

Abstract
The selection of advantageous mutations underlies tumorigenesis. The growth of a tumor is therefore a form of evolution at the somatic level, in which the population is comprised of individual cells within the tumor. Models of tumorigenesis have considered the relative importance of mutation and selection. We show that selection is more important than an increased mutation rate in the growth of a tumor. Some cancers may acquire a “mutator phenotype,” probably leading to faster growth, but mutator phenotypes are not necessary for carcinogenesis.
Keywords: tumorigenesis, evolution, selection, mutator phenotype, genomic instability
It is widely accepted that tumorigenesis is a form of somatic evolution, an idea which dates back to the writings of Boveri, James Murray, Little, and Tyzzer in the early years of this century. Tumor growth is initiated by one or more mutations which give a cell a selective advantage. The clone derived from that cell then expands. Successive advantageous mutations occur, each followed by waves of clonal expansion.
The relationship between mutation and selection in tumors has been the subject of general debate. One of the earliest models of tumorigenesis, that of Armitage and Doll (1, 2), used age-specific cancer incidence data to analyze the number of mutations necessary to convert a normal cell to a malignancy. The rate-limiting steps in solid cancers were predicted to involve about six mutations. However, no allowance was made for the selection of new mutations and the ensuing clonal expansion. Fisher (3) and Cairns (4) showed that selection of successive, advantageous new mutants could cause sequential rounds of clonal expansion at exponential rates. Knudson (5) used data from the inherited tumor retinoblastoma to suggest that the initial, rate-limiting steps in cancer were dependent on two allelic mutations, one of which might be inherited. Selection of these initial mutations led to clonal expansion; subsequent mutations might be necessary for tumor progression. Moolgavkar and colleagues (6, 7) provided mathematical analyses of Knudson’s original hypothesis. Loeb (8, 9) reviewed previous models of carcinogenesis in the light of the multiple mutations that had been detected in many cancers. He suggested that while selection and clonal expansion were undoubtedly important, exceptionally high frequencies of mutation might be necessary if more than the two mutations suggested by Knudson and Moolgavkar (6, 7) were required to initiate tumorigenesis, or if multiple mutations were necessary for tumor progression. Tomlinson and Bodmer (10) analyzed a situation in which cells gained a selective advantage through avoiding programmed death and found that clonal expansion can occur to a new equilibrium level rather than in an exponential fashion, thus explaining both benign tumors and long lag phases in tumor growth.
One of the continuing controversies in the study of cancer as somatic evolution has been the role of “mutator phenotypes” (9). In general, a cell has a mutator phenotype if it has an increased tendency to specific types of mutation, caused by mutations at loci such as those involved in DNA replication or repair. Thus, p53 mutations might lead to a mutator phenotype, because the detection of and response to DNA damage are impaired (11). Many cancers have a mutator phenotype that results from mutations at loci involved in DNA mismatch repair (MMR) (12). In the hereditary nonpolyposis colorectal cancer (HNPCC) syndrome, one MMR allele is mutant in the germ line and the other allele mutates somatically. In sporadic cancers—for example, in up to 15% of all colon cancers—somatic mutations (including allele loss) inactivate both mismatch repair alleles (13).
Despite this evidence, the role of the mutator phenotype (or “genomic instability”) in sporadic cancers remains controversial. It has variously been suggested, for example, that genomic instability is commonly an initiating event in tumorigenesis (14, 15), that a mutator phenotype is necessary for carcinogenesis to occur (9), and that it is not possible to explain the number of mutations observed in cancers unless they have a mutator phenotype (16). Below, we use mathematical models of tumorigenesis—based on colorectal cancer (Fig. 1)—to analyze the role of the mutation rate in the growth of sporadic tumors and argue that selection without increased mutation rates is sufficient to explain the evolution of tumors.
Figure 1.

Genetic model of colorectal tumorigenesis. This model illustrates the stepwise nature of tumorigenesis and the action of recessive (tumor-suppressor) and dominant (oncogene) mutations.
METHODS AND RESULTS
The Mutation Rate and the Initiation of Tumor Growth.
