43:3866–3873. [Google Scholar]
- 40.Sülze D, Gatial A, Karlsson A, Klaeboe P, Nielsen CJ. J. Mol. Struct. 1988;174:207–214. [Google Scholar]
- 41.Jensen FR, Bushweller CH, Beck BH. J. Am. Chem. Soc. 1969;91:344–351. [Google Scholar]
- 42.Bugay DE, Bushweller CH, Danehy CT, Hoogasian S, Blersch JA, Leenstra WR. J. Phys. Chem. 1989;93:3908–3911. [Google Scholar]
- 43.Subbotin OA, Sergeyev NM. J. Am. Chem. Soc. 1975;97:1080–1084. [Google Scholar]
- 44.Chu P-S, True NS. J. Phys. Chem. 1985;89:5613–5616. [Google Scholar]
- 45.Jensen FR, Bushweller CH. Adv. Alicycl. Chem. 1971;3:139. [Google Scholar]
- 46.Liang C-H, Duffield J, Romero A, Chiu Y-H, Rabuka D, Yao S, Sucheck S, Marby K, Shue Y-K, Ichikawa Y, Hwang C-K. 2004 WO 2004080391. [Google Scholar]
- 47.Judice JK, Fatheree PR, Lam BMT, Leadbetter M, Linsell MS, Mu Y, Trapp SG, Yang G, Zhu Y. 2000 WO 2000039156. [Google Scholar]
- 48.Packard GK, Rychnovsky SD. Org. Lett. 2001;3:3393–3396. doi: 10.1021/ol016617i. [DOI] [PubMed] [Google Scholar]
- 49.Nicolaou KC, Daines RA, Ogawa Y, Chakraborty TK. J. Am. Chem. Soc. 1988;110:4696–4705. [Google Scholar]
- 50.Cereghetti DM, Carreira EM. Synthesis. 2006:914–942. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Copies of spectra of all compounds, and cif files for compounds 12 and 15. This material is available free of charge via the Internet at http://pubs.acs.org.