

Abstract
An expedient synthesis of enantiomerically pure threo-β-hydroxy-α-amino acid derivatives of phenylalanine, tyrosine, histidine and tryptophan is described. The NBS-mediated radical bromination of the N,N-di-tert-butoxycarbonyl protected α-amino acids and subsequent treatment with silver nitrate in acetone provided the trans-oxazolidinones predominantly. The cesium carbonate catalyzed hydrolysis then generated the β-hydroxy amino acid derivatives in excellent overall yield from the amino acids themselves.
β-Hydroxy-α-amino acids are an interesting class of molecules due to their presence in numerous biologically active natural products. For example, β-hydroxytyrosine and β-hydroxyphenylalanine residues are found in clinically active glycopeptide antibiotics, such as vancomycin,1 bouvardin,2 orienticnic,3 phomopsins,4 ristocetin,5 actaplanin,5 and teicoplanin.5 β-Hydroxyhistidine has also been found in bleomycin,6 tallysomycin,7 exochelins MN,8a and PF244.8b These highly functionalized amino acids are also useful building blocks for the synthesis of β-lactams,9 β-fluoro-α-amino acids,10 and sugars.11
Over the years several strategies have been developed for the asymmetric syntheses of β-hydroxy-α-amino acids including asymmetric aldol reaction12 utilizing chiral oxazolidinones;13 alkylation of chiral enolates from oxazolidinones,14 oxazolidines,15 bis-lactim ethers,16 oxazolines17 and imidazolidinones;18 cycloaddition of chiral azomethine ylids;19 enzymatic transformations;20 stereoselective hydrolysis of aziridine carboxylate esters;21 Sharpless asymmetric dihydroxylations;22 asymmetric aminohydroxylations;23 asymmetric epoxidations;24 sulfonamide mediated asymmetric Strecker reaction;25 imino [1,2]-Wittig rearrangement of hydroximates26 and numerous others.25 Most of these protocols involve multiple steps and the use of chiral auxiliaries or chiral catalysts, and often proceed with less than perfect stereocontrol.
In 1990, Easton and coworkers reported a method for diastereoselective conversion of amino acids to their β-hydroxy derivatives by direct side chain bromination of the amino acid derivatives with NBS, followed by treatment with silver nitrate in aqueous acetone.27 For example, phenylalanine gave 1:1 mixture of the diastereomeric bromides and subsequently a 5:1 mixture of the syn and anti β-hydroxyphenylalanine derivatives. This side chain bromination requires an N-substituent such as phthaloyl or trifluoromethanesulfonyl, to deactivate the α-position towards hydrogen atom abstraction.28 The phthaloyl group also participates in the hydrolysis step and is thereby responsible for the observed stereodifferentiation. The main limitation of the method is the use of the phthalimido and trifluoromethanesulfonamide protecting groups with their less than ideal hydrolysis conditions. In spite of this, the methodology has been applied in the synthesis of vancomycin,22b chloramphenicol,29 cyclomarin C,30 and residues of callipeltin A.31 We report here on the use of N,N-di-tert-butoxycarbonyl protected amino acids in Easton’s protocol and on the advantages that this affords.
The methyl ester of phenylalanine (1) was converted to the di-N-tert-butoxycarbonyl derivative 232 by treatment with DMAP and (Boc)2O.33 The NBS-mediated bromination in CCl434 then afforded the bromides (3) in a 1:1 mixture, which was treated with silver nitrate in acetone to afford the trans and cis oxazolidinones 4 and 5, respectively, in 6:1 ratio and 70% yield. The oxazolidinones were completely separable by flash column chromatography and when individually subjected to hydrolysis with catalytic Cs2CO3 in methanol,26 the threo (2S,3R) and erythro (2S,3S) β-hydroxy phenylalanines 6 and 8 were obtained in 80% and 79% yields, respectively, as single diastereomers (Scheme 1). When the hydrolysis reaction of 4 was performed in MeOH-D4, no deuterium substitution at the α-centre was observed in the product 7, indicating this step to be racemization free.
Scheme 1.