Consider the initial steps in tumorigenesis, which are probably rate-limiting. We can study any progenitor cell that can become a tumor within a particular tissue, and ask whether that cell will have a raised mutation rate—by acquiring two mutations at a MMR locus—before it becomes a tumor (Fig. 2). A “normal” mutation rate per locus per generation in each cell is set. Once two mutations have occurred at the MMR locus, the intrinsic mutation rate increases to some specified figure, typically by a factor of 101 to 104. It is assumed that the normal cell must acquire two mutations at some tumor-suppressor locus before a tumor starts to grow. Thus, in the model, we ignore the possibility that the first mutation at the tumor suppressor locus confers a small selective advantage. In doing this, we are already a priori giving the “mutator theory” an advantage. For simplicity, we only consider one MMR locus and one tumor-suppressor gene, although several of each might exist in reality. In addition to this basic model, we also analyze a model in which the cell must acquire multiple tumor-suppressor mutations—six here—before tumor growth starts. For comparison, we also study tumorigenesis in an HNPCC patient; all the cells of this individual already have one MMR mutation.
Figure 2.
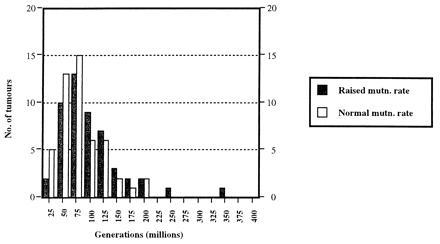
Model of tumorigenesis with two mutations required for tumor growth. The premises of the model are as follows. Given enough time, any stem cell will acquire the necessary mutations to become a tumor. In practice, organisms die before this occurs in nearly all of their stem cells. If, however, a stem cell acquires the necessary mutations exceptionally rapidly—whether by chance, after exposure to extrinsic mutagens, or as a result of an inherited tendency to cancer—a tumor will start to grow within an individual’s normal lifespan. We have taken a single stem cell and assigned to it probabilities of mutation per generation at tumor-suppressor and DNA repair loci. Here, the normal mutation rate is assumed to be 10−8 and the raised rate, 10−4 (see text). A simulation is then run to determine the time it takes for one cell to acquire the mutations needed to start tumor growth. (The computer model is available from the authors.) This simulation has been repeated for a total of 100 stem cells. [An otherwise identical model assuming a normal mutation rate of 10−6 (and thus taking far less computing time) has been run for 10,000 cells and confirms the results of this model.] The model is not quantitative (because we have not considered the many thousands of stem cells that probably exist in the human intestine, for example). Despite this, we can reasonably argue that the tumors which start to grow earliest will be those that occur most often in reality. The graph shows the number of tumors starting to grow with a raised (solid bars) and normal (open bars) mutation rate (y axis) and the cell generation number at which the tumor starts to grow (x axis; each bar representing divisions of 50 million generations, with bar 1 corresponding to 0–50 million generations, bar 2 to 51–100 million generations, etc.). While an equal number of tumors grow, in theory, with normal and raised mutation rates, the tumors that grow earliest (and therefore those that will occur in reality) usually start to grow with a normal mutation rate in this model.
The history of each progenitor cell is studied individually by simulation. On division, one daughter cell differentiates and the other remains as a progenitor cell. At each division (one generation), the cell can acquire mutations at the tumor-suppressor locus or at the MMR locus. A mutation occurs at a particular locus if a randomly generated number between 0 and 1 is less than the specified normal mutation rate. After two mutations have occurred at the MMR locus, mutations at the tumor-suppressor locus occur if the randomly-generated number between 0 and 1 is less than the specified raised mutation rate. Once the necessary two (or six) mutations have occurred at the tumor-suppressor loci, a tumor starts to grow and the simulation is stopped. If two MMR mutations have occurred by this time, then the tumor starts to grow with a raised intrinsic mutation rate and tumorigenesis is subsequently more rapid than would otherwise have been the case.
Fig. 2 illustrates the results of the simulation for what is, in our opinion, the most realistic scenario of sporadic tumorigenesis (among those we are considering), in which two mutations at a tumor-suppressor locus initiate tumor growth. There is a clear tendency for the earliest tumors—and therefore those most likely to occur in reality—to start growing with a normal mutation rate. Raised intrinsic mutation rates are not precursors to tumor growth in most cases. Sometimes, by chance, two MMR mutations will occur early. However, there is more chance of the two MMR mutations occurring first if the tumor-suppressor mutations occur (by chance) at a relatively late time; thus, perhaps contrary to expectations, raised mutation rates are more likely to occur in late rather than early-onset tumors. The reason for these results is that the expected time for a cell to acquire two mutations at the tumor-suppressor locus or the MMR locus are the same. The cell growing with the raised intrinsic mutation rate must also acquire two tumor-suppressor mutations for the tumor to grow. Thus, it is always more likely that the tumor starts to grow with a normal mutation rate, no matter what the values of the normal and raised mutation rates, or even how many DNA repair loci are assumed to exist.
If more than two tumor-suppressor mutations are needed before a tumor grows, the values of the normal and raised mutation rates and the number of tumor-suppressor mutations required will influence the proportion of tumors which start to grow with normal and raised mutation rates. Our results (details not shown) demonstrate that if six tumor-suppressor mutations are required and normal and raised mutation rates of 10−8 and 10−4, respectively, are assumed, most tumors start to grow with a raised mutation rate. In general, a raised mutation rate is more likely to play a role if many mutations are required before tumor growth is initiated and if the MMR mutations have a large effect on the mutation rate. If any of the six tumor-suppressor mutations has any selective advantage, however, the conclusions from this model are invalid and tumors are far more likely to grow without a raised mutation rate.
Finally, we consider tumorigenesis in HNPCC, assuming that two tumor-suppressor mutations are required to start growth. The amount by which the mutation rate is increased by the MMR mutations determines whether or not the mutation rate is raised when the tumor starts to grow (details not shown). If the MMR mutations have a weak effect, then even in HNPCC, some tumors start to grow with a normal intrinsic mutation rate (Fig. 3). It is probably not true, as has been suggested, that in HNPCC tumors “mutations in the APC [tumor-suppressor] gene arise only as the aftermath of [MMR gene] mutations” (17).
Figure 3.

Model of tumorigenesis in an HNPCC patient with two mutations required for tumor growth. The model is as in Fig. 2, but the cell already possesses one mutation at the DNA repair locus. Two repair mutations raise the mutation rate by only 101. A significant proportion of the earliest tumors starts to grow with a normal mutation rate.
Does the Intrinsic Mutation Rate Necessarily Become Raised During Tumorigenesis?
We envisage the typical tumor starting to grow with a normal mutation rate after two tumor-suppressor mutations. The tumor clone then expands until the next advantageous mutation(s) occur(s). This process continues in the classical step-wise model of tumorigenesis (Fig. 1). Even if cells with two MMR (or other equivalent) mutations arise, by chance, they may be so outnumbered by tumor cells with a normal mutation rate that the next mutation required for tumor progression is still more likely to occur in a cell with a normal mutation rate.
To analyze this situation, consider an incipient tumor with a normal mutation rate immediately after the first two tumor-suppressor mutations have occurred in a progenitor cell. Assume a situation relatively favorable to the tumor acquiring a raised intrinsic mutation rate, namely that one MMR mutation has occurred before the second tumor-suppressor mutation. The values of the baseline and raised intrinsic mutation rates are 10−8 and 10−4, respectively. The number of cells in the tumor expands at the rate of (1 + w) per generation. A single extra mutation—such as K-ras in colorectal tumorigenesis (Fig. 1)—is needed to give a further selective advantage. The probability that the single mutation occurs in a tumor cell that has a raised mutation rate is [1 − (1 − 10−4)N1] (where N1 is the number of cells that have descended from the one with the original pair of MMR mutations), relative to the equivalent probability for the N2 cells with normal MMR, which is [1 − (1 − 10−8)N2]. Hence, as long as approximately N2 > 104N1, the outgrowing tumor clone will probably not have a raised mutation rate. We believe that this inequality will often hold. It will do so if the tumor reaches 10,000 cells in size before any single tumor cell acquires two MMR mutations. How long does a tumor cell take to replicate to 10,000 cells? This can be accomplished easily by finding G, the number of generations after tumor growth initiated, from:
 |
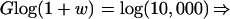 |
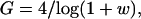 |
If
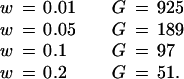 |
Thus, even with a selective advantage as low as 0.01, the tumor clone will have expanded to 10,000 cells in 925 generations. The maximum effective mutation rate in the clone occurs at its greatest size (that is, 10,000 cells) and is [1 − (1 − 10−8)]10,000 ≈ 10−4 per locus per cell per generation. The mean mutation rate as the cell grows is much lower than this. Hence, the expected generation at which the second MMR mutation occurs (and at which the intrinsic mutation rate is raised) is much greater than 925 generations (10,000-cell size). It is most likely, therefore, that the clone will have reached 10,000 cells well before any cell has acquired a raised mutation rate. Hence, we expect that N2 > 104N1.