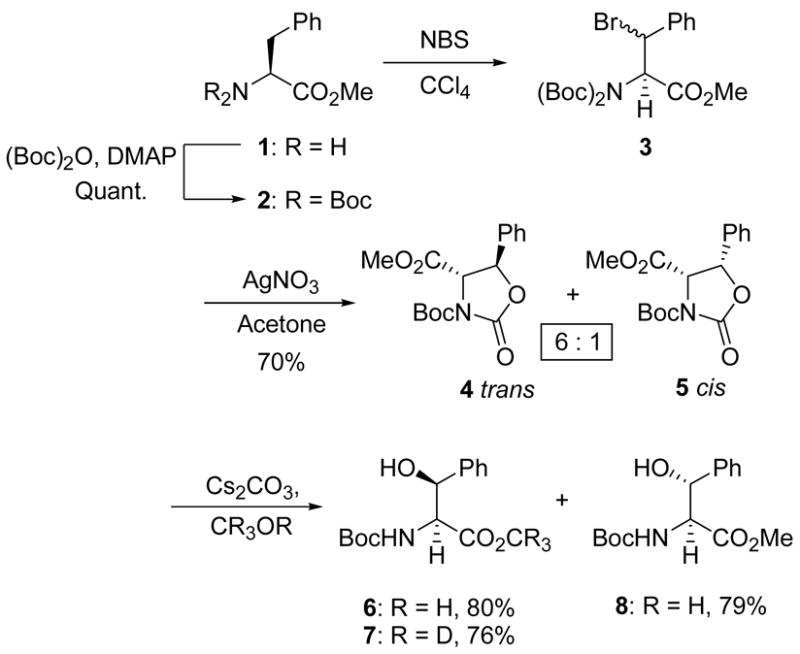
Synthesis of β-hydroxy phenylalanine.
With insoluble silver salts such as silver carbonate or silver oxide, only the erythro bromide reacted to give the trans-oxazolidinone (4) in 48% yield. The threo isomer remained unreacted and was isolated in 45% yield (Scheme 2). When the bromides (3) were treated with silver trifluoromethanesulfonate, the oxazolidinones 9 and 10 were formed in a 10:1 ratio in favor of the trans isomer, with complete cleavage of the N-tert-butoxycarbonyl group (Scheme 2).31 The drastic conditions35 required for the hydrolysis of 9 and 10 prompted their conversion to 4 and 5 with (Boc)2O and DMAP.36 The high selectivity in the conversion of 3 to 9 and 10 is offset by the lower yield and, all things considered, caused us to favor silver nitrate conditions.
Scheme 2.
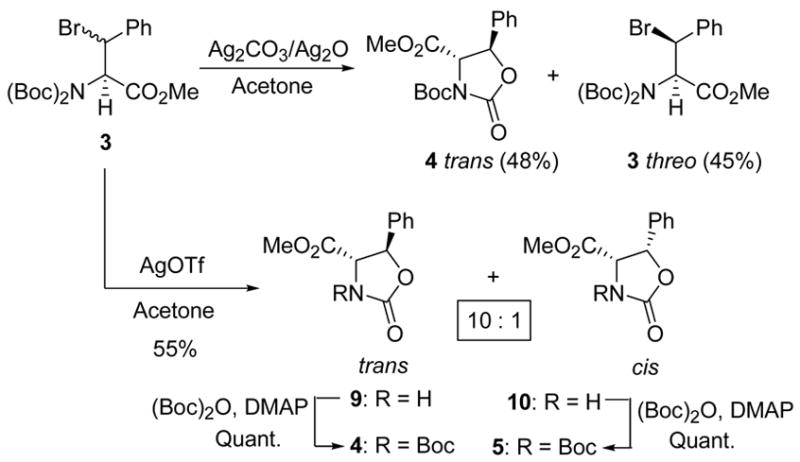
Effect of Other Silver Salts.
The NBS-mediated bromination of 12 yielded the diastereomeric bromides (13) as a 1:1 mixture, which was immediately treated with silver nitrate in acetone to afford the trans and cis oxazolidinones 14 and 15 in 65% yield and 15:1 ratio. Inspection of the 1H NMR spectrum of the diastereomeric bromides revealed that the trans oxazolidinone (14) formation was initiated even before the addition of the silver salt. The trans isomer was hydrolyzed uneventfully to β-hydroxytyrosine derivative 16 using cesium carbonate in methanol in 77% yield (Scheme 3).
Scheme 3.
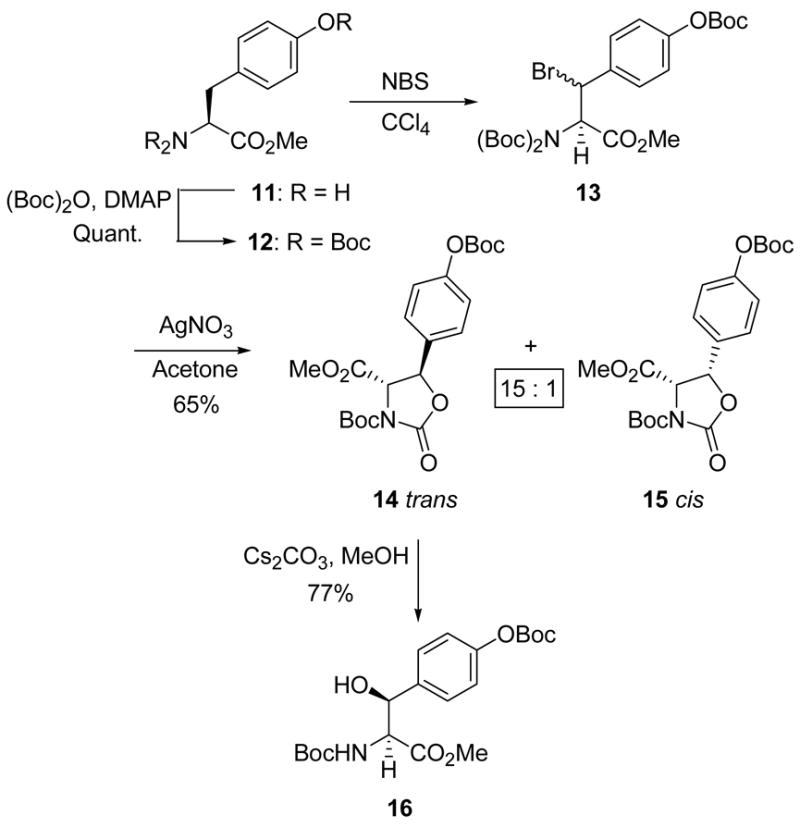
Synthesis of β-hydroxytyrosine.
The radical bromination of 1837 provided the threo bromide 19 and the trans oxazolidinone 20. This mixture was subsequently treated with silver nitrate in acetone to afford the trans oxazolidinones 20 and 21 which differ by the presence of a tert-butoxycarbonyl group, in 62% yield. Attempted hydrolysis of 20 or 21 under a wide variety of conditions produced the dehydrohistidine derivative 22. We reasoned that the free imidazole nitrogen is responsible for the elimination reaction, and that an imidazole protecting group, stable under mild basic conditions, would solve the problem. Accordingly, 21 was reacted with trityl chloride and triethylamine in dichloromethane and, after the removal of the excess trityl chloride, the reaction mixture was treated with catalytic cesium carbonate (20 mol%) in methanol leading directly to the formation of the desired (2S,3S) β-hydroxyhistidine derivative 23 in 74% yield (Scheme 4).
Scheme 4.

Synthesis of β-hydroxyhistidine.
Interestingly, the NBS-mediated radical bromination of 2532 directly formed the trans oxazolidinone 26 in 72% yield. The cesium carbonate catalyzed hydrolysis was straightforward and produced the desired product (27) in 84% yield (Scheme 5).
Scheme 5.

Synthesis of β-hydroxytryptophan.
In conclusion we have demonstrated an improved synthetic route for enantiomerically pure threo-β-hydroxy-α-amino acids from the amino acids themselves. As the aryl substituents became progressively more electron donating in nature from phenylalanine to tryptophan, the conversion of the intermediate bromides to the oxazolidinones became easier and was associated with increased stereoselectivity in this step.
Experimental Section
1. General procedure for the synthesis of oxazolidinones.27
A mixture of amino acid derivatives and NBS (1 equiv.) in CCl4 (0.05 M) was heated at reflux for 45 mins.38 under nitrogen, whilst being irradiated with a 250 W Kr lamp. The mixture was then cooled to room temperature, filtered and concentrated. To a solution of the concentrate in acetone (0.05 M), silver nitrate (1.5 equiv.) was added. The reaction mixture was stirred at room temperature in dark for 2 h. Then the reaction mixture was filtered through a celite pad and the filtrate was concentrated. The concentrate was diluted with ethyl acetate and washed with saturated NH4Cl solution, water and brine. The organic layer was dried and concentrated. Chromatographic purification afforded the oxazolidinones.