Thus, according to our model, the mutations that cause a tumor to progress probably do not occur in a cell with a raised intrinsic mutation rate. In effect, the selective advantage has more than compensated for the normal mutation rate. If one assumes different selective parameters, different normal and raised mutation rates, and different numbers of mutations required for the next round of clonal expansion to occur, the probability of a raised intrinsic mutation rate playing a role in tumorigenesis will also be different. Simulations (details not shown) suggest that if two mutations are required for the next wave of clonal expansion, a higher proportion, yet still a minority, of those mutations will occur in cells with a raised mutation rate. In general, the larger the selective parameter w, the smaller the increase in the mutation rate that the MMR mutations cause and the fewer the number of mutations required for the next stage of tumor growth, the lower will be the probability of the next clone having a raised intrinsic mutation rate.
The reasoning applied to the first wave of clonal expansion and the mutations needed to promote the expansion of the second clone also applies to subsequent clonal expansions. We emphasize the role of chance in determining when tumor-suppressor, oncogene, or DNA repair gene mutations occur and predict variation among tumors of the same type. The value of w may also vary from tissue to tissue and among individuals, depending on the tumor’s environment. Hence, some tumors may acquire a mutator phenotype. However, the model above certainly demonstrates that there is no requirement for an increased mutation rate in tumorigenesis: selection is both necessary and sufficient.
Can a Normal Mutation Rate Account for All the Mutations Observed in Cancers?
We believe that a normal mutation rate can explain the many mutations that actually occur in a cancer (16). Consider a tumor that has reached 108 cells in size. If the normal mutation rate is 10−8 per locus per cell per generation, then on average one cell in the tumor will have a mutation at any locus in that generation. Over several generations, a cell can acquire many mutations. These mutations will tend to spread throughout the cancer if they provide even a small selective advantage. Even if a mutation provides no advantage, however, it can still spread through the tumor cell population by genetic drift, especially if cell turnover is great.
Where a cancer has a generalized tendency to a specific type of mutation, it is likely that that tumor has acquired a mutator phenotype. Defects in MMR are well characterized in some cancers, and equivalent genetic defects might confer different mutator phenotypes, such as a tendency for loss of heterozygosity via mitotic recombination, chromosomal non-disjunction, or deletion.
Situations Favoring a Raised Mutation Rate in Tumors.
There seems little doubt that some sporadic cancers do have raised intrinsic mutation rates (18–20). Our models suggest four main ways in which this can occur. First, the raised mutation rate may have an inherited component, for example in heterozygote carriers of ataxia telangiectasia (ATM) (21) or Fanconi’s anemia mutations. Second, mutations which give a selective advantage will sometimes arise, by chance, in a cell that has acquired the necessary mutations for a raised intrinsic mutation rate. The stage at which the mutation rate becomes raised will vary from tumor to tumor (22, 23). Once the mutation rate is increased, the tumor will—all things being equal—evolve more rapidly than had the mutation rate been normal. Third, at some times during tumorigenesis, the number of cells in a tumor may be severely constrained by factors such as nutrient availability and blood supply. In addition, most of the mutations with large selective effects may already have occurred. Consequently, selection may be weak (low w) in these situations and only a slow increase in the numbers of any genotype will occur. In this case, the next mutation that confers a selective advantage is relatively likely to occur in a cell with a raised mutation rate (see above). Fourth, an increased intrinsic mutation rate may be a pleiotropic (or secondary) effect of a mutation primarily selected for its effects on cell proliferation or avoidance of death. If, for example, the level of free MMR proteins varies with the number of mismatches in the genome, these molecules might provide information about DNA damage to cell cycle pathways. Mutant MMR proteins might send incorrect signals to the cell cycle control, so that apoptosis does not occur. Other genes which might have these pleiotropic effects include p53 and ATM.