2. General procedure for the hydrolysis of oxazolidinones.26
A solution of the oxazolidinone in methanol was treated with Cs2CO3 (20 mol%) and stirred at room temperature for 2.5 h. Then the reaction mixture was concentrated and the concentrate was diluted with ethyl acetate and washed with saturated NH4Cl solution, water and brine. The organic layer was dried and concentrated. Chromatographic purification afforded the β-hydroxy-α-amino acid derivatives.
Methyl (4S,5R)-3-N-tert-butoxycarbonyl-5-phenyl-1,3-oxazolidin-2-oxo-4-carboxylate (4) and Methyl (4S,5S)-3-N-tert-butoxycarbonyl-5-phenyl-1,3-oxazolidin-2-oxo-4-carboxylate (5).26,14b
Following the general procedure 1 and eluting with 16–18% ethyl acetate in hexane 4 and 5 were obtained in 6: 1 ratio and 70% yield. 4: [α]22D +28.3 (c 1.6); 1H NMR (500 MHz) δ: 7.43-7.36 (m, 5H), 5.38-5.37 (d, J = 4.0 Hz, 1H), 4.63-4.62 (d, J = 4.5 Hz, 1H), 3.88 (s, 3H), 1.48 (s, 9H); 13C NMR (125 MHz) δ: 169.0, 150.7, 148.4, 137.1, 129.5, 129.2, 125.0, 84.9, 75.9, 63.7, 53.3, 27.8. 5: [α]24D +45.2 (c 1.2); 1H NMR (400 MHz) δ:7.36-7.28 (m, 5H), 5.72-5.70 (d, J = 9.2 Hz, 1H), 4.97-4.94 (d, J = 8.4 Hz, 1H), 3.23 (s, 3H), 1.48 (s, 9H); 13C NMR (100 MHz) δ: 167.3, 151.1, 148.5, 132.7, 129.5, 128.5, 126.1, 84.8, 75.5, 62.3, 52.2, 27.8.
N-tert-Butoxycarbonyl-(2S,3R)-β-hydroxy-L-phenylalanine methyl ester (6).26
Following the general procedure 2, and eluting with 20% ethyl acetate in hexane, 6 was obtained in 80% yield. [α]22D −14.8 (c 1.3); 1H NMR (400 MHz) δ: 7.35-7.23 (m, 5H), 5.41-5.39 (d, J = 8.4 Hz, 1H), 5.20 (s, 1H), 4.52-4.50 (d, J = 7.6 Hz, 1H), 3.73 (s, 3H), 3.16 (bs, 1H), 1.31 (s, 9H); 13C NMR (100 MHz) δ: 171.5, 155.7, 139.9, 128.3, 128.0, 126.0, 80.0, 73.8, 59.5, 52.6, 28.2.
Methyl (4S,5R)-3-N-tert-butoxycarbonyl-5-(4-tert-butoxycarbonyloxyphenyl)-1,3-oxazolidin-2-oxo-4-carboxylate (14) and Methyl (4S,5S)-3-N-tert-butoxycarbonyl-5-(4-tert-butoxycarbonyloxyphenyl)-1,3-oxazolidin-2-oxo-4-carboxylate (15)
Following the general procedure 1 and eluting with 16–18% ethyl acetate in hexane 14 and 15 were obtained in 15: 1 ratio and 65% yield. 14: [α]22D +31.2 (c 0.7); 1H NMR (400 MHz) δ:7.40-7.38 (d, J = 8.4 Hz, 2H), 7.25-7.23 (d, J = 8.8 Hz, 2H), 5.38-5.37 (d, J = 4.8 Hz, 1H), 4.61-4.60 (d, J = 4.8 Hz, 1H), 3.87 (s, 3H), 1.55 (s, 9H), 1.48 (s, 9H); 13C NMR (100 MHz) δ:168.9, 151.7, 151.6, 150.6, 148.3, 134.6, 126.2, 122.2, 85.0, 84.1, 75.3, 63.7, 53.3, 27.8, 27.7; ESI-HRMS Calcd for C21H27NO9 [M + Na]+ : 460.1583. Found 460.1568. 15: [α]22D +33.8 (c 1.2); 1H NMR (400 MHz) δ:7.32-7.30 (d, J = 9.2 Hz, 2H), 7.20-7.18 (d, J = 8.0 Hz, 2H), 5.72-5.70 (d, J = 8.8 Hz, 1H), 4.95-4.93 (d, J = 9.6 Hz, 1H), 3.26 (s, 3H), 1.54 (s, 9H), 1.49 (s, 9H); 13C NMR (100 MHz) δ:167.2, 151.8, 150.9, 148.5, 130.0, 127.3, 121.5, 84.9, 83.9, 74.9, 62.2, 52.4, 27.7, 27.5; ESI-HRMS Calcd for C21H27NO9 [M + Na]+ : 460.1583. Found 460.1581.
N-tert-Butoxycarbonyl-O-tert-butoxycarbonyl-(2S,3R)-β-hydroxy-L-tyrosine methyl ester (16)
Following the general procedure 2, and eluting with 26% ethyl acetate in hexane, 16 was obtained in 77% yield. [α]22D -10.5 (c 1.0); 1H NMR (400 MHz) δ: 7.37-7.35 (d, J = 8.0 Hz, 2H), 7.14-7.12 (d, J = 7.6 Hz, 2H), 5.34-5.32 (d, J = 8.8 Hz, 1H), 5.18 (s, 1H), 4.49-4.47 (d, J = 8.4 Hz, 1H), 3.73 (s, 3H), 1.53 (s, 9H), 1.32 (s, 9H); 13C NMR (100 MHz) δ:171.3, 155.7, 151.8, 150.6, 137.4, 127.1, 121.1, 83.6, 80.2, 73.3, 59.4, 52.6, 28.2, 27.7; ESI-HRMS Calcd for C20H29NO8 [M + Na]+ : 434.1791. Found 434.1772.
Methyl (4S,5S)-3-N-tert-butoxycarbonyl-5-{4-N-(tert-butoxycarbonyl)imidazolyl}-1,3-oxazolidin-2-oxo-4-carboxylate (20) and Methyl (4S,5S)-3-N-tert-butoxycarbonyl-5-imidazolyl-1,3-oxazolidin-2-oxo-4-carboxylate (21)
Following the general procedure 1 and eluting with 26–80% ethyl acetate in hexane 20 and 21 were obtained in 1.5: 1 ratio and 62% yield. 20: [α]22D +97.7 (c 0.4); 1H NMR (500 MHz) δ: 8.09 (s, 1H), 7.47 (s, 1H), 5.35-5.34 (d, J = 4.0 Hz, 1H), 5.01-5.00 (d, J = 4.0 Hz, 1H), 3.84 (s, 3H), 1.61 (s, 9H), 1.48 (s, 9H); 13C NMR (125 MHz) δ: 169.0, 150.6, 148.4, 146.4, 138.7, 138.0, 116.0, 86.7, 84.7, 71.1, 60.9, 53.2, 27.83, 27.81; ESI-HRMS Calcd for C18H25N3O8 [M + Na]+ : 434.1539. Found 434.1523. 21: [α]22D +76.5 (c 0.7); 1H NMR (500 MHz) δ: 11.39 (bs, 1H), 7.69 (s, 1H), 7.18 (s, 1H), 5.41-5.40 (d, J = 4.5 Hz, 1H), 5.14-5.13 (d, J = 4.0 Hz, 1H), 3.81 (s, 3H), 1.46 (s, 9H); 13C NMR (125 MHz) δ: 169.3, 151.8, 148.5, 136.8, 135.8, 116.1, 85.0, 71.9, 61.3, 53.2, 27.8; ESI-HRMS Calcd for C13H17N3O6 [M + Na]+ : 334.1015. Found 334.1027.