These scenarios for the role of a raised mutation rate assume that there is no selective disadvantage to a cell in having an increased number of mutations. This may not be the case: for example, a deleterious or lethal mutation may be much more likely than an advantageous mutation. More subtly, an accumulated mutational load might induce apoptosis (unless the tumor cell has escaped this control). Such factors will reduce the role of an increased intrinsic mutation rate in tumorigenesis. The normal mutation rate may actually be near-optimal in tumors, especially in the early stages. Later in tumor growth, controls such as those involving programmed cell death may have been circumvented and the tumor will have several mutations, each conferring a growth advantage over normal tissue: these factors might permit the disadvantages of a raised mutation rate to be tolerated relatively easily.
DISCUSSION
The process of tumorigenesis is a form of evolution: mutation and selection are the essential components of this process. The relative importance of these components remains controversial. Of current interest is the role of mutator phenotypes in cancer. Do cancers have mutations that raise the intrinsic mutation rate? Is a mutator phenotype necessary for carcinogenesis? We have used mathematical models of tumor growth to analyze the role of the mutation rate in carcinogenesis.
Our results show that selection is more likely than an increase in the intrinsic mutation rate to be the driving force of sporadic tumorigenesis. Most sporadic tumors start to grow with a normal mutation rate and probably continue to do so as successive mutations occur which provide a selective advantage and lead to clonal expansion. Although some tumors will acquire a mutator phenotype before they present clinically, it is not necessary to invoke an increased mutation rate to explain cancer, even in tumors which carry multiple mutations.
The evidence regarding when the intrinsic mutation rate becomes raised in tumors is conflicting. Boland et al. (24) suggested that loss of heterozygosity at specific loci occurs early in colorectal adenomas, but that widespread loss of heterozygosity was found in colorectal cancers. These data imply that the mutator phenotype does not occur early in colorectal tumorigenesis. Similarly, evidence suggests that the karyotypic instability seen in colorectal carcinomas is not an early event, occurring some time after the late adenoma/early carcinoma stage. Interestingly, Young et al. (23) found that microsatellite instability occurred at the colorectal adenoma-carcinoma boundary, although Shibata et al. (22) found microsatellite instability in early colorectal adenomas. Our own data (unpublished results) suggest that MMR defects do not occur before APC mutations in sporadic colorectal cancers.
The interpretation of data which suggest a mutator phenotype is complicated by the fact that microsatellite instability does not only result from mutations at MMR loci, but can, perhaps, result from increased cell turnover in the absence of mutations at DNA repair loci (25, 26). Many cancers with microsatellite instability may not therefore have true mutator phenotypes, but may have accumulated these microsatellite errors as a result of their high level of cell turnover. One striking piece of evidence is that DNA excision repair mutations do not appear to occur in sporadic cancers (27, 28). It would be expected that mutations in such genes would be as common as MMR mutations, given their comparable effects on the intrinsic mutation rate. There are several possible reasons for this disparity. One explanation is that MMR mutations occur in cancer in preference to excision repair mutations, because the former provide cells with a selective advantage in addition to their effects on the mutation rate.
We conclude by returning to the parallels between evolution and tumorigenesis. Evolutionary theory suggests that if the mutation rate is raised, evolution occurs more quickly. It does not follow that a raised mutation rate is required for evolution. Most sporadic tumors probably start to grow with a normal intrinsic mutation rate and subsequent clonal expansion may also occur with the mutation rate at normal levels. Some tumors may acquire a mutator phenotype during tumorigenesis, but this is not a necessary accompaniment to tumor initiation or progression.
Footnotes
Abbreviations: MMR, mismatch repair; HNPCC, hereditary nonpolyposis colorectal cancer.