N-tert-Butoxycarbonyl-4-Nim-triphenylmethyl-(2S,3S)-β-hydroxy-L-hystidine methyl ester (23)
A solution of 21 (0.19 g, 0.61 mmol) and trityl chloride (0.35 g, 1.22 mmol) in CH2Cl2 (3 mL) was treated with Et3N (170μL, 1.22 mmol) and stirred at room temperature for 1.5h. Then the reaction mixture was concentrated and the excess trityl chloride was removed by filtering through short silica gel column. The filtrate was concentrated and further dissolved in methanol. The methanolic solution was treated with cesium carbonate (0.4 g, 0.12 mmol) at room temperature for 2 h. Then the reaction mixture was concentrated and the concentrate was diluted with ethyl acetate and washed with saturated NH4Cl solution, water and brine. The organic layer was dried and concentrated. The chromatographic purification using 55% ethyl acetate in hexane afforded 23 (0.24 g, 74%). [α]22D -14.5 (c 1.0); 1H NMR (400 MHz) δ:7.36 (s, 1H), 7.34-7.29 (m, 9H), 7.12-7.09 (m, 6H), 6.79 (s, 1H), 5.71-5.69 (d, J = 8.4 Hz, 1H), 5.13 (s, 1H), 4.59-4.57 (d, J = 9.2 Hz, 1H), 4.33 (bs, 1H), 3.69 (s, 3H), 1.36 (s, 9H); 13C NMR (100 MHz) δ:171.5, 155.7, 142.2, 140.2, 138.5, 129.8, 128.1, 128.09, 118.2, 79.6, 75.5, 68.7, 58.2, 52.4, 28.3; ESI-HRMS Calcd for C31H33N3O5 [M + Na]+ : 550.2318. Found 550.2315.
Methyl (4S,5S)-3-N-tert-butoxycarbonyl-5-{N-(tert-butoxycarbonyl)indolyl}-1,3-oxazolidin-2-oxo-4-carboxylate (26)
Following the general procedure 1 and eluting with 14% ethyl acetate in hexane 26 in 72% yield. [α]22D +24.9 (c 2.3); 1H NMR (500 MHz) δ:8.21-8.19 (d, J = 8.0 Hz, 1H), 7.68 (s, 1H), 7.59-7.58 (d, J = 8.0 Hz, 1H), 7.41-7.38 (t, J = 8.0 Hz, 1H), 7.32-7.26 (t, J = 8.0 Hz, 1H), 5.67-5.66 (d, J = 3.5 Hz, 1H), 4.84-4.83 (d, J = 3.5 Hz, 1H), 3.92 (s, 3H), 1.67 (s, 9H), 1.50 (s, 9H); 13C NMR (125 MHz) δ:169.0, 150.7, 149.2, 148.5, 136.1, 126.6, 125.4, 123.7, 123.4, 118.8, 116.5, 115.8, 84.9, 84.7, 71.6, 61.8, 53.4, 28.1, 27.8; ESI-HRMS Calcd for C23H28N2O8 [M + Na]+ : 483.1743. Found 483.1725.
N-tert-Butoxycarbonyl-Nin-tert-butoxycarbonyl-(2S,3S)-β-hydroxy-L-tryptophan methyl ester (27)
Following the general procedure 2, and eluting with 22% ethyl acetate in hexane, 27 was obtained in 84% yield. [α]22D −12.5 (c 1.0); 1H NMR (400 MHz) δ:8.13-8.12 (d, J = 6.4 Hz, 1H), 7.62-7.59 (d, J = 12.0 Hz, 1H), 7.33-7.29 (t, J = 8.0 Hz, 1H), 7.25-7.21 (t, J = 7.6 Hz, 1H), 5.50-5.49 (d, J = 3.6 Hz, 1H), 5.46 (s, 1H), 4.68-4.66 (d, J = 8.0 Hz, 1H), 3.77 (s, 3H), 2.98 (bs, 1H), 1.65 (s, 9H), 1.34 (s, 9H); 13C NMR (125 MHz) δ:171.5, 155.8, 149.5, 135.6, 128.3, 124.7, 123.4, 122.8, 119.8, 119.3, 115.4, 83.9, 80.1, 68.4, 58.1, 52.7, 28.2; ESI-HRMS Calcd for C22H30N2O7 [M + Na]+ : 457.1951. Found 457.1933.
Supplementary Material
Full experimental and characterization details for 2, 3, 7–10, 12, 18, 22, and 25 and NMR spectra of all compounds. This material is free of charge via the Internet at http://pubs.acs.org.
Acknowledgments
We thank the NIH (GM 6260) for support of this work, the University of Illinois at Chicago for the award of the Moriarty Fellowship to A.B and a reviewer for helpful suggestions.
References
- 1.(a) Williams DH. Acc Chem Res. 1984;17:364. [Google Scholar]; (b) Kopecka H, Harris TM. J Am Chem Soc. 1983;105:6915. [Google Scholar]
- 2.Jolad SD, Hoffmann JJ, Torrance SJ, Wiedhopf RM, Cole JR, Arora SK, Bates RB, Gargiulo RD, Kriek GR. J Am Chem Soc. 1977;99:8040. doi: 10.1021/ja00466a043. [DOI] [PubMed] [Google Scholar]
- 3.Tsuji N, Kobayashi M, kamigauchi T, Yoshimura Y, Terui Y. J Antibiot. 1988;41:819. doi: 10.7164/antibiotics.41.819. [DOI] [PubMed] [Google Scholar]
- 4.Li Y, Kobayashi H, Tokiwa Y, Hashimoto Y, Iwasaki S. Biochem Pharmacol. 1992;43:219. doi: 10.1016/0006-2952(92)90281-m. [DOI] [PubMed] [Google Scholar]
- 5.Rao AVR, Gurjar MK, Reddy KL, Rao AS. Chem Rev. 1995;95:2135. [Google Scholar]
- 6.(a) Hecht SM. In: Cancer Chemotherapeutic Agents. Foye WO, editor. American Chemical Society; Washington, DC: 1995. pp. 369–388. [Google Scholar]; (b) Hecht SM. J Nat Prod. 2000;63:158. doi: 10.1021/np990549f. [DOI] [PubMed] [Google Scholar]
- 7.(a) Bradner WT. In: Bleomycin, Current Status and New Developments. Carter SK, Crooke ST, Umezawa H, editors. Academic Press Inc; New York: 1978. pp. 333–342. [Google Scholar]; (b) Sznaidman ML, Hecht SM. Org Lett. 2001;3:2811. doi: 10.1021/ol0101178. [DOI] [PubMed] [Google Scholar]
- 8.(a) Sharman GJ, Williams DH, Ewing DF, Ratledge C. Chem Biol. 1995;8:553. doi: 10.1016/1074-5521(95)90189-2. [DOI] [PubMed] [Google Scholar]; (b) Hancock DK, Coxon B, Wang SY, White EV, Reeder DJ. J Chem Soc, Chem Commun. 1993:468. [Google Scholar]
- 9.(a) Miller MJ. Acc Chem Res. 1986;19:49. [Google Scholar]; (b) Lotz BT, Miller MJ. J Org Chem. 1993;58:618. [Google Scholar]
- 10.For leading references see: Davis FA, Srirajan V, Titus DD. J Org Chem. 1999;64:6931. doi: 10.1021/jo990947n.
- 11.Boutin RH, Rapoport H. J Org Chem. 1986;51:5320. [Google Scholar]
- 12.(a) Blaser D, Seebach D. Liebigs Ann Chem. 1991:1067. [Google Scholar]; (b) Mettath S, Srikanth GSC, Dangerfield BS, Castle SL. J Org Chem. 2004;69:6489. doi: 10.1021/jo049283u. [DOI] [PubMed] [Google Scholar]
- 13.(a) Laib T, Chastanet J, Zhu J. Tetrahedron Lett. 1997;38:1771. [Google Scholar]; (b) Williams L, Zhang Z, Shao F, Carroll PJ, Joullie MM. Tetrahedron. 1996;52:11673. [Google Scholar]
- 14.(a) Evans DA, Weber AE. J Am Chem Soc. 1986;108:6757. [Google Scholar]; (b) Lago MA, Samanen J, Elliott JD. J Org Chem. 1992;57:3493. [Google Scholar]
- 15.Seebach D, Aebi JD. Tetrahedron Lett. 1984;25:2545. [Google Scholar]
- 16.Schollkopf U, Beulshausen T. Liebigs Ann Chem. 1989:223. [Google Scholar]
- 17.Hayashi T, Sawamura M, Ito Y. Tetrahedron. 1992;48 1999. [Google Scholar]
- 18.Seebach D, Juaristi E, Miller DD, Schickli C, Weber T. Helv Chim Acta. 1987;70:237. [Google Scholar]
- 19.Alker D, Hamblett G, Harwood LM, Robertson SM, Watkin DJ, Williams CE. Tetrahedron. 1998;54:6089. [Google Scholar]
- 20.Vassilev VP, Uchiyama T, Kajimoto T, Wong CH. Tetrahedron Lett. 1995;36:4081. [Google Scholar]