References
- 1.Armitage P, Doll R. Br J Cancer. 1954;8:1–12. doi: 10.1038/bjc.1954.1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Armitage P, Doll R. Br J Cancer. 1957;11:161–169. doi: 10.1038/bjc.1957.22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Fisher J C. Nature (London) 1958;181:651–652. doi: 10.1038/181651b0. [DOI] [PubMed] [Google Scholar]
- 4.Cairns J. Nature (London) 1975;255:197–200. doi: 10.1038/255197a0. [DOI] [PubMed] [Google Scholar]
- 5.Knudson A G. J Cancer Res Clin Oncol. 1996;122:135–140. doi: 10.1007/BF01366952. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Moolgavkar S H, Knudson A J. J Natl Cancer Inst. 1981;66:1037–1052. doi: 10.1093/jnci/66.6.1037. [DOI] [PubMed] [Google Scholar]
- 7.Moolgavkar S H. Annu Rev Public Health. 1986;7:151–169. doi: 10.1146/annurev.pu.07.050186.001055. [DOI] [PubMed] [Google Scholar]
- 8.Loeb L A, Cheng K C. Mutat Res. 1990;238:297–304. doi: 10.1016/0165-1110(90)90021-3. [DOI] [PubMed] [Google Scholar]
- 9.Loeb L A. Cancer Res. 1991;51:3075–3079. [PubMed] [Google Scholar]
- 10.Tomlinson I P M, Bodmer W F. Proc Natl Acad Sci USA. 1995;92:11130–11134. doi: 10.1073/pnas.92.24.11130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Lane D P. Nature (London) 1992;358:15–16. doi: 10.1038/358015a0. [DOI] [PubMed] [Google Scholar]
- 12.Karran P. Science. 1995;268:1857–1858. doi: 10.1126/science.7604258. [DOI] [PubMed] [Google Scholar]
- 13.Liu B, Nicolaides N C, Markowitz S, Willson J K, Parsons R E, Jen J, Papadopolous N, Peltomaki P, de la Chapelle A, Hamilton S R, Kinzler K W, Vogelstein B. Nat Genet. 1995;9:48–55. doi: 10.1038/ng0195-48. [DOI] [PubMed] [Google Scholar]
- 14.Kaufman W K. Cancer Metastasis Rev. 1995;14:31–41. doi: 10.1007/BF00690209. [DOI] [PubMed] [Google Scholar]
- 15.Morgan W F, Murnane J P. Cancer Metastasis Rev. 1995;14:49–58. doi: 10.1007/BF00690211. [DOI] [PubMed] [Google Scholar]
- 16.Christians F C, Newcomb T G, Loeb L A. Prevent Med. 1995;24:329–332. doi: 10.1006/pmed.1995.1054. [DOI] [PubMed] [Google Scholar]
- 17.Karp J E, Broder S. Nat Med. 1995;1:309–320. doi: 10.1038/nm0495-309. [DOI] [PubMed] [Google Scholar]
- 18.Carder P, Wyllie A H, Purdie C A, Morris R G, White S, Piris J, Bird C C. Oncogene. 1993;8:1397–1401. [PubMed] [Google Scholar]
- 19.Bhattacharyya N P, Skandalis A, Ganesh A, Groden J, Meuth M. Proc Natl Acad Sci USA. 1994;91:6319–6323. doi: 10.1073/pnas.91.14.6319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Harwood J, Tachibana A, Davis R, Bhattacharyya N P, Meuth M. Hum Mol Genet. 1993;2:165–171. doi: 10.1093/hmg/2.2.165. [DOI] [PubMed] [Google Scholar]
- 21.Savitsky K, Bar S A, Gilad S, Rotman G, Ziv Y, et al. Science. 1995;268:1749–1753. doi: 10.1126/science.7792600. [DOI] [PubMed] [Google Scholar]
- 22.Shibata D, Peinado M A, Ionov Y, Malkhosyan S, Perucho M. Nat Genet. 1994;6:273–281. doi: 10.1038/ng0394-273. [DOI] [PubMed] [Google Scholar]
- 23.Young J, Leggett B, Gustafson C, Ward M, Searle J, Thomas L, Buttenshaw R, Chenevix T G. Hum Mutat. 1993;2:351–354. doi: 10.1002/humu.1380020505. [DOI] [PubMed] [Google Scholar]
- 24.Boland C R, Sato J, Appelman H D, Bresalier R S, Feinberg A P. Nat Med. 1995;1:902–909. doi: 10.1038/nm0995-902. [DOI] [PubMed] [Google Scholar]
- 25.Fleischhacker M, Lee S, Spira S, Takeuchi S, Koeffler H P. Mod Pathol. 1995;8:360–365. [PubMed] [Google Scholar]
- 26.Spandidos D A, Ergazaki M, Arvanitis D, Kiaris H. Biochem Biophys Res Commun. 1996;220:137–140. doi: 10.1006/bbrc.1996.0370. [DOI] [PubMed] [Google Scholar]
- 27.Chalmers A H, Lavin M, Atisoontornkul S, Mansbridge J, Kidson C. Cancer Res. 1976;36:1930–1934. [PubMed] [Google Scholar]
- 28.Hatton D H, Mitchell D L, Strickland P T, Johnson R T. Cancer Res. 1995;55:181–189. [PubMed] [Google Scholar]