- 21.(a) Tomashini C, Vecchione A. Org Lett. 1999;1:2153. [Google Scholar]; (b) Davis FA, Liu H, Reddy GV. Tetrahedron Lett. 1996;37:5473. [Google Scholar]
- 22.(a) Shao H, Rueter JK, Goodman M. J Org Chem. 1998;63:5240. [Google Scholar]; (b) Rao AVR, Chakraborty TK, Reddy JL, Rao AS. Tetrahedron Lett. 1994;35:5043. [Google Scholar]
- 23.(a) Park H, Cao B, Joullie MM. J Org Chem. 2001;66:7223. doi: 10.1021/jo010482c. [DOI] [PubMed] [Google Scholar]; (b) Miller MJ. J Org Chem. 2002;67:4759. doi: 10.1021/jo0256078. [DOI] [PubMed] [Google Scholar]
- 24.Nagamitsu T, Sunazuka T, Tanaka H, Omura S, Sprengeler PA, Smith AB., III J Am Chem Soc. 1996;118:3584. [Google Scholar]
- 25.Davis FA, Srirajan V, Fanelli DL, Portonovo P. J Org Chem. 2000;65:7663. doi: 10.1021/jo000559h. and references therein. [DOI] [PubMed] [Google Scholar]
- 26.Miyata O, Asai H, Naito T. Chem Pharm Bull. 2005;53:355. doi: 10.1248/cpb.53.355. [DOI] [PubMed] [Google Scholar]
- 27.(a) Easton CJ, Hutton CA, Roslet PD, Tiekink ERT. Tetrahedron. 1994;50:7327. [Google Scholar]; (b) Easton CJ, Hutton CA, Tan EW, Tiekink ERT. Tetrahedron Lett. 1990;31:7059. [Google Scholar]; (c) Hutton CA. Tetrahedron Lett. 1997;38:5899. [Google Scholar]
- 28.Croft AK, Easton CJ, Kociuba K, Radom L. Tetrahedron: Asymmetry. 2003;14:2919. [Google Scholar]
- 29.Easton CJ, Hutton CA, Merrett MC, Tiekink ERT. Tetrahedron. 1996;52:7025. [Google Scholar]
- 30.Wen SJ, Yao ZJ. Org Lett. 2004;6:2721. doi: 10.1021/ol049065n. [DOI] [PubMed] [Google Scholar]
- 31.Zampella A, D’Orsi R, Sepe V, Casapullo A, Monti MC, D’Auria V. Org Lett. 2005;7:3585. doi: 10.1021/ol0513600. [DOI] [PubMed] [Google Scholar]
- 32.Mohapatra DK, Durugkar KA. ARKIVOC. 2005;xiv:20. [Google Scholar]
- 33.(a) Kokotos G, Padron JM, Martin T, Gibbons WA, Martin VS. J Org Chem. 1998;63:3741. [Google Scholar]; (b) Padron JM, Kokotos G, Martin T, Markidis T, Gibbons WA, Martin VS. Tetrahedron: Asymmetry. 1998;9:3381. [Google Scholar]
- 34.A reviewer has indicated that this type of amino acid side chain bromination with NBS also proceeds well in α,α,α-trifluorotoluene.
- 35.(a) Katz SJ, Bergmeier SC. Tetrahedron Lett. 2002;43:557. [Google Scholar]; (b) Bergmeier SC, Stanchina DM. J Org Chem. 1997;62:4449. doi: 10.1021/jo970473x. [DOI] [PubMed] [Google Scholar]
- 36.(a) DiGiovanni MC, Misiti D, Villani C, Zappia G. Tetrahedron: Asymmetry. 1996;7:2277. [Google Scholar]; (b) Burk MJ, Allen JG. J Org Chem. 1997;62:7054. [Google Scholar]
- 37.Xu J, Yadan JC. J Org Chem. 1995;60:6296. [Google Scholar]
- 38.For 18 and 25, the bromination reactions were continued for 2h and 3h, respectively.
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Full experimental and characterization details for 2, 3, 7–10, 12, 18, 22, and 25 and NMR spectra of all compounds. This material is free of charge via the Internet at http://pubs.acs.org.